
Abstract
Inflammation and fibrosis are well-defined mechanisms involved in the pathogenesis of the incurable Laminin α2-deficient congenital muscular dystrophy (MDC1A), while apoptosis mechanism is barely discussed. Our previous study showed treatment with Losartan, an angiotensin II type I receptor antagonist, improved muscle strength and reduced fibrosis through transforming growth factor beta (TGF-β) and mitogen-activated protein kinases (MAPK) signaling inhibition in the dy2J/dy2J mouse model of MDC1A. Here we show for the first time that Losartan treatment up-regulates and shifts the nuclear factor kappa B (NFκB) signaling pathway to favor survival versus apoptosis/damage in this animal model. Losartan treatment was associated with significantly increased serum tumor necrosis factor alpha (TNF-α) level, p65 nuclei accumulation, and decreased muscle IκB-β protein level, indicating NFκB activation. Moreover, NFκB anti-apoptotic target genes TNF receptor-associated factor 1 (TRAF1), TNF receptor-associated factor 2 (TRAF2), cellular inhibitor of apoptosis (cIAP2), and Ferritin heavy chain (FTH1) were increased following Losartan treatment. Losartan induced protein expression toward a pro-survival profile as BCL-2 expression levels were increased and Caspase-3 expression levels were decreased. Muscle apoptosis reduction was further confirmed using terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) assay. Thus, along with TGF-β and MAPK signaling, NFκB serves as an important regulatory pathway which following Losartan treatment promotes survival in the dy2J/dy2J mouse model of MDC1A.
Similar content being viewed by others

Main
Congenital muscular dystrophy type 1A (MDC1A) is one of the most common forms of congenital muscular dystrophies (CMDs). Clinical symptoms are severe hypotonia, muscle weakness, and delayed motor milestones.1 Typically, the children do not achieve independent ambulation and respiratory failure is followed by death in the second or third decade of life.2, 3 MDC1A is caused by mutations in the LAMA2 gene, encoding the heavy chain of laminin-2.4 Muscle biopsies are characterized by muscle fiber necrosis, inflammation, apoptosis, and fibrosis.1, 2, 3, 5 Despite extensive advances in its diagnosis, MDC1A remains an incurable disease.2, 6
The dy2J/dy2J mouse is a useful model to study the pathophysiology of MDC1A and the effect of various therapeutic agents.7, 8, 9 This mouse has a mutation in the LAMA2 gene resulting in abnormal splicing of the laminin-α2 polypeptide and a moderate to severe phenotype characterized by development of muscle weakness at about 3 weeks of age, which progressively worsens.7, 10 The pathology of dy2J/dy2J skeletal muscle is quite similar to children with MDC1A, showing muscle fiber degeneration, necrosis, and apoptosis, followed by inflammation and fibrosis.7, 10, 11, 12
Previous studies among ours have shown that the pathogenesis of muscular dystrophies involves coordinated activation of multiple key signaling pathways.9, 13, 14, 15, 16 Nuclear factor kappa B (NFκB) has been described as a significant transcription factor that regulates the expression of muscle proinflammatory cytokines.16
Early studies have shown elevated NFκB levels in skeletal muscle of mdx mice, the mouse model for Duchenne muscular dystrophy (DMD), and in inflammatory myopathies.17, 18, 19, 20, 21 NFκB activation is thought to contribute to the deterioration of skeletal muscle pathology and muscle loss in DMD.18 However, NFκB seems to have a multifaceted regulatory role and may show protective activity in different disorders. Baghdiguian et al.22, 23 have shown that a certain level of NFκB activity is required to protect myofibers from apoptosis in a Calpain-3 mouse model of Limb girdle muscular dystrophies (LGMDs). Several studies indicate that NFκB activation has a positive role in cell survival by inducing transcription of several survival genes.24, 25 However, there is only limited data regarding NFκB’s role in MDC1A.
Losartan, an angiotensin II type 1 receptor antagonist, is a commercially available and extensively used medication for hypertension with a low side effect profile, occasionally used in childhood.26 In our previous study, we showed that Losartan treatment was associated with significant impressive improvement in muscle strength and amelioration of fibrosis in the dy2J/dy2J mouse model of MDC1A, through inhibition of transforming growth factor beta (TGF-β) and the mitogen-activated protein kinases (MAPK) signaling pathway.9
Here, we demonstrate NFκB signaling pathway involvement in the pathophysiology of the dy2J/dy2J mouse model, mediating decreased apoptosis and promoting muscle cell survival following Losartan treatment. Reduced apoptosis and pro-survival NFκB target genes activation following treatment suggest a key regulatory role for the NFκB signaling pathway in this disorder.
