
Abstract
High-risk human papillomaviruses (HR-HPVs) cause nearly all cases of cervical cancer, as well as approximately 30% of head and neck cancers. HPV 16 E6, one of two major viral oncogenes, protects cells from apoptosis by binding to and accelerating the degradation of several proteins important in apoptotic signaling, including caspase 8 and p53. We proposed that blocking the interactions between HPV E6 and its partners using small molecules had the potential to re-sensitize HPV+ cells to apoptosis. To test this idea, we screened libraries of small molecules for candidates that could block E6/caspase 8 binding and identified several candidates from different chemical classes. We tested hits for dose-dependency and specificity in vitro and for toxicity in a cell-based assay and then used this information to select the two best candidates for further testing: myricetin, a flavonol, and spinacine, an imidazole amino-acid derivative of histidine. Both compounds clearly inhibited the ability of E6 to bind in vitro to both caspase 8 and E6AP, the protein that mediates p53 degradation. In addition, both compounds were able to increase the level of caspase 8 and p53 in SiHa cervical cancer cells, resulting in an increase of caspase 3/7 activity. Finally, both myricetin and spinacine sensitized HPV+ cervical and oral cancer cells, but not HPV− cervical and oral cancer cells, to apoptosis induced by the cancer-specific ligand TRAIL, as well as the chemotherapeutic agents doxorubicin and cisplatin. New therapies based on this work may improve treatment for HPV+ cancer patients.
Similar content being viewed by others
Main
High-risk types of human papillomavirus (HPV), especially types 16 and 18, are the causative agents of nearly all cases of human cervical cancer, in addition to up to 70% of head and neck cancers (HNC).1 Although the overall incidence of HNC has stabilized during the past decade, the incidence of HPV-associated cases, especially of oropharyngeal squamous cell carcinoma, has dramatically increased.2 HPVs are small, double-stranded DNA viruses that infect epithelial tissues. The HPV-encoded oncogenes E6 and E7 are responsible for cellular immortalization and transformation and, consequently, for the development of HPV-associated cancer. Although E7 is best known for the inactivation of Rb, E6 accelerates the degradation of several molecules involved in apoptosis.
Two HPV vaccines, Gardasil (MSD, Merck, Kenilworth, NJ, USA) and Cervarix (GSK, Glaxo SmithKline, London, UK), have been approved and are currently in use for the prevention of HPV infection. However, they offer no benefit to an individual who has already been infected and only protect against 2 of the 15 types of high-risk viruses, HPV-16 and -18. Surgical and ablative techniques are used to remove developed tumors; however, these approaches are invasive and cytodestructive, and lesions frequently recur following treatment. Chemotherapy, utilizing agents such as cisplatin and doxorubicin, has also been used to treat cervical cancer but with mixed results.3, 4, 5, 6, 7, 8 As researchers and clinicians have worked to move beyond these relatively non-specific and toxic agents, reagents that activate the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated, extrinsic apoptotic pathway have garnered considerable interest owing to their promise in the treatment of several types of tumors.9, 10, 11, 12, 13, 14, 15, 16, 17
Unfortunately, therapies that function by activating apoptosis, including those based on TRAIL, cisplatin and doxorubicin, are handicapped in their ability to effectively treat HPV-associated malignancies, because high-risk E6 proteins subvert both the extrinsic and intrinsic apoptotic pathways. E6 proteins from high-risk types of HPV are well known for their ability to mediate the rapid degradation of p53,18, 19, 20, 21 an important mediator of intrinsic apoptotic pathways, thereby increasing the growth and survival of transformed cells.22, 23 E6 also interacts with other partner proteins, a number of which participate in extrinsic, receptor-mediated apoptosis. For example, our laboratory found that HPV 16 E6 binds to and inactivates several molecules involved in these pathways, including TNF R1,24 Fas-associated protein with death domain (FADD),25 and caspase 8.26, 27 As a result, engagement of either the extrinsic or the intrinsic apoptotic pathways fails to result in the transduction of the intended death signal because the mediator molecules – p53 in the case of the intrinsic pathway, and FADD and caspase 8 in the case of the extrinsic pathway – are missing. Therefore, if any of these apoptosis-inducing signaling pathways are to be used as effective tools for the elimination of HPV-associated malignancies, it will be necessary to restore the missing signaling molecules.
In our previous work,28 we identified myricetin as a compound that can inhibit the E6/caspase 8 interaction in vitro. Unfortunately, myricetin is known to also inhibit a number of cellular proteins, including several tyrosine kinases, and its structure makes modification for drug development difficult. The identification of additional, more tractable inhibitors of the E6/procaspase 8 interaction was therefore pursued. Screening an ActiProbe 2 K library enabled us to identify several compounds of interest, and we further examined the ability of one particular compound, spinacine (5; see Figure 1), an unnatural amino acid that is the product of the action of formaldehyde on histidine, to inhibit specific interactions between E6 and its partner proteins. We found that both myricetin and spinacine can re-sensitize HPV+ cells to apoptosis triggered by apoptotic inducers such as TRAIL, cisplatin, and doxorubicin by increasing caspase 3/7 activity and restoring the level of apoptotic proteins in HPV+ cells. These findings identify a novel small molecule that can inhibit E6 functions and may serve as the basis for further discovery and development of effective and novel therapeutic approaches for the treatment of HPV-mediated cancers.

