
Abstract
Detachment of photoreceptors from the retinal pigment epithelium is seen in various retinal disorders, resulting in photoreceptor death and subsequent vision loss. Cell death results in the release of endogenous molecules that activate molecular platforms containing caspase-1, termed inflammasomes. Inflammasome activation in retinal diseases has been reported in some cases to be protective and in others to be detrimental, causing neuronal cell death. Moreover, the cellular source of inflammasomes in retinal disorders is not clear. Here, we demonstrate that patients with photoreceptor injury by retinal detachment (RD) have increased levels of cleaved IL-1β, an end product of inflammasome activation. In an animal model of RD, photoreceptor cell death led to activation of endogenous inflammasomes, and this activation was diminished by Rip3 deletion. The major source of Il1b expression was found to be infiltrating macrophages in the subretinal space, rather than dying photoreceptors. Inflammasome inhibition attenuated photoreceptor death after RD. Our data implicate the infiltrating macrophages as a source of damaging inflammasomes after photoreceptor detachment in a RIP3-dependent manner and suggest a novel therapeutic target for treatment of retinal diseases.
Similar content being viewed by others

Main
Retinal detachment (RD) is characterized as the separation of neurosensory retina from retinal pigment epithelium (RPE), and subsequent photoreceptor cell death is the major cause of blindness in age-related macular degeneration,1 diabetic retinopathy,2 and rhegmatogenous RD (RRD).3 The outer layers of the retina, including photoreceptors, depend on the choroidal vasculature and choriocapillaris to regulate their overall metabolic homeostasis. Once RD occurs, photoreceptor cells cannot receive sufficient metabolic supply for survival and begin to undergo apoptosis and necrosis.3, 4, 5, 6, 7 Receptor-interacting protein 3 (RIP3) has been identified as a key regulator of programmed necrosis.8, 9 Recently, we showed that RD induces RIP3-dependent programmed necrosis.5 The RIP3-dependent necrotic pathway is not only critical for the development of various retinal diseases,5, 10, 11 but also for several systemic diseases such as acute pancreatitis and antiviral immunity.8, 9, 12 Under the specific conditions where caspase-8 is inactivated, necrotic stimuli such as tumor necrosis factor α (TNFα), Fas ligand, and Toll-like receptor (TLR) trigger the formation of a complex called the necrosome, containing receptor-interacting protein 1 (RIP1), RIP3, and mixed lineage kinase domain-like protein (MLKL).8, 9, 13, 14, 15 Ultimately, upon phosphorylation of MLKL by RIP3, oligomerized MLKL is inserted into plasma membranes, resulting in membrane rupture and necrotic cell death.16, 17, 18 Additionally, it is known that dead cells release endogenous molecules, such as high mobility group box 1 (HMGB1), ATP, and uric acid.19, 20, 21, 22 These molecules, termed damage-associated molecular patterns (DAMPs), can alert the innate immune response through pattern-recognition receptors. It should be noted that some DAMPs such as HMGB1 are released only from necrotic cells and not from apoptotic cells.19
The inflammasome is a molecular platform that regulates the activation of caspase-1, which leads to the cleavage of interleukin-1β (IL-1β).23, 24, 25 Recent studies have revealed that inflammasomes are stimulated by DAMPs (such as ATP and uric acid crystals) through the NOD-like receptor family, pyrin domain containing-3 (NLRP3) protein.26, 27 Once NLRP3 is activated, it oligomerizes with apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and pro-caspase-1, followed by the autocleavage of caspase-1.
It is reported that necrotic cells, not apoptotic cells, trigger the NLRP3 inflammasome.28 Other recent reports show evidence that DAMPs such as HMGB1 and histones are increased in the vitreous of RD patients.29, 30 Recently, endogenous inflammasome activation in retinal disorders has been implicated as either protective or detrimental to neuronal cell death.31, 32, 33, 34 In this study, we investigated whether inflammasomes are induced in mouse and human eyes with photoreceptor detachment, and whether loss of RIP3 affects inflammasome activation in a mouse model of RD. We also characterized the cellular origin of IL-1β production during RD. IL-1β is known as a central mediator of the immune response against acute and chronic diseases, but there is still debate on whether it is a neurotoxic or neuroprotective agent in vivo.32, 34 Here, we revealed that inhibition of IL-1β by caspase-1 inhibitor, neutralization of IL-1β, and NLRP3 depletion can protect against photoreceptor cell death after RD.
Results
RD triggers the activation of inflammasomes, which is diminished by Rip3 deletion
We previously demonstrated that RD leads to both caspase-dependent apoptosis and RIP3-dependent necrosis of photoreceptor cells, and that Rip3 deletion diminishes photoreceptor cell death during RD.5 Furthermore, necrotic cells (rather than apoptotic cells) have been reported to be the major triggers of the NLRP3 inflammasome.35 Therefore, we investigated whether photoreceptor cell death during RD is associated with activation of inflammasomes in humans and in a mouse model of RD.
