
Abstract
Resistance to ‘apoptotic’ cell death is one of the major hallmarks of cancer, contributing to tumor development and therapeutic resistance. Damage-associated molecular patterns (DAMPs) are molecules released or exposed by dead, dying, injured, or stressed non-apoptotic cells, with multiple roles in inflammation and immunity. Release of DAMPs not only contributes to tumor growth and progression but also mediates skewing of antitumor immunity during so-called immunogenic tumor cell death (ICD). Autophagy is a lysosome-mediated homeostatic degradation process in which cells digest their own effete organelles and macromolecules to meet bioenergetic needs and enable protein synthesis. For tumor cells, autophagy is a double-edged sword. Autophagy, in balance with apoptosis, can function as a tumor suppressor; autophagy deficiency, associated with alterations in apoptosis, initiates tumorigenesis in many settings. In contrast, autophagy-related stress tolerance generally promotes cell survival, which enables tumor growth and promotes therapeutic resistance. Most anticancer therapies promote DAMP release and enhance autophagy. Autophagy not only regulates DAMP release and degradation, but also is triggered and regulated by DAMPs. This interplay between autophagy and DAMPs, serving as ‘strange attractors’ in the dynamic system that emerges in cancer, regulates the effectiveness of antitumor treatment. This interplay also shapes the immune response to dying cells upon ICD, culling the least fit tumor cells and promoting survival of others. Thus, DAMPs and autophagy are suitable emergent targets for cancer therapy, considering their more nuanced role in tumor progression.
Similar content being viewed by others
Facts
-
Dysfunction of cell death pathways participates in the pathogenesis of many diseases, including cancer and autoimmunity.
-
The interplay between autophagy and damage-associated molecular pattern (DAMPs) regulates cell death and shapes the immune response to dying cells.
-
Autophagy has pro- and antitumor roles depending on the cellular context and early (anti-) and late (pro-) settings of tumorigenesis and tumor progression.
-
Immunogenic tumor cell death (ICD) involves the release of DAMPs, often necrotic death, and the subsequent elicitation of antitumor immunity; tolerogenic cell death (TCD) involves clearance of apoptotic cells or release of many of these same DAMP factors such as high mobility group box 1 (HMGB1), oxidized within the tissues.
Open Questions
-
What are the distinct roles of individual DAMPs in autophagy and tumor immunity?
-
What controls the network of autophagy signaling pathways?
-
Why are the effects of cell death on the immune system so complex?
-
Do dying cells directly induce DAMP secretion from immune cells following ICD or TCD?
-
What is the role of ICD/TCD in tumorigenesis and other diseases?
Randomness, with its own underlying order, is not a bad definition of an emergent cancer – wild and chaotic, yet bound by metabolic, bioenergetic, and spatial limitations with selection by immune effectors, ‘culling’ tumor cells and shaping their emergent behavior. In 2011, Hanahan and Weinberg1 updated their 10 hallmarks of cancer that define tumor cells, distinguishing them from normal cells. Of note, resistance to ‘apoptotic’ cell death is a major hallmark of cancer, which has a profound effect on the malignant phenotype and contributes to the development and progression of cancer. Necrosis releases multiple pro-inflammatory factors, including damage-associated molecular pattern molecules (DAMPs) within the surrounding microenvironment, which actively promotes tumor growth or mediates immune responses to dying cells. Autophagy, which had been proposed as a third mode of cell death, is primarily an intracellular degradation system and stress response required for survival. Autophagy has dual roles in cancer, acting as a tumor suppressor or promoter in a context-dependent manner.2 Apoptotic, autophagic, and necrotic tumor cells all release DAMPs, which interact with DAMP receptors on immune cells. This in part largely determines whether cell death is immunogenic (termed immunogenic tumor cell death (ICD)) or tolerogenic (termed tolerogenic cell death (TCD)).3 Here, we provide an overview of the cell death process and the release and function of DAMPs and focus on the immune response to cell death along with the functional interplay between autophagy and DAMPs in tumor therapy and immunity.