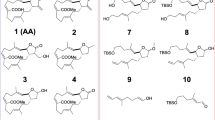
Structures of the tested potential E6 ligands
Results
Imidazole derivatives specifically inhibit the interaction of HPV E6 with caspase 8
We previously reported that HPV E6 binds to caspase 829 and that myricetin can block the E6/caspase 8 interaction in vitro.28 In that study, we utilized a bead-based assay based on AlphaScreen Technology (Perkin-Elmer, Waltham, MA, USA) to identify inhibitors of the E6/caspase 8 interaction.28 In the current work, we extended our search by screening the ActiProbe 2 K (2000 compounds) library using the same approach. In our primary screen, 118 (5.9%) compounds demonstrated an ability to inhibit the E6/caspase 8 interaction. Seventy-nine of these 118 compounds presented EC50 values <10 μM and were therefore chosen for further analysis. Twenty-three of these 79 compounds also demonstrated specific inhibition of the E6/caspase 8 interaction, in that they were unable to block formation of the caspase 8/caspase 8 homodimer (counter-screen). These 23 compounds represented three different chemical classes, of which the imidazoles and the related benzimidazoles showed the best inhibition of E6/caspase 8 binding. Analysis of the structures and physical/chemical characteristics of those molecules led to the selection of five additional compounds: 3-(1H-indol-1-yl)propan-1-amine methanesulfonate (1), benzimidazole (2), 1H-benzimidazole-1-methanol (3), 2-(methoxymethyl)-1H-benzimidazole (4), and spinacine (5) for further study (Figure 1).
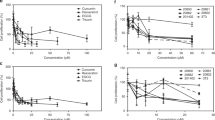
To determine the dose-responsiveness of these five compounds, variable concentrations of those small molecules were first tested for their ability to inhibit E6/caspase binding. The results showed that only compounds 1, 2, and 5 demonstrated dose-dependent inhibition of E6/caspase 8 binding (Figure 2a). We then asked whether these molecules, which had been selected for their ability to interfere with E6/caspase 8 binding, could also affect other interactions of E6, such as its binding to E6-associated protein (E6AP). The E6/E6AP interaction is required for the E6-mediated acceleration of p53 degradation.21 We therefore tested the ability of these five compounds to inhibit E6/E6AP binding and found that compounds 1, 2, and 5, the same agents that had inhibited E6/caspase 8 binding, could also inhibit the E6/E6AP interaction and could do so nearly as well as they had inhibited the E6/caspase 8 interaction (Figures 2a and b).

3-(1H-indol-1yl)propan-1-amine (1), three benzimidazole derivatives (2–4), and spinacine (5) inhibit protein–protein interactions. Five compounds at the indicated concentrations (1.4 μM–3.2 mM) were tested using our bead-based screening assay for their ability to inhibit three different protein/protein interactions: (a) GST-E6/His-caspase 8; (b) GST-E6/HisE6AP; and (c) GST-caspase 8/His-caspase 8. Binding in the presence of 1.4 μM of the test compound was set at 100%. Experiments were performed in triplicate, and error bars indicate the S.D.
To ask whether this inhibition was specific, we employed a counter-screening assay in which we assessed the ability of candidates to inhibit the binding of GST-caspase 8 to His-caspase 8; those that did were to be eliminated. In this counter-screening assay, the materials in each well were the same as in the primary screen, with the exception that GST-caspase 8 replaced GST-E6. Compounds 1 and 2 were able to inhibit the caspase 8/caspase 8 interaction about as well as they inhibited the E6/caspase 8 interaction, indicating that their actions were non-specific. However, spinacine (5) did not significantly inhibit the binding of caspase 8 to itself, providing evidence of its specificity for E6 (Figure 2c). These results are summarized in Table 1, which shows that, of the compounds tested, spinacine displayed the lowest EC50 value for the inhibition of E6/caspase 8 and E6/E6AP interactions, while not inhibiting caspase 8/caspase 8 binding.
Myricetin and spinacine sensitize SiHa cells to TRAIL-induced apoptosis
We next asked which of the molecules that block E6/caspase 8 binding in vitro could also act in the context of a cell. SiHa cells are an HPV+ cell line, derived from a cervical carcinoma, which serves as a commonly used model for HPV-associated malignancies. To determine whether HPV+ SiHa cells are resistant to TRAIL-induced apoptosis, SiHa cells were treated with TRAIL and cell viability was assessed. TRAIL-sensitive, HPV− U2OS cells served as a positive control. The results (Figure 3a) demonstrate that, in comparison to U2OS cells, SiHa cells are relatively resistant to treatment with TRAIL, as predicted. Furthermore, both myricetin and spinacine displayed low toxicity to SiHa cells in the absence of TRAIL (Figure 3b). We next asked whether myricetin and/or spinacine could sensitize these HPV+ cells to TRAIL-induced apoptosis. The indicated concentrations (0 –125 μM) of myricetin, (3) (negative control), and spinacine (5) were added to SiHa cells in the presence of TRAIL, and Figure 3c shows that, in the presence of TRAIL, myricetin and spinacine were both able to reduce the viability of SiHa cells to 30–40% at a dose of 125 μM, in contrast to the negative control, 3. Table 2 lists and compares the concentrations of the three tested compounds required to induce death in 50% of SiHa cells in the presence or absence of TRAIL and demonstrates that both myricetin and spinacine induced more cell death (an increase of 3.5-fold for myricetin and of 8.6-fold for spinacine) in the presence than in the absence of TRAIL (Table 2). These results provide strong evidence that inhibiting the E6/caspase 8 interaction can restore the sensitivity of HPV+ cells to apoptotic signals.

(a) HPV+ SiHa cells are resistant to TRAIL-induced apoptosis as compared with the human osteosarcoma cell line U2OS. Cells (2 × 104 cells per well) were seeded into a 96-well plate, allowed to incubate overnight, and then treated with the indicated concentration of TRAIL in the presence of cycloheximide (5 μg/ml). The viability of cells was measured after overnight incubation using the MTT assay; the viability of cells untreated with TRAIL was set at 100% for each group. (b) The indicated concentrations of three small molecules were added to SiHa cells, and an MTT assay was preformed after overnight incubation. The viability of cells untreated with small molecules was set as 100%. (c) The indicated concentrations of myricetin or spinacine were added 4 h prior to TRAIL (100 ng/ml) in the presence of cycloheximide (5 μg/ml) and incubated at 37 °C overnight. The viability of cells untreated with small molecules was set at 100%. (d and e) SiHa cells (2 × 104 cells per well) were seeded into a 96-well plate and allowed to incubate overnight. In all, 100 μM of myricetin and 50 μM of spinacine were added 4 h prior to either (d) doxorubicin or (e) cisplatin. Cell viability was measured after overnight incubation using the MTT assay, and the viability of cells untreated with either myricetin or spinacine was set at 100%. Experiments were performed in triplicate, and error bars indicate the S.D.