In human eyes with RD, various cytokines have been reported to be increased in the subretinal or vitreous fluid.36, 37, 38 However, alterations of the inflammasome cytokine IL-1β have not been previously investigated. We assessed the subretinal fluid and the vitreous of patients in the early phase of RRD and the vitreous of control patients (macular hole and vitreomacular traction syndrome) (Table 1). The High Sensitivity ELISA kit (eBioscience) enabled us to detect extremely low concentrations of IL-1β (such as 0.05 pg/ml) in samples compared with the general ELISA kit (Supplementary Figure S1). The median of IL-1β concentration in the subretinal fluid of RRD patients (0.25 pg/ml, range: 0.10–1.97 pg/ml) was significantly higher than the vitreous fluid of control patients (0.02 pg/ml, range: 0.01–0.03 pg/ml) (Figure 1). IL-1β in the vitreous fluid of RRD patients (0.06 pg/ml, range: 0.001–0.21 pg/ml) was also elevated, but not to the levels of the subretinal fluid of RRD patients, probably due to dilution. These data suggest that RD triggers upregulation of IL-1β in human eyes, and that IL-1β is concentrated in the subretinal space. This elevation was somewhat specific, since TNFα showed no difference among the three groups in accordance with a previous report in human eyes (Supplementary Figure S2a).39 In contrast, IL-6 was elevated both in the subretinal fluid and in the vitreous of RRD patients (Supplementary Figure S2b) in accordance with a previous report.36

Concentration of IL-1β in human eyes with rhegmatogenous retinal detachment (RRD). IL-1β levels in vitreous fluid of control eyes (n=7) versus subretinal fluid (SRF) of eyes with RRD (n=8) and vitreous fluid of eyes with RRD (n=8). Horizontal line indicates median in each group. ***P<0.001
To further characterize the activation of inflammasomes in photoreceptor injury, we examined the time course of IL-1β levels in tissues containing subretinal fluid, retina, RPE, and choroid in a mouse model of RD (Figure 2a). IL-1β was increased 4.6-fold as early as 6 h after RD, compared with control eyes, and peaked at 12 h. After 24 h, the level of IL-1β declined to some extent, although it remained significantly higher (3.4 times) than control eyes. In the same samples, IL-6 was significantly upregulated, whereas vascular endothelial growth factor (VEGF) was modestly elevated (Supplementary Figures S3a and b). TNF was below the detection limit.

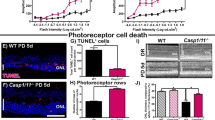
Retinal detachment (RD) induces activation of IL-1β, which is diminished in Rip3-deficient mice. (a) The data show IL-1β levels detected by ELISA at 0 h (non-treated eyes, n=6) and from 6 to 120 h after induction of RD. n=6–9. (b) Western blotting image shows representative data of cleaved IL-1β and β-actin in wild-type (WT) and Rip3−/− mouse eyes after RD. (-): non-treated eyes. (c) Densitometry analyses of the western blotting data of (b) normalized to the intensity of β-actin (n=4). Data are presented as mean±s.d. *P<0.05, **P<0.01, ***P<0.001
Since ELISA cannot reliably distinguish cleaved IL-1β from pro IL-1β, we used western blotting to better assess levels of cleaved IL-1β. As seen in Figures 2b and c, RD induced the production of cleaved IL-1β with a peak at 24 h in both wild-type (WT) and Rip3−/− mice, although Rip3−/− mice had significantly lower cleaved IL-1β compared with WT mice (69.4 and 64.8% less at 12 and 24 h, respectively) (Figures 2b and c). In contrast to the significant changes in the mature form of IL-1β, we did not detect significant differences in the protein levels of pro IL-1β or NLRP3 between WT and Rip3−/− mice at each time point (Figures 2b and c) despite modest changes in mRNA level at 12 h after RD (Supplementary Figures S4a and b), suggesting that RIP3 has a limited role in priming pro IL-1β after RD.
Similar to Il1b, RD also led to upregulation of Tnfa and Il6 mRNA (Supplementary Figures S4c and d). Rip3 deficiency did not affect caspase-3 activation, as cleaved caspase-3 tended to increase in both WT and Rip3−/− mice at 24 h after RD to a similar extent (Supplementary Figures S5a and b).
Taken together, these results demonstrate that inflammasomes are activated after photoreceptor separation from the RPE in both human and mouse eyes, and that Rip3 deficiency inhibits inflammasome activation directly or indirectly.
Infiltrating subretinal cells produce IL-1β
In many retinal degenerative diseases, including RD, macrophages infiltrate the diseased tissue in response to chemokines such as monocyte chemoattractant protein 1 (MCP-1).40, 41 It is generally thought that IL-1β is produced and secreted by monocytes/macrophages, and recent studies have revealed that other non-immune cells may contain NLRP3 and activate NLRP3 inflammasomes.31, 42 Thus, we assessed the expression of Il1b mRNA via laser capture microdissection (LCM) in the outer nuclear layer (ONL), which contains the photoreceptor cell bodies; the subretinal space, where macrophages are mostly seen during RD; and the RPE/choroid complex. Each part of the eye was microdissected as indicated in Figure 3a. In non-RD eyes, there was no Il1b mRNA expression detected in either the ONL or the RPE/choroid complex (Figure 3b). RD led to a significant increase of Il1b mRNA expression in the subretinal space compared with the ONL. Although the sample from the RPE/choroid complex in RD eyes also showed an increase in Il1b mRNA, it was not statistically significant. These data suggest that IL-1β largely originates from cells migrating into the subretinal space during RD.

IL-1β is produced by the cells migrating into subretinal space. (a) ONL, subretinal space, and RPE including choroid were circled as indicated in the left column (red circles), followed by cutting with LCM. The right column shows the images after cutting out the samples from the sections. Nuclei were stained with Toluidine blue. INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium. Scale bars: 100 μm. (b) The graph shows the Il1b mRNA expression in each layers with (+) RD (n=12) or without (−) RD (n=6). ND, not detectable. Data are presented as mean±s.d. *P<0.05
Macrophage recruitment in the detached retina is not altered in Rip3−/− mice
To assess whether inhibition of cleaved IL-1β in Rip3−/− mice during RD is due to decreased macrophage recruitment to the site of detachment, we analyzed the numbers of infiltrating macrophages labeled with an anti-CD11b antibody in the detached retina, including the ONL and the subretinal space (Figures 4a and b). Macrophage infiltration through the entire subretinal space was noted as early as 12 h after RD, and peaked at 24 h. Interestingly, the number of macrophages in Rip3−/− mice was not significantly lower than WT mice, suggesting that the decrease in inflammasomes and cleaved IL-1β is not due to impaired macrophage recruitment in Rip3−/− mice after RD.