Cell Death Mechanisms in Cancer Therapy
The earliest classification of cell death was proposed in 1973, in which cell death is divided into apoptosis (type I), autophagic cell death (type II), and necrotic cell death (type III) based on morphological characteristics. Since then, new forms of cell death have been described, as biomedicine and technology have advanced from the bench to the bedside.4 Cancer is still best and most successfully treated by surgical excision with removal of the tumor. Nonsurgical therapies include chemotherapy, radiation therapy, hormonal therapy, and immunotherapy, which primarily inhibit cancer growth by promoting cell death with removal of the cellular corpse in situ. Apoptosis and necrosis are the two major types of tumor therapy-induced cell death, and induction of autophagy within the tumor is often significantly enhanced during tumor therapy. Clearly, apoptosis, necrosis, and autophagy can all be initiated by common stimuli and their regulatory mechanisms appear to be multiple and overlapping. In general, apoptosis and necrosis are both observed during effective anticancer activity, whereas induction of autophagy increases tumor cell resistance to treatment. The switch from one type of death response to another determines an individual tumor cell’s fate – either survival or death – following anticancer therapy.
Apoptosis: Programmed Cell Death
Apoptosis is the process of programmed cell death and includes ‘extrinsic’ and ‘intrinsic’ pathways, as well as the cytolytic mechanisms mediated by T and natural killer (NK) cells (Figure 1). All three cause characteristic cellular changes, including cell shrinkage and fragmentation into membrane-bound apoptotic bodies that are efficiently cleared by phagocytes. The extrinsic pathway begins outside the cell and involves transmembrane death receptors (DRs) that are members of the tumor necrosis factor receptor gene superfamily, which are activated by corresponding death ligands. In addition, dependence receptors (for example, unc-5 homolog A and deleted in colorectal carcinoma) mediate the alternative extrinsic pro-apoptotic pathway through the assembly of a caspase-9-activating platform or by the dephosphorylation-mediated activation of death-associated protein kinase 1, respectively. The intrinsic apoptotic pathway alters mitochondrial permeability and subsequent mitochondrial protein release. Most apoptosis is accompanied by a complex cascade of intracellular events that may include the activation of pro-apoptotic Bcl-2 family members, caspases, and several nucleases. However, the intrinsic apoptotic pathway can also function in a caspase-independent manner by translocation of apoptosis-inducing factor and endonuclease G from the mitochondria to the nucleus, thereby mediating large-scale DNA fragmentation. In addition, Omi/HTRA2, a mitochondrial serine protease, also contributes to caspase-independent apoptosis primarily by cleaving cytoskeletal proteins.

Extrinsic and intrinsic apoptosis. There are two intracellular pathways to control apoptosis. A third pathway, mediated by cytolytic effectors including T and NK cells, delivers pro-apoptotic granzymes through perforin-mediated pores, culling the least fit cells, and promoting survival of the remaining cells through autophagy. The extrinsic pathway is mediated by DRs including tumor necrosis factor family members such as Fas/CD95 and dependence receptors like deleted in colorectal carcinoma or unc-5 homolog A. The intrinsic mitochondrial pathway is controlled by Bcl-2 family proteins such as Bax and Bak. When lethal signals prevail, mitochondrial outer membrane permeabilization occurs and leads to release of the mitochondrial proteins such as Cyt C, SMAC, HTRA2, endonuclease G (ENDOG), apoptosis-inducing factor (AIF), and HTRA2. Among these, Cyt C, SMAC, and HTRA2 contribute to caspase-dependent apoptosis, whereas ENDOG, AIF, and HTRA2 contribute to caspase-independent apoptosis
Interestingly, increased apoptosis in tumor tissue is observed early in tumorigenesis. A possible reason for this is that rapid reparative epithelial proliferation, possible fusion events with inflammatory macrophages, and selection by lymphoid cells promotes early genetic instability required for tumor transformation and progression. The associated DNA damage in turn activates the intrinsic apoptosis pathway. Excessive apoptosis can lead to immune dysfunction within the tumor microenvironment. Recent studies indicate that loss of pro-apoptotic genes, such as CD95/Fas and PUMA, promotes tumorigenesis by activation of JNK, or sustains tumor stem cell survival.5, 6, 7 These findings raise the possibility that apoptosis promotes early tumorigenesis.8
Necrosis: More than an Accident