Results
Losartan treatment modifies TNF-α expression
The tumor necrosis factor alpha (TNF-α) serum level was significantly increased in treated dy2J/dy2J mice compared with untreated dy2J/dy2J mice (5.1±0.96 pg/ml versus 2.05±0.58 pg/ml; *P<0.05). TNF-α serum level was also significantly increased in Losartan-treated WT mice (treated: 5.69±0.49 pg/ml versus untreated: 2.42±0.56 pg/ml; **P<0.0005). These results are presented in Figure 1a. As these results were unexpected, we further examined the effect of Losartan on TNF-α transcript levels, using quantitative real-time PCR (TaqMan). The TNF-α mRNA level was unchanged in untreated dy2J/dy2J mice compared with WT groups, but was significantly increased in treated dy2J/dy2J mice hind limb muscles (*P<0.05; Figure 1b). Since TNF-α induces NFκB target gene expression, we analyzed NFκB activity and its downstream effects following Losartan treatment in more detail.

TNF-α activity in dy2J/dy2J and WT mice following Losartan treatment. (a) Losartan significantly increased serum TNF-α level in treated compared with untreated dy2J/dy2J mice (*P<0.05). Losartan also increased TNF-α levels in treated compared with untreated WT mice (**P<0.0005). Each bar represents the mean±S.E.M. of nine mice at 19 weeks of age. (b) Total RNA was extracted from Hind limb muscles of WT and dy2J/dy2J mice. Quantitative real-time PCR (TaqMan) of TNF-α mRNA expression levels was determined. Significant increased mRNA level of TNF-α was noted upon Losartan treatment in dy2J/dy2J mice. Expression levels were normalized to the housekeeping gene, TATA box binding protein (TBP) mRNA level (*P<0.05). Results represent the mean±S.E.M. of five mice
Losartan treatment altered NFκB activation
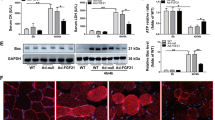
Using western blot analysis we found significantly decreased expression of the classic NFκB inhibitor, IκB-α protein, in hind limb muscles of both treated and untreated dy2J/dy2J mice compared with WT mice (untreated dy2J/dy2J: 0.74±0.07-fold versus untreated WT: 1±0.08-fold; P<0.005; and treated dy2J/dy2J: 0.72±0.05-fold versus treated WT: 0.99±0.13-fold; P<0.005). However IκB-α protein expression was unchanged in dy2J/dy2J mice following Losartan treatment, indicating pre-existing NFκB activation in untreated dy2J/dy2J mice (Figure 2a).

NFκB signaling pathway activity in dy2J/dy2J and WT mice. (a) Representative western blot gel and densitometry graph of NF-kappa-B inhibitor alpha (IkB-α) expression in WT and dy2J/dy2J mice. Significant reduction in IkB-α was noted in untreated and treated dy2J/dy2J mice compared with WT groups (*P<0.005). (b) Representative western blot gel and densitometry graph of NF-kappa-B inhibitor beta (IkB-β) expression in WT and dy2J/dy2J mice. Significant reduction in IkB-β was noted in treated compared with untreated dy2J/dy2J mice (*P<0.05). Losartan treatment was also associated with decreased IkB-β in treated WT mice compared with the untreated group (**P<0.005). Results of IkB-α and IkB-β levels were obtained from densitometric analysis and expressed as ratio of IkB-α-β/GAPDH and as change fold over control (WT group). These results represent three independent experiments. Each bar represents the mean±S.E.M. of 12 mice for IkB-α and 11 mice for IkB-β. (c) Intracellular localization of p65 using Immunofluorescence analysis. Expression of p65 was analyzed using anti-Alexa-647 (yellow fluorescence) antibody. Dystrophin staining as a skeletal muscle marker was analyzed using anti-cy2 (red fluorescence) antibody, and DAPI staining (blue fluorescence) was used as nuclear staining. When p65 protein is localized to the muscle nucleus, Alexa-647, cy2, and DAPI are merged. The quadriceps muscle of untreated and treated dy2J/dy2J mice showed nuclear localization of p65 demonstrating NFkB activation. Lack of p65 in the nucleus was illustrated in untreated and treated WT groups. Scale bar, 50 μm. Each bar represents the mean±S.E.M. of five fields per mice of six mice in the WT groups and seven mice in the dy2J/dy2J groups (*P<0.00001)
We next measured protein expression of IκB-β, an additional classic NFκB inhibitor. Only following Losartan treatment was IκB-β expression significantly decreased in both dy2J/dy2J and WT mice (treated dy2J/dy2J:0.32±0.05-fold versus untreated dy2J/dy2J: 0.53±0.07-fold; *P<0.05; treated WT: 0.37±0.04-fold versus untreated WT: 1±0.15-fold; **P<0.005) (Figure 2b). Thus in untreated dy2J/dy2 NFκB activation follows decreased expression of IκB-α, however after Losartan treatment NFκB activation follows decreased expression of both inhibitors; IκB-α and IκB-β.
In the next step, immunofluorescence staining of quadriceps muscles revealed significantly higher accumulation and co-localization of NFkB p65 subunit in the muscle nucleus of treated and untreated dy2J/dy2J mice, compared with WT mice (untreated dy2J/dy2J: 20.57±1.25% versus WT: 0.166±0.166%; P<0.00001 and treated dy2J/dy2J: 20.71±2.59% versus WT: 0.66±0.49%; P<0.00001) (Figure 2c). These findings confirm NFκB activation in dy2J/dy2J mice with and without treatment.