Macrophage recruitment is not altered in Rip3−/− mice. (a) The images show macrophages stained with CD11b (green) that have migrated into the subretinal space after 12 and 24 h in WT mice and Rip3−/− mice. Nuclei staining: TO-PRO-3 (blue). Scale bar: 50 μm. (b) The number of macrophages in the subretinal space at 12 and 24 h after RD (n=6). (-): non-treated eyes. Data are presented as mean±s.d. *P<0.05
Inhibition of IL-1β diminishes photoreceptor cell death through NLRP3 inflammasome after RD
The exact role of IL-1β in neuronal diseases is still unclear. In the ischemic injury model, inhibition of IL-1β is effective in preventing neuronal damage.32 In contrast, inhibition of IL-1β does not affect photoreceptor cell loss in the light injury model.34 Thus, we examined whether cleaved IL-1β exacerbates photoreceptor cell death during RD. The caspase-1 inhibitor Ac-Tyr-Val-Ala-Asp-chloromethylketone (YVAD) was administered to the detached lesion to reduce production of cleaved IL-1β, and the efficiency of inhibition was determined by western blotting (Figures 5a and b). YVAD treatment dampened the production of cleaved IL-1β by 61.9% compared with eyes treated with vehicle. Next, we analyzed the effect of YVAD treatment during RD using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) as a marker of cell death. The number of TUNEL-positive cells in the ONL was reduced by 55.3% in WT mice treated with YVAD compared with WT mice treated with vehicle at 24 h after RD (Figures 5c and d). The efficiency of YVAD treatment was similar to that observed in Rip3−/− mice at 12 and 72 h after RD (Figure 6d).

Inhibition of caspase-1 reduces photoreceptor cell death after RD. (a) Inhibitor of caspase-1 (YVAD) was administered into the subretinal space, and cleaved IL-1β was measured with western blotting to evaluate the efficiency of caspase-1 inhibition. (b) Densitometry analyses of the western blotting data of (a) normalized to the intensity of β-actin (n=4). (c) TUNEL staining (green) in WT and Rip3−/− mouse retinal sections. The eyes were treated with vehicle and YVAD and analyzed at 24 h after RD. Nuclei staining: TO-PRO-3 (blue), scale bar: 50 μm. (d) Quantification of TUNEL-positive cells in the ONL after RD. n=4–10 at each groups. (a and b) Data are presented as mean±s.d. *P<0.05, **P<0.01

Inhibition of IL-1β through Nlrp3 depletion or a neutralizing antibody diminishes photoreceptor cell death after RD. (a) Cleaved IL-1β was assessed at 24 h after RD in WT and Nlrp3−/− mice eyes with western blotting. (b) Densitometry analyses of the western blotting data of (b) normalized to the intensity of β-actin (n=4). (c and d) TUNEL staining (green) in WT and Nlrp3−/− mouse retinal sections at 24 h after RD (c) and quantification of TUNEL-positive cells in the ONL (d). WT: n=8, Nlrp3−/− mice: n=7. (e and f) Neutralizing anti-IL-1β antibody (10 and 100 μg/ml) and control IgG were administered into subretina and analyzed at 24 h after RD. TUNEL staining (green) in mouse retinal sections treated with antibody (e) and quantification of TUNEL-positive cells in the ONL (f). n=8. Nuclei staining: TO-PRO-3 (blue), scale bar: 50 μm. Data are presented as mean±s.d. *P<0.05, **P<0.01
Since YVAD inhibits caspase-4/11 in addition to caspase-1 (albeit to a lesser extent), we wanted to further investigate the role of IL-1β in RD-induced photoreceptor cell death by using a neutralizing anti-IL-1β antibody and Nlrp3−/− mice. As seen in Figures 6a and b, neutralizing anti-IL-1β antibody administered into the subretinal space (10 and 100 μg/ml) resulted in significantly reduced numbers of TUNEL-positive cells compared with control IgG (100 μg/ml). Although we used a high dose of neutralizing antibody, it is still possible that we failed to completely neutralize all IL-1β activity. For this reason, we used Nlrp3−/− mice, which showed almost complete lack of cleaved IL-1β compared with WT mice after RD (Figures 6c and d). Similar to YVAD treatment and IL-1β-neutralizing antibody, Nlrp3 deletion led to a significant but not complete reduction in TUNEL-positive cells compared with WT mice at 24 h after RD (Figures 6e and f). These data suggest that NLRP3 is important for inflammasome activation and production of IL1-β after RD, and that inhibition of cleaved IL-1β protects photoreceptors partially but not completely from RD induced cell death. These data indicate that more mechanisms in addition to inflammasome are important for overall photoreceptor cell loss after RD.
Discussion
In this study, we demonstrate that inflammasome activation takes place in mouse and human eyes after RD, and is an important mediator of photoreceptor cell loss. We further show that Rip3 deletion attenuates inflammasome activation in a mouse model of RD, and that the major sources of inflammasomes appear to be infiltrating macrophages rather than dying photoreceptors (summarized in Figure 7).

Model of mechanism in RD-induced photoreceptor cell death. After photoreceptor separation from RPE, caspase and RIPK mediated cell death ensues as well as release of inflammatory mediators and DAMPs. Ensuing macrophage recruitment results in inflammasome activation and release of IL-1b. Rip3 deletion attenuates both directly and indirectly inflammasome activation. Neutralization of IL-1b or inhibition of inflammasome activation partially diminishes cell death
In human eyes with RRD, liquefied vitreous fluid passes into the potential subretinal space (newly formed space between photoreceptors and RPE) through the retinal breaks. This separation of the photoreceptors from the RPE by the subretinal fluid results in photoreceptor cell death in part due to lack of oxygenation and other trophic factors.3, 43 Since subretinal fluid does not occur in normal eyes, we could only compare the levels of inflammatory cytokines in the subretinal fluid of RRD patients with levels in the vitreous of control patients. Our data showed significantly higher concentrations of IL-1β in the subretinal fluid of RRD patients compared with the vitreous fluid of control patients. Although IL-1β in the vitreous fluid of RRD patients was higher than that of control patients, IL-1β in the vitreous fluid of RRD patients was not as high as the subretinal fluid of RRD patients, suggesting that IL-1β is primarily concentrated in the subretinal space after RD, rather than the vitreous fluid in RD patients. There is a lack of published studies that have examined IL-1β in the phase of photoreceptor cell death after RD. A study that examined late scarring after RD (a late complication) found no relationship between IL-1β and the development of proliferative vitreoretinopathy (scarring) after RD.38 Thus, upregulation of IL-1β in the subretinal fluid appears to be an early rather than late inflammatory cytokine signal after RD.