The term ‘necrosis’ was used to describe irreversible tissue damage in pathological circumstances and was considered a merely accidental cell death mechanism lacking the morphological characteristics of apoptosis or autophagy. The term ‘necroptosis’ has recently been used to describe programmed necrosis when cells lack the capacity to activate caspase-8 following DR ligation.9 Generation of the kinase receptor-interacting protein 1 (RIP1)/RIP3 containing ‘necrosomes’ activates downstream necroptotic signaling, which can be inhibited by necrostatin 1 (Figure 2). Morphologically, necrosis is characterized by disruption of the cellular membrane, swelling of the cytoplasm and mitochondria, and breakdown of organelles. DNA in apoptotic cells is degraded specifically, whereas DNA in necrotic cells is usually degraded randomly by extracellular DNAseI or by lysosomal DNAse II. In necrosis, cellular contents leak into the extracellular environment where they may act as a ‘danger signal’ that promotes inflammation. The fundamental causes of necrosis include calcium overload, ROS generation, cellular energy depletion, and membrane lipid injury. Poly (ADP-ribose) polymerase (PARP) is activated by DNA strand breaks, thereby facilitating DNA repair and enzyme access to damaged DNA. Induced overactivation of PARP-1 causes ATP depletion, which leads to necrosis in a TRAF2/RIP1/JNK-dependent manner. Activation of necrosis often occurs through increased ROS production, calpain activation, lysosomal destabilization, and cathepsin release. Necrotic death is induced in cancer cells by photodynamic treatment (PDT), several DNA alkylating agents, and other cytotoxic agents, including β-lapachone, apoptolidin, and honokiol.

Pathways of necrosis. Necrosis can be elicited by a wide range of stimuli. Activation of DRs such as Fas and TNFR or cellular stresses induces the interaction and activation of the kinases RIP1 and RIP3. Necrotic death stimuli can also activate PARP-1, which can potentially induce necrosis either through activation of the RIP kinases or another signaling pathway. Activation of necrosis by RIP1/RIP3 and PARP-1 is thought to occur through cathepsin release, increased ROS production, ATP depletion, and calpain activation. Necrostatin 1 is a small molecule inhibitor of RIP1
Autophagy: an Intracellular Degradation System
The primary autophagy mechanisms, including macroautophagy, microautophagy, and chaperone-mediated autophagy, are the evolutionarily highly conserved catabolic pathways involving degradation of cellular components.10 The main identified form of autophagy is macroautophagy (hereafter referred to as autophagy), which initiates with engulfment of cytosolic material by the phagophore, resulting in the formation of autophagosomes. Autophagosomes then fuse with lysosomes to form autolysosomes, which lead to degradation of the engulfed material by lysosomal enzymes and production of amino acids and fatty acids to meet bioenergetic needs and protein synthesis (Figure 3a). This dynamic process is primarily controlled by members of the autophagy-related gene (Atg) family and share regulators derived from other trafficking and cell death pathways.

The process and types of autophagy. (a) Autophagy is an intracellular bulk degradation system through which cytoplasmic components are delivered to lysosomes to be degraded. The main process of autophagy includes formation and maturation of the phagophore, autophagosome, and autolysosome. LC3, a mammalian homolog of yeast Atg8, is localized in autophagosome membranes after processing to LC3-II and can be degraded by the autolysosome. Autophagy provides nucleic, amino, and fatty acids for the synthesis of DNA/RNA, protein, and ATP. (b) Types of selective autophagy. Autophagy can also target selective cargo for degradation such as organelles, proteins, microbes, and RNA
During starvation, mammalian target of rapamycin 1 dissociates from the unc-51-like kinase 1 (ULK1) complex, allowing it to initiate autophagosome formation. There are at least three class III phosphatidylinositol 3 kinase (PI3KC3)–beclin-1 complexes that are involved in the regulation of autophagosome formation. The Atg14L (Atg14L–beclin-1–hVps34–p150) and UVRAG (UVRAG–beclin-1–hVps34–p150) complexes are required for autophagy, whereas the Rubicon complex (Rubicon–UVRAG–beclin-1–hVps34–p150) negatively regulates autophagy. Moreover, discovery and identification of beclin-1-binding protein increases the complexity of this process.11 Microtubule-associated protein light chain 3 (LC3)-II as an autophagosome marker ultimately degrades in the autolysosome. Impairment of the early and late steps of autophagy can have opposing effects on the number of autophagy-related vacuoles, as well as LC3-II expression. Autophagy is not only a nonselective bulk process in response to starvation but also a highly selective quality control mechanism in regulation of organelle turnover, cellular debris disposal, and pathogen clearance (Figure 3b).