Myricetin and spinacine increase the sensitivity of SiHa cells to doxorubicin and cisplatin
As noted earlier, both myricetin and spinacine can also inhibit the binding of E6 to E6AP, an E3 ligase involved in the degradation of p53, in vitro. Based on this observation, we next asked whether myricetin and/or spinacine would increase the sensitivity of SiHa cells to two drugs, doxorubicin and cisplatin, that are thought to act by inducing intrinsic apoptosis through the activation of pathways that involve p53. We found that, as compared with the negative control, both myricetin and spinacine increased the sensitivity of SiHa cells to doxorubicin and cisplatin (Figures 3d and e). These results suggest that the ability of both myricetin and spinacine to inhibit the interaction between E6 and E6AP, resulting in a predicted increase in p53, results in the sensitization of HPV+ cells to inducers of p53-mediated apoptosis.
Myricetin and spinacine increase caspase 3/7 activity
Activation of caspase 3/7 is an essential marker for both extrinsic and intrinsic apoptosis and can be used to determine whether cell death occurs through the apoptotic pathway. Our previous data (Figure 2) demonstrates that both myricetin and spinacine can inhibit the E6/caspase 8 and E6/E6AP interactions in vitro, resulting in increased cell death. If this cell death occurred through apoptosis, one prediction is that pretreatment of cells with either myricetin or spinacine should increase the level of TRAIL-, cisplatin-, and doxorubicin-induced activation of caspase 3/7. To test this prediction, caspase 3/7 activity was measured in SiHa cells following TRAIL, cisplatin, and doxorubicin treatment in the presence or absence of myricetin or spinacine. As shown in Figure 4a, we found that the caspase 3/7 activity increased when myricetin or spinacine were combined with TRAIL as compared with controls. Similar results were obtained following treatment with cisplatin and doxorubicin (Figures 4b and c). These data demonstrate that the increased cell death induced by the combination of apoptotic inducers and myricetin/spinacine occurs through the apoptotic pathway.

Myricetin and spinacine (5) increase caspase 3/7 activity in SiHa cells following treatment with TRAIL and chemotherapy drugs. SiHa cells (2 × 104 cells per well) were seeded into a 96-well plate and allowed to incubate overnight and then pretreated with 100 μM of myricetin or 50 μM of spinacine for 4 h. (a) TRAIL (100 ng/ml), along with cycloheximide (5 μg/ml), (b) cisplatin (50 μM), or (c) doxorubicin (2 μM) were added respectively. Caspase 3/7 activity was measured after 0, 1.5, 3, and 6 h using the CellTiter-Glo assay. Activity at 0 h of treatment was set at 100% for each group. Experiments were performed in triplicate, and error bars indicate the S.D.
Myricetin and spinacine re-sensitize HPV+ cells to TRAIL-induced apoptosis by blocking the binding of HPV E6 to caspase 8
To determine whether myricetin and spinacine re-sensitize SiHa cells to TRAIL-induced apoptosis by specifically blocking the binding of E6 to caspase 8, we compared responses in the HPV+ SiHa cells with those from the HPV− human cervical carcinoma cell, C33A, as these cells do not express E6. We found that, although the HPV+ SiHa cells displayed resistance to TRAIL at concentrations up to 100 ng/ml, their sensitivity increased dramatically in the presence of 100 μM myricetin. In contrast, the HPV− C33A cells remained resistant to TRAIL-mediated apoptosis even in the presence of myricetin (Figure 5a). Similar results were also found following pretreatment with 50 μM spinacine (Figure 5b). Together, these results suggest that myricetin and spinacine can re-sensitize HPV+ cells to TRAIL-mediated apoptosis by specifically blocking the binding of HPV E6 to apoptotic proteins, such as caspase 8.

Myricetin and spinacine (5) can re-sensitize HPV+, but not HPV−, cells to treatment with TRAIL. SiHa (2 × 104 cells per well) and C33A cells (1 × 104 cells per well) were seeded into a 96-well plate and allowed to incubate overnight, and then cells were pretreated in the presence or absence of (a) myricetin (100 μM) or (b) spinacine (50 μM) for 4 h. The indicated concentration of TRAIL along with cycloheximide (5 μg/ml) was added, and the cells were allowed to incubate overnight. Cell viability was measured by MTT assay, and the viability of cells untreated with TRAIL was set at 100%. Experiments were performed in triplicate, and error bars indicate the S.D.