Losartan treatment upregulates several pro-survival NFκB target genes
Since NFkB appears to be involved in the regulation of both apoptosis and cell survival, we examined the effect of Losartan on NFkB target genes using quantitative real-time PCR (TaqMan). Anti-apoptotic NFkB target genes TNF receptor-associated factor 1 and TNF receptor-associated factor 2 (TRAF1 and TRAF2), cellular inhibitor of apoptosis (cIAP2), and Ferritin heavy chain (FTH1) were analyzed. We found that mRNA expression of TRAF1, an adaptor protein required for optimal anti-apoptotic NFκB activation, was significantly increased in treated WT and in both treated and untreated dy2J/dy2J compared with the untreated WT mice (*P<0.01,**P<0.05; Figure 3a). Because TRAF1 recruits TRAF2 and cIAPs to activate the anti-apoptotic process, we next measured the transcript levels of TRAF2 and cIAP2 genes. TRAF2 and cIAP2 genes were significantly increased in hind limb muscles of both dy2J/dy2J and WT mice following treatment (TRAF2; *P<0.005, **P<0.05 and cIAP2; *P<0.01, **P<0.05) (Figures 3b and c). FTH1 gene expression was significantly increased following treatment in dy2J/dy2J mice (*P<0.01), with no significant increase in the WT mice (Figure 3d). All of these findings suggest that Losartan upregulates NFκB pro-survival target genes.

Losartan modified pro-survival/anti-apoptotic NFκB signaling target genes. Total RNA was extracted from Hind limb muscles of WT and dy2J/dy2J mice. Quantitative real-time PCR (TaqMan) of (a) TRAF1, (b) TRAF2, (c) cIAP2, and (d) FTH1 mRNA expression levels were determined. A significantly increased mRNA level of the anti-apoptotic gene TRAF1 was noted in treated WT and in both treated and untreated dy2J/dy2J compared with untreated WT mice (*P<0.01,** P<0.05). Significantly increased mRNA levels of the anti-apoptotic genes, TRAF2 and cIAP2, were noted in Losartan treated WT and dy2J/dy2J mice (TRAF2; *P<0.005, **P<0.05 and cIAP2; *P<0.01, **P<0.05). Significantly increased mRNA levels of the anti-apoptotic genes FTH1 were noted in Losartan-treated dy2J/dy2J mice (*P<0.01). Expression levels were normalized to the housekeeping gene, TATA box binding protein (TBP) mRNA level. Results represent the mean±S.E.M. of five mice for TRAF1, TRAF2, cIAP2, and FTH1
Losartan treatment increases anti-apoptotic protein BCL-2 expression and decreases the pro-apoptotic protein Caspase-3 expression
Next using western blot analysis we examined the hind limb expression of B-cell lymphoma 2 (BCL-2), an anti-apoptotic protein. BCL-2 expression was significantly higher in Losartan treated compared with untreated dy2J/dy2J mice (treated dy2J/dy2J: 0.90±0.052-fold versus untreated dy2J/dy2J: 0.59±0.071-fold; *P<0.01). There was no significant difference in BCL-2 expression between treated and untreated WT mice (Figure 4a).

Increased protein expression of anti-apoptotic BCL-2 and decreased protein expression of pro-apoptotic Caspase 3 following Losartan treatment. (a) Representative western blot gel and densitometry graph of BCL-2 expression in WT and dy2J/dy2J mice. A significantly higher BCL-2 protein expression level was noted in treated compared with untreated dy2J/dy2J mice (*P<0.01). (b) Representative western blot gel and densitometry graph of Caspase-3 expression in WT and dy2J/dy2J mice. A significantly lower Caspase-3 protein expression level was noted in treated compared with untreated dy2J/dy2J mice (*P<0.0001). Results of BCL-2 and Caspase-3 levels were obtained from densitometric analysis and expressed as ratio of BCL-2 and Caspase-3 to GAPDH and as change fold over control (WT group). These results represent three independent experiments. Each bar represents the mean±S.E.M. of 12 mice for BCL-2 (*P<0.01) and 12 mice for Caspase-3 (*/**P<0.0001)
As for the protein expression level of the pro-apoptotic protein Caspase-3, Losartan treatment reduced significantly its expression in treated compared with untreated dy2J/dy2J mice (treated dy2J/dy2J: 3.9±1.3-fold versus untreated dy2J/dy2J: 10±1.9-fold; *P<0.0001) and in treated compared with untreated WT mice (treated WT: 0.3±0.1-fold versus untreated WT: 1±0.02-fold; **P<0.0001 Figure 4b). Taken together, these results suggest that Losartan treatment modifies NFκB signaling toward pro-survival/anti-apoptotic pathway.
Losartan reduces TUNEL-positive muscle cells
In order to confirm NFκB involvement in apoptosis signaling, we used i n situ DNA nick-end labeling (TUNEL), DNA fragmentation assay TUNEL analysis (Figure 5) showed significant reduction of TUNEL-positive cells in quadriceps muscles of Losartan treated compared with untreated dy2J/dy2J mice, indicating apoptosis (treated dy2J/dy2J: 2.6±0.35% versus untreated dy2J/dy2J: 10.14±1%; *P<0.0001). Almost no TUNEL-positive cells were found in untreated and treated WT groups.