IL-1β is an important proinflammatory mediator produced at sites of injury or infection to mediate the immune system’s cytokine network and mount an appropriate cellular response. Upregulation of IL-1β in the central nervous system (CNS) after ischemic and traumatic brain injury has been reported in animals.44, 45 Increased IL-1β is also reported in ischemic retinal injury and RD models in rats.32, 41 In this study, we found an increase in both pro- and cleaved IL-1β during the early phase of RD in the mouse model. Total (pro- and cleaved) IL-1β levels were observed to peak 12 h after RD, whereas the mature cleaved form of IL-1β peaked later, at 24 h. The kinetics of pro- and mature IL-1β follows the time course of photoreceptor cell death after RD, peaking at 24 h. Synthesis of pro-IL-1β is stimulated through TLRs, TNF receptor, or IL-1 receptor. Prior studies have shown TNFα to peak early (6 h) after RD,41 suggesting that TNFα may be involved in priming IL-1β. Following priming of IL-1β, production of cleaved mature IL-1β occurs through activation of inflammasomes by DAMPs released from dead cells. In contrast to previous reports by us and others,4, 46, 47, 48 our new modified model of RD49, 50, 51 exhibits the peak of photoreceptor cell death around 24 h after RD, likely due to a larger and more consistent volume of RD compared with previous studies. Thus, it is reasonable that the peak of mature IL-1β appears close to the peak of photoreceptor cell death (at 24 h).
We found that Rip3−/− mice show attenuated inflammasome activation compared with WT mice during RD. There are various explanations for this. One possibility is lower protein synthesis of both pro-IL-1β and NLRP3 in Rip3−/− mice. However, despite modest changes at the early time point of mRNA expressions for IL-1β and Nlrp3 in Rip3−/− mice, the protein levels of pro-IL-1β and NLRP3 were not altered. Alternatively, different amount of DAMPs may be released from dying photoreceptor cells during RD between WT mice and Rip3−/− mice. Our group previously reported that Rip3-deficient mice undergo relatively less photoreceptor cell death than WT mice.5 We therefore expect that DAMPs, which activate inflammasomes, will also be lower in Rip3−/− mice compared with WT mice. A third explanation is that RIP3 deletion in macrophages might affect inflammasome activation. Recent in vitro evidence suggests that there are RIP3-dependent pathways that activate the NLRP3 inflammasome under specific conditions, such as depletion of inhibitor of apoptosis proteins (IAPs) or caspase-8 deficiency.52, 53 However, more research is necessary to understand the interaction between inflammasomes and the RIP3 pathway.
Most inflammasome studies have been focused on the myeloid lineage, such as monocyte/macrophages, dendritic cells, and neutorophils. However, there are several reports that non-myeloid cells such as the RPE and keratinocytes also activate inflammasomes.31, 42, 54 By means of LCM, we demonstrated that the major origin of IL-1β in RD is not from the dying photoreceptors cells but rather from the CD11b-positive cells migrating into the subretinal space after RD. Kawano et al.30 showed evidence that DAMPs increase after RD; furthermore, they showed that histones, one class of DAMPs, are preferentially located in the subretinal space, which supports our findings of inflammasome activation in subretinal macrophages after RD. Several studies have shown that macrophages are recruited to the subretinal space through Bruch’s membrane and the RPE during RD.55, 56 Our results showed a trend of increasing in Il1b mRNA expression in the RPE/choroid complex after RD, though there was no significant difference compared with the ONL and subretinal space. Recent studies have shown that the RPE also activates the NLRP3 inflammasome and induces IL-1β.31, 42 It is unclear how much of the Il1b mRNA in the RPE/choroid complex is derived from the RPE itself compared with the choroid, since the choroid contains circulating leukocytes in the choriocapillaris, which may serve as a potential source of Il1b. Moreover, there are macrophages migrating from the choroid toward the subretinal space through the RPE during RD.55 Results from in vitro experiments performed by our group have shown that the production of mature IL-1β from RPE cells is several orders of magnitude less than macrophages (Supplementary Figures S6a and b).
In the CNS, IL-1β shows both neurotoxicity and neuroprotection, depending on cell type, microenvironment, and cytokine concentrations.57, 58, 59 As in the CNS, the role of IL-1β in retinal diseases differs in various models. IL-1β prevents photoreceptor cell death or at least induces no toxicity in the light injured retina model.34, 60 In contrast, IL-1β is neurotoxic to photoreceptor cells in the retinal ischemic model, as an IL-1 receptor antagonist confers neuroprotection against glutamate toxicity.32 Under hypoxic or hypoglycemic stress, IL-1β induces neuronal cell death in mixed cortical cell cultures, while there is no effect during normoxia.61 During RD, after separation from the RPE, photoreceptors are in a relatively hypoxic and hypoglycemic state.6 In our model of RD, caspase-1 inhibitor YVAD suppressed IL-1β production and diminished cell death, although YVAD is also know to inhibit (albeit to a lesser extend) caspase-4/11. For this reason we used a neutralizing anti-IL-1β antibody, resulting in the suppression of photoreceptor cell death after RD. Although we used a high dose of neutralizing IL-1β antibody, it is still possible that we might have failed to completely neutralize all IL-1β. Thus, we employed Nlrp3−/− mice, which showed almost complete lack of IL-1β after RD. These Nlrp3−/− mice, similar to the mice treated with caspase-1 inhibitor or IL-1β neutralizing antibody showed reduced but not complete inhibition of cell loss after RD. Taken together, these data suggest that IL-1β contributes partially but not completely to photoreceptor cell death after RD. This is in contrast to previous studies that have shown that administration of IL-1β into the subretinal space does not increase photoreceptor cell death during RD.41 This could be a result of endogenous IL-1β signaling having already reached maximum levels, saturating its effectors, and thus the addition of exogenous IL1-β may no longer lead to additional detrimental effect.