Myricetin and spninacine increase the levels of caspase 8 and p53 in SiHa cells
The data described above demonstrate that myricetin and spinacine are able to increase caspase 3/7 activity (Figure 4) and to sensitize SiHa cells to TRAIL (Figures 3a and c and 5) and p53 (Figures 3d and e) induced apoptosis. If this ability is due to inhibition of E6 functions, then an increase in the levels of caspase 8 and p53 should be observable in SiHa cells following treatment with myricetin and spinacine. To test this prediction, we treated SiHa cells with 50, 100, and 200 μM of myricetin or spinacine for 24 h and then measured the level of caspase 8 by immunoblot. The results (Figure 6a) demonstrate that treatment with myricetin caused an increase in caspase 8 levels at doses of 100 and 200 μM, while spinacine caused an increase in caspase 8 at doses from 50 to 200 μM. To ask how p53 levels were affected, we employed an ELISA assay. For comparison, C33A cells, which lack HPV E6 and express mutant p53, were also treated with the same concentrations of spinacine and myricetin. Mitomycin C was included in the assay to cause DNA damage and therefore increase p53 levels. The results from this experiment (Figure 6b) demonstrate that treatment of SiHa cells with myricetin caused a 4.4-fold increase in p53 at a dose of 200 μM and that treatment with spinacine caused a 3.4-fold increase in p53 at a dose of 100 μM, as compared with untreated cells. Furthermore, there was no significant change in p53 levels in C33A cells, confirming our earlier conclusion that myricetin and spinacine impact apoptotic pathways only in the presence of E6. Together, these results suggest that these small molecules are able to increase the sensitivity of SiHa cells to both TRAIL- and p53-induced apoptosis by blocking the ability of E6 to bind to its cellular partners, thereby reducing the degradation of caspase 8 and p53 in HPV+ SiHa cells.

Treatment with myricetin and spinacine (5) increases cellular levels of caspase 8 and p53. (a) SiHa cells (1 × 106 per well) were seeded into a six-well plate and allowed to incubate overnight. The indicated concentrations of myricetin and spinacine were added, and then cells were incubated for 24 h. Cells were then washed in 1 × PBS and then harvested, and the resulting level of caspase 8 was measured by immunoblot. (b) SiHa and C33A cells (1 × 106 per well) were seeded into a six-well plate and allowed to incubate overnight. In all, 200 μM of myricetin and 100 μM spinacine were added together with 4 μg/ml mitomycin C and incubated for 24 h. The resulting level of p53 was measured by enzyme-linked immunosorbent assay, and the level of p53 found in untreated cells was set at 100%. Experiments were performed in triplicate, and error bars indicate the S.D.
Myricetin and spinacine sensitize HPV+ head and neck squamous cell carcinoma (HNSCC) cells to TRAIL-induced cell death
As shown above, myricetin and spinacine sensitize SiHa cells to TRAIL and chemotherapy drugs. These data provide proof-of-principle that small-molecule inhibitors can block HPV E6 functions and trigger HPV+ cervical cancer cell lines to undergo apoptosis. We next examined whether other types of HPV+ cancer cells, and in particular, HNSCC cells can also be sensitized in this manner. All selected cell lines demonstrated resistance to TRAIL at concentrations as high as 100 ng/ml (Figure 7a). For the combination experiments, 50 ng/ml TRAIL was applied in combination with either myricetin (0–200 μM) or spinacine (0–100 μM). As shown in Figure 7b, the HPV− HNSCC cell lines (UMSCC29, #29; and SCC84, #84) were resistant to TRAIL in both the presence and absence of myricetin or spinacine. In contrast (Figure 7c), TRAIL-induced cell death was enhanced 40% by 40 μM myricetin in the HPV+ cell line UMSCC47, #47; by 30% in UPCI-SCC90-UP-Clone 35, #90; and by 70% in the UM-SCC47-TC-Clone 3, 47CL3. Furthermore, 40 μM spinacine was able to enhance TRAIL-induced cell death in the HPV+ #47 and #90 cell lines up to 50% and near 60% in the HPV+ #47CL3 cell line. Together, these results indicate that both myricetin and spinacine sensitize HPV+, but not HPV−, HNSCC cell lines to TRAIL-induced apoptosis, confirming our previous results that these small molecules block HPV E6 functions.


Myricetin and spinacine (5) can re-sensitize HPV+, but not HPV−, head and neck cancer cell lines to treatment with TRAIL. (a) Both HPV− and HPV+ HN cancer cell lines display resistance to TRAIL treatment. (b) HPV− (UMSCC 29 and SCC 84) and (c) HPV+ (UMSCC 47, UPCI-SCC90-UP-Clone 35, and UM-SCC47-TC-Clone 3) head and neck cancer cell lines (2 × 104 cells per well) were seeded into 96-well plates and allowed to incubate overnight, and then cells were pretreated with myricetin (0–200 μM) or spinacine (0–100 μM) for 4 h. In all, 50 μM of TRAIL along with cycloheximide (5 μg/ml) was added, and cells were allowed to incubate overnight. Cell viability was measured by the MTT assay, and the viability of cells untreated with small molecules was set at 100%. Experiments were performed in triplicate, and error bars indicate the S.D.
Myricetin and spinacine sensitize HPV+ HNSCC cells to doxorubicin
The results described above indicate that myricetin and spinacine sensitize HPV+ HNSCC cells to TRAIL-induced apoptosis. To ask whether this sensitization also occurs in the context of chemotherapeutic agents, several HNSCC cell lines were tested for their responses to combination treatments. As shown in Figure 8a, the HPV− HNSCC cell lines UMSCC 29 (#29) and SCC 84 (#84) were resistant to doxorubicin in both the presence and absence of myricetin and spinacine. However, HPV+ HNSCC cancer cell lines, such as UPCI-SCC90-UP-Clone 35 (#90), UMSCC 47 (#47), and UM-SCC47-TC-Clone 3 (#47 CL3), displayed an increase in cell death when treated by combinations of 2 μM doxorubicin plus the indicated small molecules (Figure 8b). In these experiments, spinacine demonstrated a higher efficiency than did myricetin, sensitizing #47, #47CL3, and #90 to doxorubicin by up to 50% at a concentration of 12.5 μM. Together, these data demonstrate that myricetin and spinacine can sensitize both cervical and oral HPV+ cancer cells to chemotherapeutic agents such as doxorubicin.