Losartan decreased apoptosis in dy2J/dy2J mice muscle. Expression of apoptosis was analyzed using TUNEL assay with an In Situ Cell Death Detection Kit. TUNEL-positive cells were stained in yellow. Dystrophin, as muscle marker, was analyzed using anti-Alexa-647 antibody (red), and DAPI staining (blue) was used as nuclear staining. The quadriceps muscle of Losartan-treated dy2J/dy2J mice showed significant reduction in TUNEL-positive cells compare with the untreated mice. Almost no TUNEL-positive cells were illustrated in untreated and treated WT groups. Scale bar, 50 μm. Each bar represents the mean±S.E.M. of five fields per mice of six mice in the WT groups and seven mice in the dy2J/dy2J groups (*P<0.0001)
Discussion
CMDs are genetically heterogeneous diseases, which result in severe disability and premature death. Muscle fibrotic tissue accumulation and progressive skeletal muscle strength reduction characterize both children and the dy2J/dy2J mouse model of MDC1 A, one of the most frequent forms of CMD. We previously showed that Losartan treatment significantly increased both fore and hind limb muscle strength, with reduced collagen accumulation and fibrotic markers, in dy2J/dy2J mice skeletal muscle. This clinical and histological improvement was associated with TGF-β and MAPK signaling pathway inhibition, as Losartan was associated with reduced expression of the regulatory Smad; P-Smad2 and 3 and increased expression of the inhibitory Smad; Smad7. Furthermore, Losartan was associated with significant reduction of the three parallel MAPK signaling pathways P-ERK1/2, P-JNK, and P-p38.9
Our current study sheds new light on the NFκB signaling pathway and its involvement in the pathophysiology of the dy2J/dy2J mouse model of MDC1A, mainly through a new insight into the apoptosis pathway. In addition, these data reveal a new role for Losartan with regard to NFκB signaling and apoptosis in this disorder.
In this study, while evaluating Losartan's effect on cytokine levels in mice serum, we found an unexpected significant increase in the TNF-α level following treatment in both WT and dy2J/dy2J mice (Figure 1a). TNF-α is known to mediate a variety of cellular responses including inflammation, necrosis, fibrosis, and apoptosis.27 One major role of TNF-α is stimulation of the NFκB signaling pathway.28, 29 We therefore further investigated the NFκB signaling pathway involvement in MDC1A pathology.
NFκB is a transcription factor that in its resting state binds to inhibitory IκB proteins, keeping it inactive and localized to the cytoplasm. Following stimulation NFκB detaches from its inhibitors. The resulting free NFκB translocates into the nucleus where it activates or represses target genes.17, 30 Here we demonstrated NFκB activation in both treated and untreated dy2J/dy2J mice through IκB-α protein reduction and p65 (NFkB subunit) transcription factor accumulation in the nucleus of skeletal muscle cells (Figures 2a and c). Losartan-treated mice showed NFkB activity via reduction of an additional inhibitor, IκB-β (Figure 2b). These results may indicate different branches in the NFkB pathway are activated upon Losartan treatment compared with untreated dy2J/dy2J mice.
Previous studies have addressed Losartan’s role in inhibition of NFκB inflammatory processes. They showed Losartan reduces and inhibits NFκB activity in muscle cells from porcine coronary artery,31 and suppresses inflammation in aged rat kidney.32 However, NFκB signaling regulates transcriptional programs that are essential for the development and maintenance of the skeletal system,33 epithelium,34 and immune system,35 which in turn impacts differentiation, proliferation, cell death, and survival.36, 37 Therefore, NFκB signaling pathways have a multifaceted regulatory role that can either mediate apoptosis or anti-apoptotic routes. NFκB signaling can also engage the Caspase signaling pathway to mediate cell apoptosis.38, 39 On the other hand, it can activate TNF receptor-associated factors (TRAFs) and cellular inhibitor of apoptosis (CIAPs) to suppress cell death.21, 24, 25, 40
In Duchenne muscular dystrophy, NFκB activation is perceived as contributing to the deterioration of skeletal muscle pathology and skeletal muscle loss.18 For example, deletion of a single allele of NFkB (RelA/p65 subunit) was sufficient to considerably reduce infiltration of macrophages, fiber necrosis and calcification in dystrophic muscle in mdx mice (DMD mouse model). In addition, NFκB inhibition augmented the regeneration of mdx mice myofibers.41, 42 Furthermore, overexpression in skeletal muscle of A20 protein, a potent negative regulator of NFκB, reduced chronic inflammation and muscle degeneration in mdx mice.43, 44
However, in the current study, NFκB activity following Losartan treatment maintained survival and anti-apoptotic effects in dy2J/dy2J mice. Losartan treatment was associated with increased mRNA expression of pro-survival genes TRAF1, TRAF2, CIAP2, and FTH-1 (Figures 3a and d). These findings support other studies findings showing gene expression of TRAFs and CIAPs following NFκB activation, lead to inhibition of TNF-α induced cell death.29, 36, 45, 46 Cells lacking c-IAPs through genetic ablation or treated with IAPs antagonists have been shown to be more sensitive to TNF-α induced cell death through decreased NFκB survival mechanism.25 Losartan's role in apoptosis inhibition was further demonstrated by a significant increase of anti-apoptotic BCL-2 protein expression in skeletal muscle of treated dy2J/dy2J mice (Figure 4a). BCL-2 has been shown to have an important role in increasing lifespan, growth rate, and reducing apoptosis following muscle-specific overexpression in Lama2 null mice.47, 48 Furthermore, overexpression of BCL-2 was found to ameliorate muscle weakness and reduce apoptosis in oculopharyngeal muscular dystrophy (OPMD) mouse model.49 As for DMD, transgenic overexpression of BCL-2 did not improve muscle pathology in mdx mice,48, 50 and indeed the effect of Losartan in mdx mice is less pronounced than in dy2J/dy2J mice.15
We further investigated the anti-apoptotic effect of Losartan. Losartan treatment significantly decreased the pro-apoptotic protein Caspase-3 in dy2J/dy2J and WT mice (Figure 4b), and overall apoptosis was significantly decreased in Losartan-treated dy2J/dy2J mice as demonstrated by TUNEL-positive skeletal muscle cell reduction (Figure 5).