In contrast to caspase-1 inhibition in this study, previous reports utilizing pan-caspase inhibitor did not show significant protection for photoreceptor cell death after RD.5, 46 This may be related to the fact that inhibition of certain caspases lead to activation of alternative cell death pathways. It has been reported that when caspase-8 is inhibited, the RIP3-dependent necrotic pathway is more activated than the apoptotic pathway.8, 9, 62
The composition of subretinal fluid differs between human RRD and animal models of RD. In humans, subretinal fluid is primarily liquefied vitreous, serum transudate, and contribution from aqueous humor,63, 64 whereas in the animal model a more viscous fluid (1% sodium hyaluronate) is introduced. Sodium hyaluronate is the predominant glycosaminoglycan of human vitreous, and the concentration of hyaluronan in adult human vitreous has reported to be between 65 and 400 μg/ml (~0.0065–0.04%).65 In spite of these differences, we found similarities in the inflammatory responses such as activation of IL-1β and macrophage infiltration between human RD and the animal model.66, 67
In summary, our study shows for the first time that photoreceptor injury by separation from RPE, as seen in many human retinal diseases, induces inflammasomes in a RIP3-dependent manner. It also shows for the first time that IL-1β production is mainly derived from migrating macrophages and not from dying photoreceptors. We also demonstrate that inhibition of IL-1β maturation by several mechanisms (caspase-1 inhibition, neutralizing antibody, and NLRP3 deletion) can partially protect photoreceptor cell death after RD, suggesting a novel therapeutic target for treatment of retinal and other neuronal diseases.
Materials and Methods
Patients and high sensitivity ELISA
The research protocol followed the tenets of the Declaration of Helsinki and was approved by the Nagoya University Hospital Ethics Review Board. All enrolled patients provided written informed consent before the surgical procedure. All samples were collected at Nagoya University Hospital during surgical treatments. Subretinal fluid samples were collected during scleral buckling surgery from patients with primary RRD by making a small incision of the sclera and choroid with diathermy coagulation as a routine surgical procedure. The vitreous samples from primary RRD patients and control vitreous samples from patients with either macular hole or vitreomacular traction syndrome were collected by 25-gauge pars plana vitrectomy without dilution at the start of vitrectomy. All the samples were immediately stored at −80 °C. The subretinal fluid and vitreous samples were spun down at 17 000 × g for 5 min, followed by the measurement of IL-1β, TNFα, and IL-6 with High Sensitivity ELISA (eBioscience, San Diego, CA, USA) according to the manufacturer’s protocol.
Animal models
All experimental procedures and research protocols were approved by the Animal Care Committee of the Massachusetts Eye and Ear Infirmary. C57BL/6 mice were purchased from Charles River Laboratories International, Inc. (Wilmington, MA, USA). Rip3−/− mice were provided by DM Dixit (Genentech).5 Nlrp3−/− mice and their control mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). RD was created in the eyes of 6- to 8-week-old mice, according to the protocol described previously with slight modifications.49 In brief, 4 μl of sodium hyaluronate (Provisc; Alcon, Fort Worth, TX, USA) was injected into the subretinal space through the sclera and choroid of mouse eyes. The scleral wound was sealed with octyl/butyl cyanoacrylate (GLUture; Abbott Laboratories, Irving, TX, USA) to prevent leakage of sodium hyaluronate. For inhibition of caspase-1, Ac-Tyr-Val-Ala-Asp-chloromethylketone (YVAD; Sigma-Aldrich, St. Louis, MO, USA) was diluted with sodium hyaluronate to a final concentration of 300 μM. For neutralization of IL-1β, anti-mouse IL-1β antibody and control IgG (AF-401-NA and AB-108-C, R&D Systems, Minneapolis, MN, USA) were diluted with sodium hyaluronate to a final concentration as indicated. The eyes were collected at each time point as indicated, with western blotting, ELISA, and TUNEL staining performed as per the protocols outlined below.
ELISA for mouse experiments
To prepare sample lysates, the mouse eyes were lysed with M-PER lysis buffer (Thermo Scientific, Rockford, IL, USA) containing Halt Phosphatase Inhibitor Cocktail (Thermo scientific) and cOmplete Protease Inhibitor Cocktail (Roche, Indianapolis, IN, USA) after removing the cornea, lens, and iris. Lysates were spun down at 17 000 × g for 5 min, and mouse IL-1β, TNFα, IL-6, and VEGF in the lysates were measured with ELISA kits (MLB00C, MTA00B, M6000B, and MMV00; R&D Systems), according to the manufacturer’s protocol.
Western blotting
In all, 20 mM HEPES buffer (Sigma-Aldrich) containing Halt Phosphatase Inhibitor Cocktail and cOmplete Protease Inhibitor Cocktail was used as a lysis buffer. The eyes (with the cornea, lens, and iris removed) were homogenized and ultracentifuged for 60 min at 50 000 × g to collect soluble proteins as supernatants. The samples were then electrophoresed through NuPAGE 4-12% Bis-Tris Gels (Life Technologies, Grand Island, NY, USA), and transferred onto PVDF membranes. The following antibodies were used: rat anti-mouse IL-1β (MAB4011; R&D Systems), rabbit anti-NLRP3 (5447; ProSci, Poway, CA, USA), rabbit anti-cleaved caspase-3 (9661; Cell Signaling Technology, Danvers, MA, USA), and rabbit anti-β-actin (4970; Cell Signaling Technology). The images of the blots were taken with ChemiDoc MP and analyzed with Image Lab Software version 4.1 (Bio-Rad Laboratories, Hercules, CA, USA).