Myricetin and spinacine (5) can re-sensitize HPV+, but not HPV−, HNSCC cell lines to treatment with doxorubicin. Two HPV− (UMMSCC 29 and SCC 84) and three HPV+ (UMSCC 47, UPCI-SCC90-UP-Clone 35, and UM-SCC47-TC Clone 3) head and neck cancer cell lines (2 × 104 cells per well) were seeded into 96-well plates and allowed to incubate overnight, and then cells were pretreated with myricetin or spinacine (0–12.5 μM) for 4 h. In all, 2 μM of doxorubicin was added, and the cells were allowed to incubate overnight. Cell viability was measured by MTT assay, and the viability of cells untreated with small molecules was set at 100%. Experiments were performed in triplicate, and error bars indicate the S.D.
Discussion
Currently, no small molecules targeting any of the HPV proteins are available for clinical use, although a few groups are examining the possibility of developing these sorts of small, inhibitory molecules to expand and enhance the limited therapeutic options currently available for HPV-associated malignancies.30, 31 E6 and E7 oncogenes are expressed at relatively high levels during cancer development and therefore have the potential to serve as useful targets. For example, inhibition of E6 is predicted to lead to increased cell death, as E6 normally functions to block both intrinsic and extrinsic apoptotic pathways.32
One approach to blocking the interactions between E6 and its cellular partners is to use peptides, and previous work from our laboratory and others has identified peptide inhibitors that can specifically inhibit the interactions between E6 and caspase 8 and FADD33 or between E6 and E6AP.33, 34 As compared with peptide inhibitors, however, small molecules possess numerous advantages because they are more stable, penetrate target cells more easily, and can more readily be modified and optimized by organic chemists during drug development. Some progress has been made in this area, as published studies have identified several compounds (some with EC50 values between 17 and 29 μM) that could inhibit the interaction between E6 and E6AP.30, 35 However, no structure–activity relationship (SAR) has been carried out for those compounds because of the limited data set, and none of these findings have yet led to clinically useful interventions.
Our laboratory previously reported that the flavonol myricetin can inhibit interactions between E6 and caspase 8 in vitro, and preliminary results (data not shown) indicate that myricetin binds to E6 at a ratio of 2 : 1. Unfortunately, myricetin is known to inhibit a number of cellular proteins (including several kinases), indicating a lack of sufficient specificity. In addition, the high EC50 value observed in the context of a cell-based model system (Table 2) suggests that its ability to negatively affect tumors may be limited, perhaps because its polar nature limits its ability to enter cells. As polar groups are an important feature in assessing SARs, optimization of this series of compounds would be difficult without resorting to a pro-drug approach. Therefore, we engaged in additional screening efforts with libraries containing more ‘lead-like’ members that are expected to provide better platforms for optimization.
Of the five tested compounds (Figure 1), spinacine (5) was best able to specifically inhibit the interactions of E6 with both caspase 8 and E6AP (Figures 2a and b), demonstrating the lowest EC50 values for both E6/Caspase 8 and E6/E6AP binding. However, it did not inhibit caspase 8/caspase 8 binding, thus demonstrating the desired specificity (Table 1). In the experiments that involved treating SiHa cells with TRAIL, the EC50 values demonstrate that, as compared with myricetin, spinacine may be better able to penetrate into cells and thus re-sensitize cells more efficiently to TRAIL-induced apoptosis (Table 2).
Therapies based on TRAIL-mediated apoptosis, alone or in combination with other agents, are attractive possibilities for the treatment of many different types of cancer.14, 15, 16, 17 However, these treatments are unlikely to be helpful in the context of HPV on their own, owing to the ability of E6 to compromise apoptotic pathways. Our study found that the combination of TRAIL with small molecules can re-sensitize E6-expressing cells to TRAIL-induced apoptosis, potentially filling this gap. In addition to sensitizing cells to TRAIL, we found that both myricetin and spinacine sensitized cells to intrinsic apoptosis triggered by chemotherapy drugs, such as doxorubicin and cisplatin. This may be clinically relevant, because cervical cancer tends to be relatively resistant to chemotherapeutic treatments, such as cisplatin.36 We note that myricetin required a higher concentration (100 μM) than did spinacine (50 μM) to inhibit binding and to sensitize cells, suggesting that spinacine may be a more efficient agent. Taken together, these data suggest that myricetin and spinacine can block the binding of E6 to multiple apoptotic proteins, including caspase 8 and E6AP/p53, thereby reactivating the E6-compromised apoptotic pathways and rendering HPV+ cells sensitive to both intrinsic and extrinsic inducers of apoptosis.
The ability of myricetin and spinacine to sensitize HPV+ SiHa cells to apoptosis induced by TRAIL and chemotherapy agents led us to ask whether the same effects could be seen in HPV+ HNC cells. We found that the result from our HNC cells closely mirrored those from the cervical cells, indicating that our results should have broad applicability to HPV-associated malignancies regardless of the site of origin.
Our data suggest a mode of action in which the small molecule interacts directly with E6, either destabilizing the virus protein and changing its conformation, or blocking the interactions by direct interference. If the latter, one possibility is that myricetin/spinacine binds to a region on E6 required by both E6AP and caspase 8. Further work is necessary to develop a detailed model for the interactions between E6 and E6AP/caspase 8, which can then be used to design one or more highly specific inhibitors.
Overall, these results regarding the combination of small molecules with TRAIL/chemotherapeutic agents suggest a promising and novel therapeutic approach for the effective and selective killing of HPV+ cancer cells and may provide the basis for developing an effective therapeutic strategy to treat HPV-mediated cancers.
Materials and Methods
Protein purification
The construction of the pGEX-E6, pTriEx-E6AP, and pTriEx-Caspase-8 DED plasmids has been reported.33 Expression and purification of GSTE6, His-E6AP, and His-Caspase-8 DED were carried out as previously described.33, 37 GST-tagged and His-tagged proteins were diluted into GST dilution buffer (PBS pH 8.0, 5% glycerol, 2 mM DTT) and His dilution buffer (20 mM Hepes pH 7.4, 150 mM NaCl, 2 mMKCl, 5% glycerol, 2 mM DTT), respectively. Protein concentration was measured using Coomassie Plus – The Better Bradford Assay Reagent (Thermo Scientific, Waltham, MA, USA). Purity of the isolated proteins was estimated following separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie staining.