All of the above data together propose an important role for NFκB signaling in the pathophysiology of MDC1A, mainly in terms of its contribution to the apoptosis process. On the basis of current findings, we suggest that Losartan treatment shifts NFκB signaling to favor the survival route versus inflammation, fibrosis and apoptosis/damage in the dy2J/dy2J mouse model of MDC1A.
We therefore suggest a model regarding NFκB signaling activity following Losartan treatment. Losartan treatment results in increasing TNF-α, which in turn activates NFκB through IκB-α and IκB-β degradation, p65 nuclear accumulation and upregulation of pro-survival genes and proteins to mediate the anti-apoptotic effect of NFκB in the dy2J/dy2J mouse model of MDC1A (Figure 6).

Proposed model for NFkB as a key regulator in the survival path of muscle following Losartan treatment. Losartan treatment increases TNF-α, which in turn activates NFκB through IκB-α and IκB-β degradation, p65 nuclear accumulation and upregulation of the pro-survival genes: TRAF1, TRAF2, CIAP2, and FTH1 in addition to anti-apoptotic BCL-2 protein, to mediate the anti-apoptotic effect of NFκB in skeletal muscle of the dy2J/dy2J mouse model of MDC1A. In addition, Losartan treatment results in decreased expression of the pro-apoptotic protein Caspase-3
In muscular dystrophies there are indications that apoptosis, beside necrosis, may contribute to muscle loss and dysfunction. In human and mouse models of muscular dystrophy signs of muscle cell death by apoptosis have been documented but not discussed in detail.16, 21, 51, 52 In this study, we show for the first time that apoptosis has an important role in dy2J/dy2J muscle pathology. Therefore, it seems that therapies designed to include apoptosis inhibition might be beneficial for patients with congenital muscular dystrophy. These new findings support our previous data that demonstrated significant improvement in animal muscle strength and reduced fibrosis following 12 weeks of Losartan treatment. A future more prolonged Losartan study will provide additional information regarding long-term benefit and survival in dy2J/dy2J mice. This trial provides further support for a Losartan therapeutic trial in children with MDC1A.
Materials and Methods
Mice population and treatment
Muscle tissues for this study were obtained from the mice used in a previous Losartan study9 as follows; C57BL/6J Lama2dy-2J heterozygote mice (Jackson Laboratories, Bar Harbor, ME, USA) were bred at the Hebrew University specific pathogen-free animal housing facility. The joint ethics committee of Hebrew University and Hadassah Medical Center (accredited by AAALAC) approved the study protocol for animal welfare (permit number: 122.03-04). Mice were maintained under standard conditions, 23±1 °C, 12- h light cycle (0700–1900 h), with ad libitum access to food and drink. Delineation between the Lama2dy-2 J (dy2J/dy2J) affected mice, heterozygous for the LAMA2 gene mutation, and wild-type C57BL/6 J (WT) mice was detected by PCR.53 WT and dy2J/dy2J mice received 0.6 g/l Losartan (Merck Sharp & Dohme, West Point, PA, USA) in their drinking water or placebo as a control, The mice were treated for 12 weeks from 6 weeks of age (n=12/group; each group consisted of 6 male and 6 female mice).
Cytokines
A commercial BDTM Cytometric Bead Array Mouse Th1/Th2/Th17 Cytokine Kit (CBA) (lot: 77184, BD Biosciences, San Jose, CA, USA) was used to determine the levels (pg/ml) of TNF-α in serum samples, according to the manufacturer’s instructions. Fluorescence was analyzed using a flow cytometer (BDTM LSR II flow cytometer system; BD Biosciences) and the cytokine level was determined using a BD CBA Software (BD Biosciences).