Laser capture microdissection
LCM was performed with the Leica LMD7000 system and LMD application version 7.5 (Leica Microsystems, Buffalo Grove, IL, USA). Enucleated mouse eyes were frozen in O.C.T Compound (Sakura Finetek, Torrance, CA, USA) and cut into 20 μm sections on Frame slides (Leica Microsystems). The sections were fixed with 75% ethanol for 30 s, washed with nuclease-free water (Life Technologies) for 30 s, stained with 0.02% toluidine blue solution (Sigma-Aldrich) for 20 s, and washed with nuclease-free water for 30 s. The sections were then dehydrated with 75 and 95% ethanol for 30 s each, and twice with 100% ethanol for 30 s. The tissues from the ONL, subretina (where the sensory retina detached from the RPE), and the RPE (with choroid) were cut by laser and collected into 0.5 ml tubes containing RNAlater (Life Technologies).
RNA extraction and RT-PCR
RNA extraction was performed with RNeasy plus micro kit for the samples collected with LCM and RNeasy mini kit for the whole mouse eye samples excluding the cornea, lens, and iris (Qiagen, Valencia, CA, USA) according to manufacturer’s protocol. cDNA was synthesized with SuperScript III Reverse Transcriptase and Oligo(dT) Primer (Life Technologies). Real-time PCR was performed by StepOnePlus Real-Time PCR System (Life Technologies) and analyzed with StepOne Software ver. 2.0 (Grand Island, NY, USA). Reactions were performed with TaqMan Fast Universal PCR Master Mix and primers (18s rRNA: Mm03928990_g1; Il1b: Mm00434228_m1; Nlrp3: Mm00840904_m1; Tnfa: Mm99999068_m1; Il6: Mm00446190_m1, Life Technologies). The relative quantity of mRNA expression was calculated by ΔΔCT method normalized to 18s rRNA as endogenous control.
TUNEL staining and immunohistochemistry
Mouse eyes frozen in O.C.T Compound were cut into 8 μm sections. TUNEL staining was performed with ApopTag Fluorescein In Situ Apoptosis Detection Kit (S7110; EMD Millipore, Billerica, MA, USA) according to the manufacturer’s protocol. For immunohistochemistry, the frozen sections were postfixed with 4% paraformaldehyde for 15 min, blocked with 5% goat serum in phosphate-buffered saline (PBS). They were then incubated with rat anti-CD11b antibody (BD Biosciences, San Jose, CA, USA) at 4 °C overnight, followed by incubation with Alexa Fluor 488-conjugated goat anti-rat IgG antibody (Life Technologies) and TO-PRO-3 Iodide (Life Technologies) for 30 min. All images obtained by AxioVision microscope (Zeiss, Chester, VA, USA) were masked and randomized before analysis with ImageJ 1.44o (NIH, Bethesda, MD, USA). TUNEL-positive cells in the center of the detached retina were counted from at least four sections per eye and adjusted to the numbers of cells per 1 mm2 of the ONL. The CD11b-positive cells were counted in areas measuring 500 μm × 500 μm that included the ONL and subretinal space in the center of the detached retina from two retinal sections per eye.
Statistical analyses
All statistical analyses were analyzed with GraphPad Prism 5.0c (GraphPad software, La Jolla, CA, USA). For mouse experiments, one-way ANOVA followed by Dunnett’s multiple comparison test (Figure 2a, compared with the data at 0 h), one-way ANOVA followed by Tukey’s test (Figures 3b, 5b, d, and 6b) and Student’s t-test (Figures 2c, 4b, 6d and f) were performed. For human samples, Kruskal–Wallis test was used for Figure 1. Kruskal–Wallis test, Fisher’s exact test, and Mann–Whitney U test were performed as indicated in Table 1, since the human data were not normally distributed. The difference was considered as significant at P<0.05.
Abbreviations
- IL-1β:
-
interleukin-1β
- RD:
-
retinal detachment
- RPE:
-
retinal pigment epithelium
- RRD:
-
rhegmatogenous RD
- RIP1:
-
receptor-interacting protein 1
- RIP3:
-
receptor-interacting protein 3
- TNFα:
-
tumor necrosis factor α
- HMGB1:
-
high mobility group box 1
- MLKL:
-
mixed lineage kinase domain-like protein
- DAMPs:
-
damage associated molecular patterns
- NLRP3:
-
NOD-like receptor family, pyrin domain containing-3
- ASC:
-
apoptosis-associated speck-like protein containing a caspase recruitment domain
- WT:
-
wild type
- MCP-1:
-
monocyte chemoattractant protein 1
- LCM:
-
laser capture microdissection
- VEGF:
-
vascular endothelial growth factor
- ONL:
-
outer nuclear layer
- TUNEL:
-
terminal deoxynucleotidyl transferase dUTP nick end labeling
- CNS:
-
central nervous system
References
Bird A, Bressler N, Bressler S, Chisholm IH, Coscas G, Davis M et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. Surv Ophthalmol 1995; 39: 367–374.
Aiello L . Perspectives on diabetic retinopathy. Am J Ophthalmol 2003; 136: 122–135.
Arroyo J, Yang L, Bula D, Chen D . Photoreceptor apoptosis in human retinal detachment. Am J Ophthalmol 2005; 139: 605–610.
Cook B, Lewis G, Fisher S, Adler R . Apoptotic photoreceptor degeneration in experimental retinal detachment. Invest Ophthalmol Vis Sci 1995; 36: 990–996.
Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck C et al. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci USA 2010; 107: 21695–21700.
Mervin K, Valter K, Maslim J, Lewis G, Fisher S, Stone J . Limiting photoreceptor death and deconstruction during experimental retinal detachment: the value of oxygen supplementation. Am J Ophthalmol 1999; 128: 155–164.
Sakai T, Lewis G, Linberg K, Fisher S . The ability of hyperoxia to limit the effects of experimental detachment in cone-dominated retina. Invest Ophthalmol Vis Sci 2001; 42: 3264–3273.
Cho Y, Challa S, Moquin D, Genga R, Ray T, Guildford M et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112–1123.
He S, Wang L, Miao L, Wang T, Du F, Zhao L et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–1111.
Murakami Y, Matsumoto H, Roh M, Suzuki J, Hisatomi T, Ikeda Y et al. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc Natl Acad Sci USA 2012; 109: 14598–14603.
Murakami Y, Matsumoto H, Roh M, Giani A, Kataoka K, Morizane Y et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ 2013; 21: 270–277.
Upton J, Kaiser W, Mocarski E . Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010; 7: 302–313.
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000; 1: 489–495.
He S, Liang Y, Shao F, Wang X . Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA 2011; 108: 20054–20059.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012; 148: 213–227.
Chen X, Li W, Ren J, Huang D, He W, Song Y et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 2013; 24: 105–121.
Cai Z, Jitkaew S, Zhao J, Chiang H, Choksi S, Liu J et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014; 16: 55–65.
Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014; 54: 133–146.
Scaffidi P, Misteli T, Bianchi M . Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418: 191–195.
Di Virgilio F . Dr. Jekyll/Mr. Hyde: the dual role of extracellular ATP. J Auton Nerv Syst 2000; 81: 59–63.
Bianchi M . DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 2007; 81: 1–5.
Shi Y, Evans J, Rock K . Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003; 425: 516–521.
Srinivasula S, Poyet J-L, Razmara M, Datta P, Zhang Z, Alnemri E . The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem 2002; 277: 21119–21122.
Thornberry N, Bull H, Calaycay J, Chapman K, Howard A, Kostura M et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 1992; 356: 768–774.
Martinon F, Burns K, Tschopp J . The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10: 417–426.
Mariathasan S, Weiss D, Newton K, McBride J, O'Rourke K, Roose-Girma M et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440: 228–232.
Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J . Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440: 237–241.
Iyer S, Pulskens W, Sadler J, Butter L, Teske G, Ulland T et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA 2009; 106: 20388–20393.
Arimura N, Ki-i Y, Hashiguchi T, Kawahara K, Biswas K, Nakamura M et al. Intraocular expression and release of high-mobility group box 1 protein in retinal detachment. Lab Invest 2009; 89: 278–289.
Kawano H, Ito T, Yamada S, Hashiguchi T, Maruyama I, Hisatomi T et al. Toxic effects of extracellular histones and their neutralization by vitreous in retinal detachment. Lab Invest 2014; 94: 569–585.
Tarallo V, Hirano Y, Gelfand B, Dridi S, Kerur N, Kim Y et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 2012; 149: 847–859.
Yoneda S, Tanihara H, Kido N, Honda Y, Goto W, Hara H et al. Interleukin-1beta mediates ischemic injury in the rat retina. Exp Eye Res 2001; 73: 661–667.
Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med 2012; 18: 791–798.
Lavalette S, Raoul W, Houssier M, Camelo S, Levy O, Calippe B et al. Interleukin-1β inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am J Pathol 2011; 178: 2416–2423.
Takeuchi O, Akira S . Pattern recognition receptors and inflammation. Cell 2010; 140: 805–820.
Yoshimura T, Sonoda KH, Sugahara M, Mochizuki Y . Comprehensive analysis of inflammatory immune mediators in vitreoretinal diseases. PLoS One 2009; 4: e8158.
Ricker L, Kijlstra A, Jager W, Liem A, Hendrikse F, Hiji E . Chemokine levels in subretinal fluid obtained during scleral buckling surgery after rhegmatogenous retinal detachment. Invest Ophthalmol Vis Sci 2010; 51: 4143–4150.
Ricker L, Kijlstra A, Kessels A, Jager W, Liem A, Hendrikse F et al. Interleukin and growth factor levels in subretinal fluid in rhegmatogenous retinal detachment: a case-control study. PLoS One 2011.
Rasier R, Gormus U, Artunay O, Yuzbasioglu E, Oncel M, Bahcecioglu H . Vitreous levels of VEGF, IL-8, and TNF-alpha in retinal detachment. Curr Eye Res 2010; 35: 505–509.
Nakazawa T, Hisatomi T, Nakazawa C, Noda K, Maruyama K, She H et al. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci USA 2007; 104: 2425–2430.
Nakazawa T, Matsubara A, Noda K, Hisatomi T, She H, Skondra D et al. Characterization of cytokine responses to retinal detachment in rats. Mol Vis 2006; 12: 867–878.
Tseng W, Thein T, Kinnunen K, Lashkari K, Gregory M, D'Amore P et al. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci 2013; 54: 110–120.
Ghazi N, Green W . Pathology and pathogenesis of retinal detachment. Eye (London, England) 2002; 16: 411–421.
Minami M, Kuraishi Y, Yabuuchi K, Yamazaki A, Satoh M . Induction of interleukin-1 beta mRNA in rat brain after transient forebrain ischemia. J Neurochem 1992; 58: 390–392.
Fan L, Young P, Barone F, Feuerstein G, Smith D, McIntosh T . Experimental brain injury induces expression of interleukin-1 beta mRNA in the rat brain. Brain Res Mol Brain Res 1995; 30: 125–130.
Hisatomi T, Sakamoto T, Murata T, Yamanaka I, Oshima Y, Hata Y et al. Relocalization of apoptosis-inducing factor in photoreceptor apoptosis induced by retinal detachment in vivo. Am J Pathol 2001; 158: 1271–1278.