Small-molecule library and acquisition of additional compounds
The 2000-compound small-molecule library (ActiProbe 2K) was acquired from TimTec, LLC (Newark, DE, USA) and was chosen because it encompasses a highly diverse selection of lead-like compounds. Five additional derivatives were purchased from Sigma (St. Louis, MO, USA) (benzimidazole, 2-(methoxymethyl)-1H-benzimidazole, and 3-(1H-Indol-1-yl)propan-1-amine methanesulfonate) and TimTec (1H-benzimidazole-1-methanol and 4,5,6,7-tetrahydro-1H-imidazo[4,5–c]pyridine-6-carboxylic acid). (Note: The material provided by TimTec as 6,7-dihydro-1H-imidazo[4,5–c]pyridine-6-carboxylic acid was analyzed by 1H-NMR and MS and found to be instead 4,5,6,7-tetrahydro-1H-imidazo[4,5–c]pyridine-6-carboxylic acid whose common name is spinacine. Furthermore, comparison with the spectra of authentic spinacine (purchased and synthesized) confirms this identification.)
Screening of the small-molecular library
AlphaScreen Technology was used to assess the interactions between GST-E6, GST-caspase 8, His-E6AP, and His-caspase 8. Binding assays were performed in white 384-well plates (Perkin-Elmer) in a total volume of 25 μl as previously described.33 Briefly, 5 μl (50 ng) of GST-E6 and 5 μl (87.5 ng) of His-caspase 8 were included in each reaction mixture with 5 μl blocking buffer (0.5 mg BSA, 0.5% Tween 20 in PBS) in the absence or presence of 10 μM of each test chemical. Members of the library were present at 10 μM in DMSO. After a 1-h incubation of the mixture at room temperature, 5 μl donor beads and 5 μl acceptor beads (Perkin-Elmer) were added to each well according to the manufacturer’s protocol. The mixture was incubated in the dark at room temperature overnight, and the emitted signal was detected using the Envision Multilabel plate reader (Perkin-Elmer). In the presence of test chemicals, the binding affinity was calculated as a percentage of the binding in the presence of carrier only (DMSO).
Cell culture
U2OS, SiHa, and C33A cells were obtained from the America Type Culture Collection (Manassas, VA, USA) and cultured in Eagle's minimal essential medium (Invitrogen, Carlsbad, CA, USA) supplemented as described previously.33 HNSCC cell lines were obtained from several sources: UM-SCC47-TC-Clone 3 (#47CL3), UD-SCC2-TC-Clone 5(#2TC), UPCI-SCC90-UP-Clone 35 (#90), and SCC 84 were a gift from Dr. John Lee, Sanford Research (South Dakota, USA). UMSCC 47 (#47), UMSCC 29 (#29), and UMSCC 104 (#104) were a gift from Dr. Thomas Carey, University of Michigan (Michigan, USA). HNSCC cells were cultured in Dulbecco's Modified Eagle Medium (Mediatech, Manassas, VA, USA) supplemented with 10% of FBS.
Cell viability assay
The extracellular domain of human TRAIL was cloned into a pTriEx expression plasmid containing N-terminal Hisx6 tag. His-TRAIL was expressed in the E. coli BL-21 pLys strain and purified as previously described. Doxorubicin and cisplatin were purchased from Sigma. His-TRAIL, doxorubicin, and cisplatin were diluted in PBS to the desired concentration before using. To measure cell survival following treatment with TRAIL, cisplatin and doxorubicin, SiHa (2 × 104/well), C33A (1 × 104/well), and HNSCC cells (2 × 104/well) were seeded into 96-well plates and allowed to adhere overnight. Small molecules at the desired concentration were added and incubated at 37 °C for 4 h. As indicated, TRAIL, cisplatin, or doxorubicin was then added. The TRAIL treatment group contained cycloheximide (5 μg/ml) to inhibit de novo protein synthesis, and the cells were incubated for 16 h prior to measuring cell viability by the MTT assay preformed as described previously.38 All experiments were repeated at least three times (three biological replicates, carried out on different days), with each experimental group measured in triplicate within each of these individual experiments. Data presented are from a representative experiment.
Caspase activity assay
Cells were plated into 96-well plates at a density of 2 × 104 cells per well and incubated overnight. Small molecules were then added and incubated at 37 °C for 4 h. TRAIL (100 ng/ml), cisplatin (50 μM), and doxorubicin (2 μM) were then added. Cycloheximide (5 μg/ml) was added along with TRAIL at the indicated time points. Caspase 3/7 activity was measured using the flourogenic substrate CellTitier-Glo for caspase 3/7 activity (Promega, Fitchburg, WI, USA) following the manufacturer's instructions. Briefly, cells were lysed by the addition of 20 μl of 5 × passive lysis buffer (Promega). The plate was put on an orbital shaker and incubated for 10 min at room temperature. In all, 20 μl of cell lysates were transferred to white plates, and either substrate alone or substrate plus the caspase 3/7 inhibitor was added to the appropriate wells. After a 10-min incubation, the released fluorophore was measured using a plate-reading fluorimeter (Flx800, Bio-Tek Instrument Co., Winooski, VT, USA). The activity in wells treated with inhibitor was subtracted from the activity in wells lacking inhibitor. The resulting difference was expressed as a percentage of the caspase activity of the untreated cells.