Western blot analysis
Western blot analysis was performed as previously described.8 Immunoblotting was performed using the following antibodies: anti IκB-α and IκB-β (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-BCL-2 (Santa Cruz Biotechnology) and anti-Caspase-3 (Sigma-Aldrich, St Louis, MO, USA). The antibodies were used according to standard procedures. Equal protein loading of blots was confirmed by immunoblotting of glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology). Densitometry of the bands was obtained by chemi DOC XRS+ image lab software (Bio-Rad Laboratories, Hercules, CA, USA). Results are presented as fold of change over control (untreated WT group), which is defined as 1.
Real-time quantitative PCR
RNA extraction was performed with Tri Reagent (Sigma-Aldrich) according to the manufacturer’s instruction. cDNA was reverse transcribed from total RNA with random primers using the High-Capacity cDNA Reverse Transcription Kit with RNase inhibitor according to the manufacturer’s instruction (Applied Biosystems, Foster City, CA, USA). mRNA expression was quantified by TaqMan (Applied Biosystems) using the Illumina high-performance Eco Real-Time PCR system. Quantitative real-time PCR (TaqMan) assays were conducted in triplicates, and standard deviation was used to calculate error bars.
Immunofluorescence
Quadriceps muscles were isolated and fixed in acetone for 1 h. The quadriceps muscle tissues were sliced into 8 μm cross sections after being embedded in OCT. The mounted sections were washed three times in phosphate-buffered saline (PBS), blocked with 1% bovine serum albumin and probed with anti-p65 rabbit antibody (Abcam, Cambridge, UK) and with anti-dystrophin mouse antibody (Santa Cruz Biotechnology). After three washes in PBS, the sections were incubated with Alexa fluor 647-conjugated affini-pure donkey anti-rabbit IgG (H+L) and with Cy2-conjugated affini-pure donkey anti-mouse IgG (H+L) (Jackson ImmunoResearch, West Grove, PA, USA). After three more washes in PBS, coverslips were mounted on glass slides with a DAPI-containing mounting medium (Vector Laboratories, Burlingame, CA, USA). Fluorescence analysis was performed using a Zeiss LSM 710 confocal laser scanning system (Carl Zeiss MicroImaging GmbH, Jena, Germany). The P65-positive fluorescent cells were counted under a fluorescent microscope, and the numbers were expressed as the percentage of total P65 area cells±S.D. A negative control without anti-p65 rabbit antibody (Abcam) was also performed.
Tunel staining
Quadriceps muscles sections were assessed by terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) assay with an In Situ Cell Death Detection Kit (Roche Diagnostic, Indianapolis, IN, USA). Muscles were isolated and fixed in acetone for 1 h; the tissues were sliced into 8 μm cross sections after being embedded in OCT. Afterwards washed twice in PBS. Each slice was embedded with 100 μl permeabilisation solution, 0.1% Triton X-100 for 2 min, washed twice in PBS and the tissue sections were labeled and stained with the TUNEL reaction mixture (label plus enzyme solutions) for 60 min at room temperature and washed twice with PBS. Then the slices were probed with anti-dystrophin mouse antibody (Santa Cruz Biotechnology), and after three more washes in PBS the sections were incubated with Alexa fluor 647-conjugated affini-pure donkey anti-mouse IgG (H+L) (Jackson ImmunoResearch). After three more washes in PBS, coverslips were mounted on glass slides with a DAPI-containing mounting medium (Vector Laboratories). The apoptotic fluorescent cells were counted under a fluorescent microscope, and the numbers were expressed as the percentage of total Tunel area cells±S.D. A negative control without enzyme treatment and a positive control with DNase I (Roche Diagnostic) treatment were also performed.
Statistical analysis
All data are expressed as mean and standard error of the mean. Statistical analysis for direct comparison between two groups was performed by unpaired Student's t test. Significance was set at P<0.05 for all comparisons.
Abbreviations
- MDC1A:
-
congenital muscular dystrophy type 1A
- CMD:
-
congenital muscular dystrophy
- DMD:
-
Duchenne muscular dystrophy
- OPMD:
-
oculopharyngeal muscular dystrophy
- LGMDs:
-
Limb girdle muscular dystrophies
- NFκB:
-
Nuclear factor kappa B
- TGF-β:
-
transforming growth factor beta
- MAPK:
-
Mitogen- activated protein kinases
- TRAF1:
-
TNF receptor-associated factor 1
- TRAF2:
-
TNF receptor-associated factor 2
- cIAP2:
-
cellular inhibitor of apoptosis
- FTH1:
-
Ferritin heavy chain
- TBP:
-
TATA box binding protein
- TNF-α:
-
tumor necrosis factor alpha
- IkB-α:
-
NF-kappa-B inhibitor alpha
- IkB-β:
-
NF-kappa-B inhibitor beta
- BCL-2:
-
B-cell lymphoma 2
References
Reed UC . Congenital muscular dystrophy. Part II: a review of pathogenesis and therapeutic perspectives. Arq Neuropsiquiatr 2009; 67: 343–362.
Gawlik KI, Durbeej M . Skeletal muscle laminin and MDC1A: pathogenesis and treatment strategies. Skelet Muscle 2011; 1: 9.
Muntoni F, Voit T . The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004; 14: 635–649.