Autophagy is primarily an anti-oncogenic mechanism during tumorigenesis because deletion of autophagy genes or regulators such as beclin-1, UVRAG, ATG5, and Bif in mice increases tumor development by their ability to shape inflammatory responses, bioenergetics, and oxidative stress. In addition, autophagy has a pro-tumor growth effect in some cancers. For example, loss of ATG3 in mice prevents BCR-Abl-mediated leukemogenesis. Similar to apoptosis, these findings suggest that autophagy has a complex role in tumorigenesis and may have opposing roles in the early and late cancer development stages.2
Immune Responses to Cell Death: Immunogenicity and Tolerogenicity
Apoptosis is usually considered intrinsically tolerogenic, whereas necrosis is immunogenic and promotes inflammation.12 A characteristic of apoptotic cell clearance is that no pro-inflammatory responses are induced. It is widely accepted that removal of dying cells is achieved either by phagocytes or neighboring cells.
The macrophage is the major phagocyte to clear dying cells, and it has a preference for clearing apoptotic rather than necrotic cells, although the post-engulfment functional consequences are significantly different. Phagocytosis of apoptotic cells by macrophages triggers the release of anti-inflammatory cytokines, whereas the production of pro-inflammatory cytokines is suppressed. Phagocytosis of necrotic cells does not induce pro-inflammatory cytokine production in macrophages. Phagocytosis of autophagic cells induces inflammasome activation and IL-1β secretion in macrophages, suggesting that autophagy is not immunologically silent.
The dendritic cell (DC) is another cell type derived from the mononuclear phagocyte. Uptake of necrotic tumor cells induces maturation of DCs with the capacity to induce antigen-specific CD4+ and CD8+ T cells and the immune response. In contrast, phagocytosis of apoptotic cells by DCs fails to induce maturation and causes tolerance to tumor antigens by generating helpless CD8+ T cells that produce TRAIL to kill activated T cells. Phagocytosis of apoptotic cells, however, may lead to T-cell immunity if followed by an additional maturation signal provided by DAMPs, pathogen-associated molecular patterns (PAMPs), inflammatory products, and CD40L-CD40 interactions. DC type and tissue localization are also important in shaping immune responses to cell death.13 In addition, the redox status of DAMPs from dying cells determines whether cell death is ICD or TCD. For example, the production of ROS in apoptotic cells can oxidize cysteine 106 in high mobility group box 1 (HMGB1). This oxidized HMGB1 cannot activate DCs and has tolerogenic activities.14
Currently, it is clear that the immunogenic characteristics of dying cells are mainly mediated by DAMPs, which will be further discussed in the section below, ‘DAMPs, Immunogenic Cell Death, and Cancer Therapy’. It is important to note that both apoptotic and necrotic cells have the ability to release DAMPs, but necrotic cells may release much of their content and have varied and increased DAMP activity. In addition, autophagy dysfunction also contributes to cell death-associated immune responses through regulation of phagocytosis, DC activation and maturation, and DAMP release and degradation, which will also be further discussed in the sections ‘Autophagy, Immunity, and Tumor Cell Death’ and ‘Interplay between DAMPs and Autophagy’. Taken together, the crosstalk between dying and immune cells determines outcome, whether or not the death of the cell is ICD or TCD.
DAMPs, Immunogenic Cell Death, and Cancer Therapy
DAMPs are molecules that are secreted, released, or undergo surface exposure by dying, stressed, or injured cells.15 These molecules are mainly recognized by pattern recognition receptors (PRRs). Many of the individual PRRs also detect PAMPs, suggesting similar activity between DAMPs and PAMPs in the regulation of immunity.16 The most studied DAMPs are HMGB1, the S100 calcium-binding protein family, heat shock proteins (HSPs), ATP, uric acid, and DNA. The list of DAMPs is rapidly increasing, with recent new additions such as histone, mitochondrial DNA, mitochondrial transcription factor A, peroxiredoxin, and cold-inducible RNA-binding protein.