Immunoblot analysis
Cells (1 × 106) were collected and washed with PBS. All liquid was removed by centrifugation at 3000 r.p.m. for 5 min at 4 °C. The cell pellet was lysed in 100 μl lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, with one tablet of protease inhibitor mixture (Roche Molecular Biochemicals, Basel, Switzerland) per 10 ml of buffer added just prior to use) for 10 min on ice. Lysates (10–40 μg total protein/lane) were then subjected to 10 or 12% SDS-PAGE and transferred to PVDF membranes using the iBlot dry blotting system (Invitrogen). Anti-caspase 8 monoclonal antibodies (BD Pharmingen, Franklin, NJ, USA) and anti-β-actin monoclonal antibodies (Sigma) were applied at 1 : 5000 dilutions. The secondary antibodies used were goat anti-mouse IRDye800 (LI-COR Biosciences, Lincoln, NE, USA) for caspase 8 and goat anti-mouse Dy680 (LI-COR Biosciences) for β-actin at 1 : 30 000 dilutions. Signals were measured using the Odyssey Infrared Imaging system (LI-COR Biosciences) and expressed in relative light units.
p53 ELISA
The p53 ELISA was performed as described previously,26 with some modifications. Antibodies secreted by clone pAb122 (hybridoma obtained from ATCC, antibodies purified from the culture medium using protein-A Sepharose) were used as monoclonal capture antibodies. This antibody has a broad specificity, binding to both human and mouse forms of p53 and to both wild-type and mutant forms of the protein. These antibodies were absorbed (50 μl per well, 4 μg/ml) onto the surfaces of a Nunc-immuno plate, MaxiSorp Surface (NalgeNunc International, Rochester, NY, USA) by incubation overnight at 4 °C. The plates were then washed on an Auto Strip Washer, ELX50 (Bio-Tek Instruments, Inc.) using PBST (PBS plus 0.1% Tween 20) six times. Nonspecific binding sites were blocked by incubation with 200 μl per well of PBS, including 10% calf serum (Invitrogen) (blocking buffer) for 2 h at room temperature, followed by washing as described above. In all, 100 μl of each cell lysate to be tested were then added to the coated wells and allowed to incubate overnight at 4 °C. After washing, 100 μl of a solution containing 4 μg/ml biotinylated anti-p53 antibodies (polyclonal, produced in sheep, Roche) diluted into blocking buffer was added to each well and allowed to incubate for 45 min at room temperature. The plates were washed, and then the avidin–peroxidase conjugate (Sigma; 100 μl/well, 2.5 μg/ml diluted into blocking buffer) was added and allowed to incubate for 30 min at room temperature. After washing, 100 μl of the substrate (0.3 mg/ml ABTS (2,2’-azino-di-(3-ethylbenzthiazolin sulfonate) dissolved into 0.1 M citric acid, pH 4.35, with 1 μl/ml 30% H2O2 added just before use) was added to each well and allowed to incubate for approximately 30 min. The absorbance at 405 nm was read with a microplate reader (Dynex Technologies; MRX Revelation software, Chantilly, VA, USA). The protein concentration of each lysate was also measured using the BCA method (Pierce, Chantilly, VA, USA) and used to normalize the measured p53 values for possible variations in the number of cells per well. Each p53 value (obtained from the ELISA assay) was divided by the protein concentration to obtain a normalized p53 value (ng p53 per mg total protein). The average and S.D. of the replicates (a minimum of three) were calculated, normalized to the control, and used to prepare the graph.
Abbreviations
- HPV:
-
human papillomavirus
- E6AP:
-
E6-associated protein
- TRAIL:
-
tumor necrosis factor-related apoptosis-inducing ligand
- FADD:
-
Fas-associated protein with death domain
- HNSCC:
-
head and neck squamous cell carcinoma
References
Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol 2011; 29: 4294–4301.
Adams AK, Wise-Draper TM, Wells SI . Human papillomavirus induced transformation in cervical and head and neck cancers. Cancers (Basel) 2014; 6: 1793–1820.
Bonomi P, Blessing JA, Stehman FB, DiSaia PJ, Walton L, Major FJ . Randomized trial of three cisplatin dose schedules in squamous-cell carcinoma of the cervix: a Gynecologic Oncology Group study. J Clin Oncol 1985; 3: 1079–1085.
Thigpen T, Shingleton H, Homesley H, Lagasse L, Blessing J . Cis-platinum in treatment of advanced or recurrent squamous cell carcinoma of the cervix: a phase II study of the Gynecologic Oncology Group. Cancer 1981; 48: 899–903.
Thigpen JT, Blessing JA, DiSaia PJ, Fowler WC Jr, Hatch KD . A randomized comparison of a rapid versus prolonged (24 hr) infusion of cisplatin in therapy of squamous cell carcinoma of the uterine cervix: a Gynecologic Oncology Group study. Gynecol Oncol 1989; 32: 198–202.
Scatchard K, Forrest JL, Flubacher M, Cornes P, Williams C . Chemotherapy for metastatic and recurrent cervical cancer. Cochrane Database Syst Rev 2012; 10: CD006469.
Lee TY, Jeung YJ, Lee CJ, Kim HY, Kim SH, Kim WG . Promising treatment results of adjuvant chemotherapy following radical hysterectomy for intermediate risk stage 1B cervical cancer. Obstet Gynecol Sci 2013; 56: 15–21.
Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L . Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 2004; 56: 185–229.
Buchsbaum DJ, Zhou T, Grizzle WE, Oliver PG, Hammond CJ, Zhang S et al. Antitumor efficacy of TRA-8 anti-DR5 monoclonal antibody alone or in combination with chemotherapy and/or radiation therapy in a human breast cancer model. Clin Cancer Res 2003; 9: 3731–3741.
Younes A, Kadin ME . Emerging applications of the tumor necrosis factor family of ligands and receptors in cancer therapy. J Clin Oncol 2003; 21: 3526–3534.
Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med 2001; 7: 954–960.
Chuntharapai A, Dodge K, Grimmer K, Schroeder K, Marsters SA, Koeppen H et al. Isotype-dependent inhibition of tumor growth in vivo by monoclonal antibodies to death receptor 4. J Immunol 2001; 166: 4891–4898.