Zacks DN, Hänninen V, Pantcheva M, Ezra E, Grosskreutz C, Miller JW . Caspase activation in an experimental model of retinal detachment. Invest Ophthalmol Vis Sci 2003; 44: 1262–1267.
Yang L, Bula D, Arroyo J, Chen D . Preventing retinal detachment–associated photoreceptor cell loss in bax-deficient mice. Invest Ophthalmol Vis Sci 2004; 45: 648–654.
Matsumoto H, Miller J, Vavvas D . Retinal detachment model in rodents by subretinal injection of sodium hyaluronate. J Vis Exp 2013; 79: e50660.
Matsumoto H, Murakami Y, Kataoka K, Lin H, Connor K, Miller J et al. Mammalian STE20-like kinase 2, not kinase 1, mediates photoreceptor cell death during retinal detachment. Cell Death Dis 2014; 29: e1269.
Matsumoto H, Kataoka K, Tsoka PA, Connor KM, Miller JW, Vavvas DG . Strain difference in photoreceptor cell death after retinal detachment in mice. Invest Ophthalmol Vis Sci 2014; 55: 4165–4174.
Vince J, Wong W, Gentle I, Lawlor K, Allam R, O'Reilly L et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012; 36: 215–227.
Kang T-B, Yang S-H, Toth B, Kovalenko A, Wallach D . Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 2013; 38: 27–40.
Watanabe H, Gaide O, Pétrilli V, Martinon F, Contassot E, Roques S et al. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol 2007; 127: 1956–1963.
Hisatomi T, Sakamoto T, Sonoda K-H, Tsutsumi C, Qiao H, Enaida H et al. Clearance of apoptotic photoreceptors: elimination of apoptotic debris into the subretinal space and macrophage-mediated phagocytosis via phosphatidylserine receptor and integrin alphavbeta3. Am J Pathol 2003; 162: 1869–1879.
Johnson N, Foulds W . Observations on the retinal pigment epithelium and retinal macrophages in experimental retinal detachment. Br J Ophthalmol 1977; 61: 564–572.
Thornton P, Pinteaux E, Gibson R, Allan S, Rothwell N . Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J Neurochem 2006; 98: 258–266.
Rothwell N, Strijbos P . Cytokines in neurodegeneration and repair. Int J Dev Neurosci 1995; 13: 179–185.
Pinteaux E, Trotter P, Simi A . Cell-specific and concentration-dependent actions of interleukin-1 in acute brain inflammation. Cytokine 2009; 45: 1–7.
LaVail M, Unoki K, Yasumura D, Matthes M, Yancopoulos G, Steinberg R . Multiple growth factors, cytokines, and neurotrophins rescue photoreceptors from the damaging effects of constant light. Proc Natl Acad Sci USA 1992; 89: 11249–11253.
Fogal B, Hewett J, Hewett S . Interleukin-1beta potentiates neuronal injury in a variety of injury models involving energy deprivation. J Neuroimmunol 2005; 161: 93–100.
Vandenabeele P, Berghe V, Festjens N . Caspase inhibitors promote alternative cell death pathways. Sci Signal 2006; 2006: pe44.
Smith J, Douty E . Electrophoresis of subretinal fluid. Arch Ophthalmol 1960; 64: 114–119.
Heath H, Beck T, Foulds W . Chemical composition of subretinal fluid. Br J Ophthalmol 1962; 46: 385–396.
Bishop P . The biochemical structure of mammalian vitreous. Eye 1996; 10: 664–670.
Lam K, Constable I, Schepens C . Subretinal fluid: Isoenzymes and cytologic studies. Invest Ophthalmol Vis Sci 1972; 11: 1037–1043.
Feeney L, Burns R, Mixon R . Human subretinal fluid: its cellular and subcellular components. Arch Ophthalmol 1975; 93: 62–69.
Acknowledgements
We thank Tetsu Asami, MD, PhD, Takeshi Iwase, MD, PhD, and Yasuki Ito, MD, PhD, for their assistance in the collection of patients’ samples. We also thank Wendy Chao, PhD for her support in critical review. This work was supported by: NEI R21EY023079-01A1 (DGV); The Yeatts Family Foundation (DGV, JWM); The Loefflers Family Fund (DGV, JWM); the 2013 Macula Society Research Grant award (DGV); Bausch & Lomb Vitreoretinal Fellowship (HM); a Physician Scientist Award (DGV), an unrestricted grant (JWM) from the Research to Prevent Blindness Foundation; NIH R01EY022084–01/S1 (KMC), and NEI grant EY014104 (MEEI Core Grant).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
JWM, DGV, and YM are named inventors on patent applications on methods and compositions for preserving photoreceptor viability not related to this work.
Additional information
Edited by T Kaufmann
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kataoka, K., Matsumoto, H., Kaneko, H. et al. Macrophage- and RIP3-dependent inflammasome activation exacerbates retinal detachment-induced photoreceptor cell death. Cell Death Dis 6, e1731 (2015). https://doi.org/10.1038/cddis.2015.73
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.73
This article is cited by
-
Anti-inflammatory potential of simvastatin and amfenac in ARPE-19 cells; insights in preventing re-detachment and proliferative vitreoretinopathy after rhegmatogenous retinal detachment surgery
International Ophthalmology (2024)
-
Increased intravitreal glucose in rhegmatogenous retinal detachment
Eye (2023)
-
UVA induces retinal photoreceptor cell death via receptor interacting protein 3 kinase mediated necroptosis
Cell Death Discovery (2022)
-
An allosteric interleukin-1 receptor modulator mitigates inflammation and photoreceptor toxicity in a model of retinal degeneration
Journal of Neuroinflammation (2020)
-
Insulin inhibits inflammation-induced cone death in retinal detachment
Journal of Neuroinflammation (2020)