Bonnemann CG, Wang CH, Quijano-Roy S, Deconinck N, Bertini E, Ferreiro A et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord 2014; 24: 289–311.
Roland EH . Muscular dystrophy. Pediatr Rev 2000; 21: 233–237 quiz 238.
Jimenez-Mallebrera C, Brown SC, Sewry CA, Muntoni F . Congenital muscular dystrophy: molecular and cellular aspects. Cell Mol Life Sci 2005; 62: 809–823.
Vainzof M, Ayub-Guerrieri D, Onofre PC, Martins PC, Lopes VF, Zilberztajn D et al. Animal models for genetic neuromuscular diseases. J Mol Neurosci 2008; 34: 241–248.
Dadush O, Aga-Mizrachi S, Ettinger K, Tabakman R, Elbaz M, Fellig Y et al. Improved muscle strength and mobility in the dy(2 J)/dy(2 J) mouse with merosin deficient congenital muscular dystrophy treated with Glatiramer acetate. Neuromuscul Disord 2010; 20: 267–272.
Elbaz M, Yanay N, Aga-Mizrachi S, Brunschwig Z, Kassis I, Ettinger K et al. Losartan, a therapeutic candidate in congenital muscular dystrophy: studies in the dy(2 J) /dy(2 J) mouse. Ann Neurol 2012; 71: 699–708.
Guo LT, Zhang XU, Kuang W, Xu H, Liu LA, Vilquin JT et al. Laminin alpha2 deficiency and muscular dystrophy; genotype-phenotype correlation in mutant mice. Neuromuscul Disord 2003; 13: 207–215.
Xu H, Wu XR, Wewer UM, Engvall E . Murine muscular dystrophy caused by a mutation in the laminin alpha 2 (Lama2) gene. Nat Genet 1994; 8: 297–302.
Sunada Y, Bernier SM, Utani A, Yamada Y, Campbell KP . Identification of a novel mutant transcript of laminin alpha 2 chain gene responsible for muscular dystrophy and dysmyelination in dy2J mice. Hum Mol Genet 1995; 4: 1055–1061.
Burks TN, Andres-Mateos E, Marx R, Mejias R, Van Erp C, Simmers JL et al. Losartan restores skeletal muscle remodeling and protects against disuse atrophy in sarcopenia. Sci Transl Med 2011; 3: 82ra37.
Meinen S, Lin S, Ruegg MA . Angiotensin II type 1 receptor antagonists alleviate muscle pathology in the mouse model for laminin-alpha2-deficient congenital muscular dystrophy (MDC1A). Skelet Muscle 2012; 2: 18.
Nelson CA, Hunter RB, Quigley LA, Girgenrath S, Weber WD, McCullough JA et al. Inhibiting TGF-beta activity improves respiratory function in mdx mice. Am J Pathol 2011; 178: 2611–2621.
Bhatnagar S, Kumar A . Therapeutic targeting of signaling pathways in muscular dystrophy. J Mol Med (Berl) 2009; 88: 155–166.
Shin J, Tajrishi MM, Ogura Y, Kumar A . Wasting mechanisms in muscular dystrophy. Int J Biochem Cell Biol 2013; 45: 2266–2279.
Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest 2007; 117: 889–901.
Hnia K, Gayraud J, Hugon G, Ramonatxo M, De La Porte S, Matecki S et al. L-arginine decreases inflammation and modulates the nuclear factor-kappaB/matrix metalloproteinase cascade in mdx muscle fibers. Am J Pathol 2008; 172: 1509–1519.
Messina S, Bitto A, Aguennouz M, Minutoli L, Monici MC, Altavilla D et al. Nuclear factor kappa-B blockade reduces skeletal muscle degeneration and enhances muscle function in Mdx mice. Exp Neurol 2006; 198: 234–241.
Monici MC, Aguennouz M, Mazzeo A, Messina C, Vita G . Activation of nuclear factor-kappaB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology 2003; 60: 993–997.
Baghdiguian S, Martin M, Richard I, Pons F, Astier C, Bourg N et al. Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IkappaB alpha/NF-kappaB pathway in limb-girdle muscular dystrophy type 2 A. Nat Med 1999; 5: 503–511.
Baghdiguian S, Richard I, Martin M, Coopman P, Beckmann JS, Mangeat P et al. Pathophysiology of limb girdle muscular dystrophy type 2 A: hypothesis and new insights into the IkappaBalpha/NF-kappaB survival pathway in skeletal muscle. J Mol Med (Berl) 2001; 79: 254–261.
Karin M, Lin A . NF-kappaB at the crossroads of life and death. Nat Immunol 2002; 3: 221–227.
Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem 2008; 283: 24295–24299.
Burnier M, Wuerzner G . Pharmacokinetic evaluation of losartan. Expert Opin Drug Metab Toxicol 2011; 7: 643–649.
Van Herreweghe F, Festjens N, Declercq W, Vandenabeele P . Tumor necrosis factor-mediated cell death: to break or to burst, that's the question. Cell Mol Life Sci 2010; 67: 1567–1579.
Hayden MS, Ghosh S . Signaling to NF-kappaB. Genes Dev 2004; 18: 2195–2224.
Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr. . NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998; 281: 1680–1683.
Smale ST . Hierarchies of NF-kappaB target-gene regulation. Nat Immunol 2011; 12: 689–694.
Zahradka P, Werner JP, Buhay S, Litchie B, Helwer G, Thomas S . NF-kappaB activation is essential for angiotensin II-dependent proliferation and migration of vascular smooth muscle cells. J Mol Cell Cardiol 2002; 34: 1609–1621.
Kim JM, Heo HS, Choi YJ, Ye BH, Mi HaY, Seo AY et al. Inhibition of NF-kappaB-induced inflammatory responses by angiotensin II antagonists in aged rat kidney. Exp Gerontol 2011; 46: 542–548.
Novack DV . Role of NF-kappaB in the skeleton. Cell Res 2010; 21: 169–182.
Wullaert A, Bonnet MC, Pasparakis M . NF-kappaB in the regulation of epithelial homeostasis and inflammation. Cell Res 2010; 21: 146–158.
Oeckinghaus A, Hayden MS, Ghosh S . Crosstalk in NF-kappaB signaling pathways. Nat Immunol 2011; 12: 695–708.
Hayden MS, Ghosh S . NF-kappaB in immunobiology. Cell Res 2011; 21: 223–244.
Tergaonkar V . NFkappaB pathway: a good signaling paradigm and therapeutic target. Int J Biochem Cell Biol 2006; 38: 1647–1653.
Tang G, Yang J, Minemoto Y, Lin A . Blocking caspase-3-mediated proteolysis of IKKbeta suppresses TNF-alpha-induced apoptosis. Mol Cell 2001; 8: 1005–1016.
Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007; 131: 669–681.
Beg AA, Baltimore D . An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 1996; 274: 782–784.
Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS Jr. . NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 2000; 289: 2363–2366.
Li H, Malhotra S, Kumar A . Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med (Berl) 2008; 86: 1113–1126.
Charan RA, Hanson R, Clemens PR . Deubiquitinating enzyme A20 negatively regulates NF-kappaB signaling in skeletal muscle in mdx mice. FASEB J 2012; 26: 587–595.
Charan RA, Niizawa G, Nakai H, Clemens PR . Adeno-associated virus serotype 8 (AAV8) delivery of recombinant A20 to skeletal muscle reduces pathological activation of nuclear factor (NF)-kappaB in muscle of mdx mice. Mol Med 2012; 18: 1527–1535.
Lee NK, Lee SY . Modulation of life and death by the tumor necrosis factor receptor-associated factors (TRAFs). J Biochem Mol Biol 2002; 35: 61–66.
Lee SY, Kaufman DR, Mora AL, Santana A, Boothby M, Choi Y . Stimulus-dependent synergism of the antiapoptotic tumor necrosis factor receptor-associated factor 2 (TRAF2) and nuclear factor kappaB pathways. J Exp Med 1998; 188: 1381–1384.
Girgenrath M, Dominov JA, Kostek CA, Miller JB . Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J Clin Invest 2004; 114: 1635–1639.
Dominov JA, Kravetz AJ, Ardelt M, Kostek CA, Beermann ML, Miller JB . Muscle-specific BCL2 expression ameliorates muscle disease in laminin {alpha}2-deficient, but not in dystrophin-deficient, mice. Hum Mol Genet 2005; 14: 1029–1040.
Davies JE, Rubinsztein DC . Over-expression of BCL2 rescues muscle weakness in a mouse model of oculopharyngeal muscular dystrophy. Hum Mol Genet 2011; 20: 1154–1163.
Abmayr S, Crawford RW, Chamberlain JS . Characterization of ARC, apoptosis repressor interacting with CARD, in normal and dystrophin-deficient skeletal muscle. Hum Mol Genet 2004; 13: 213–221.
Miller JB, Girgenrath M . The role of apoptosis in neuromuscular diseases and prospects for anti-apoptosis therapy. Trends Mol Med 2006; 12: 279–286.
Mehuron T, Kumar A, Duarte L, Yamauchi J, Accorsi A, Girgenrath M . Dysregulation of matricellular proteins is an early signature of pathology in laminin-deficient muscular dystrophy. Skelet Muscle 2014; 4: 14.
Vilquin JT, Vignier N, Tremblay JP, Engvall E, Schwartz K, Fiszman M . Identification of homozygous and heterozygous dy2J mice by PCR. Neuromuscul Disord 2000; 10: 59–62.
Acknowledgements
Losartan was kindly provided by Merck Sharp & Dohme (MSD). This project was supported by Association Francaise contre les Myopathies (AFM) (grant number 12752), the Israeli Ministry of Health, Chief Scientist’s Office (grant number 3763) and by Teva Pharmaceutical Industries Ltd. under the Israeli National Network of Excellence in Neuroscience (NNE) established by Teva.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou.
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Elbaz, M., Yanay, N., Laban, S. et al. Life or death by NFκB, Losartan promotes survival in dy2J/dy2J mouse of MDC1A. Cell Death Dis 6, e1690 (2015). https://doi.org/10.1038/cddis.2015.60
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.60