Tanaka S, Sugimachi K, Shirabe K, Shimada M, Wands JR . Expression and antitumor effects of TRAIL in human cholangiocarcinoma. Hepatology 2000; 32: 523–527.
Bellail AC, Qi L, Mulligan P, Chhabra V, Hao C . TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev Recent Clin Trials 2009; 4: 34–41.
El-Zawahry A, McKillop J, Voelkel-Johnson C . Doxorubicin increases the effectiveness of Apo2L/TRAIL for tumor growth inhibition of prostate cancer xenografts. BMC Cancer 2005; 5: 2.
Mom CH, Verweij J, Oldenhuis CN, Gietema JA, Fox NL, Miceli R et al. Mapatumumab, a fully human agonistic monoclonal antibody that targets TRAIL-R1, in combination with gemcitabine and cisplatin: a phase I study. Clin Cancer Res 2009; 15: 5584–5590.
Naka T, Sugamura K, Hylander BL, Widmer MB, Rustum YM, Repasky EA . Effects of tumor necrosis factor-related apoptosis-inducing ligand alone and in combination with chemotherapeutic agents on patients' colon tumors grown in SCID mice. Cancer Res 2002; 62: 5800–5806.
Huibregtse JM, Scheffner M, Howley PM . A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J 1991; 10: 4129–4135.
Scheffner M, Huibregtse JM, Vierstra RD, Howley PM . The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993; 75: 495–505.
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM . The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990; 63: 1129–1136.
Huibregtse JM, Scheffner M, Howley PM . A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J 1991; 10: 4129–4135.
Crook T, Tidy JA, Vousden KH . Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and trans-activation. Cell 1991; 67: 547–556.
Lechner MS, Laimins LA . Inhibition of p53 DNA binding by human papillomavirus E6 proteins. J Virol 1994; 68: 4262–4273.
Tungteakkhun SS, Filippova M, Fodor N, Duerksen-Hughes PJ . The full-length isoform of human papillomavirus 16 E6 and its splice variant E6* bind to different sites on the procaspase 8 death effector domain. J Virol 2010; 84: 1453–1463.
Filippova M, Parkhurst L, Duerksen-Hughes PJ . The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J Biol Chem 2004; 279: 25729–25744.
Filippova M, Johnson MM, Bautista M, Filippov V, Fodor N, Tungteakkhun SS et al. The large and small isoforms of human papillomavirus type 16 E6 bind to and differentially affect procaspase 8 stability and activity. J Virol 2007; 81: 4116–4129.
Garnett TO, Filippova M, Duerksen-Hughes PJ . Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis. Cell Death Differ 2006; 13: 1915–1926.
Yuan CH, Filippova M, Tungteakkhun SS, Duerksen-Hughes PJ, Krstenansky JL . Small molecule inhibitors of the HPV16-E6 interaction with caspase 8. Bioorg Med Chem Lett 2012; 22: 2125–2129.
Garnett TO, Filippova M, Duerksen-Hughes PJ . Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis. Cell Death Differ 2006; 13: 1915–1926.
D'Abramo CM, Archambault J . Small molecule inhibitors of human papillomavirus protein - protein interactions. Open Virol J 2011; 5: 80–95.
Phelps WC, Barnes JA, Lobe DC . Molecular targets for human papillomaviruses: prospects for antiviral therapy. Antiviral Chem Chemother 1998; 9: 359–377.
Hebner C, Beglin M, Laimins LA . Human papillomavirus E6 proteins mediate resistance to interferon-induced growth arrest through inhibition of p53 acetylation. J Virol 2007; 81: 12740–12747.
Tungteakkhun SS, Filippova M, Neidigh JW, Fodor N, Duerksen-Hughes PJ . The interaction between human papillomavirus type 16 and FADD is mediated by a novel E6 binding domain. J Virol 2008; 82: 9600–9614.
Liu Y, Liu Z, Androphy E, Chen J, Baleja JD . Design and characterization of helical peptides that inhibit the E6 protein of papillomavirus. Biochemistry 2004; 43: 7421–7431.
Baleja JD, Cherry JJ, Liu Z, Gao H, Nicklaus MC, Voigt JH et al. Identification of inhibitors to papillomavirus type 16 E6 protein based on three-dimensional structures of interacting proteins. Antiviral Res 2006; 72: 49–59.
Rein DT, Kurbacher CM . The role of chemotherapy in invasive cancer of the cervix uteri: current standards and future prospects. Anticancer Drugs 2001; 12: 787–795.
Tungteakkhun SS, Filippova M, Fodor N, Duerksen-Hughes PJ . The full-length isoform of human papillomavirus 16 E6 and its splice variant E6* bind to different sites on the procaspase 8 death effector domain. J Virol 2010; 84: 1453–1463.
Filippova M, Parkhurst L, Duerksen-Hughes PJ . The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J Biol Chem 2004; 279: 25729–25744.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Baehrecke
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yuan, CH., Filippova, M., Krstenansky, J. et al. Flavonol and imidazole derivatives block HPV16 E6 activities and reactivate apoptotic pathways in HPV+ cells. Cell Death Dis 7, e2060 (2016). https://doi.org/10.1038/cddis.2015.391
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.391
This article is cited by
-
Effects of HPV16 E6 protein on Daxx-induced apoptosis in C33A cells
Cellular & Molecular Biology Letters (2020)
-
Identification of potential binding pocket on viral oncoprotein HPV16 E6: a promising anti-cancer target for small molecule drug discovery
BMC Molecular and Cell Biology (2019)
-
Nb-induced stabilisation of p53 in HPV-infected cells
Scientific Reports (2019)
-
A quantitative LumiFluo assay to test inhibitory compounds blocking p53 degradation induced by human papillomavirus oncoprotein E6 in living cells
Scientific Reports (2018)