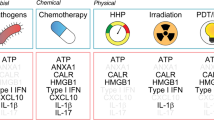
More recently, increasing evidence suggests that particular DAMPs serve as powerful immunological adjuvants and mediate ICD in cancer therapy.17, 18 ICD is the process by which DAMPs from dying cells contribute to immune-mediated eradication of tumors during chemotherapy (for example, anthracyclines), radiotherapy, or PDT (for example, hypericin-based photodynamic therapy, Hyp-PDT) (Figure 4). ICD is characterized by the exposure to and/or release of calreticulin,19 HMGB1,20 HSP70/HSP90,21, 22 and ATP23 from pro-apoptotic, post-apoptotic, and/or necrotic cells. Calreticulin, HMGB1/HSPs, and ATP then interact with the receptors CD91, Toll-like receptor 4(TLR4), and purinergic P2X7 receptors, respectively, which are present on the surface of DCs. CD91, TLR4, and P2X7 are present on DCs and promote engulfment of dying cells, presentation of tumor antigens, and production of inflammasome-dependent IL-1β release, respectively. A tumor-specific immune response, such as the cytotoxic T lymphocyte or NK response, is triggered to kill (and cull) cancer cells during cancer therapy. Apart from these DAMPs and signaling, a recent study suggested that accumulation of cation-independent mannose-6-phosphate receptor on the tumor cell surface is also important for ICD.24 Of note, the signaling pathways elicited by distinct ICD inducers (for example, anthracyclines and Hyp-PDT) overlap but are not identical. Interestingly, ICD-mediated tumor clearance following chemotherapy works only in tumor cell line transplantation models or the immunogenic 3-methylcholanthrene fibrosarcoma model but not in spontaneous tumor models.25 Therefore, it appears that ICD is dependent on additional factors such as tumor type and convergent signals in the tumor microenvironment. Below, we highlight examples of DAMPs as immunogenic effectors to ICD and their multiple functions in cancer.

The process of immunogenic cell death (ICD). Cancer cells responding to ICD inducers expose calreticulin and HSP70/HSP90 on the outer leaflet of their plasma membranes at a preapoptotic stage and secrete ATP during apoptosis and autophagy. In addition, cells undergoing ICD release HMGB1 during necrosis. Consequently, these DAMPs interact with receptors on immature DCs such as CD91, P2X7, TLR4, and with unknown surface molecules. These DAMPs cause maturation of DCs, characterized by cell-surface upregulation of MHC-II, CD86, CD83, and CD80 and a distinctly pro-inflammatory cytokine pattern as indicated. Altogether, these processes result in IL-1β-dependent activation of IL-17-producing γδT cells and increased proliferation of IFN-γ-producing CD4+ or CD8+ αβ T cells, which eventually can lead to the eradication of therapy-resistant tumor cells
Calreticulin
Calreticulin was discovered in 1974 and is a highly conserved and unique ER luminal resident protein (Figure 5a). Within the ER lumen, calreticulin has two major functions: chaperoning and regulating calcium metabolism.26 As a chaperone, it has a critical role in correctly folding many proteins and glycoproteins, including ion channels, surface receptors, integrins, and transporters. Calreticulin affects the Ca2+ capacity of the ER stores by interacting with other chaperones, specific substrates, and other ER proteins including SERCA2b, IP3 receptor, and ERp57. Studies on calreticulin-knockout mice indicate that the protein is essential in early cardiac development. Calreticulin has been identified in many other cellular compartments, including the cytosol or nucleus or both, the cell membrane, and the extracellular matrix, which supports calreticulin’s other functions outside the ER, such as regulation of nuclear transport, gene transcription, proliferation, migration, adhesion, and phagocytosis.26

Calreticulin structure and exposure. (a) Linear representation of calreticulin domains is shown. The protein contains an N-terminal amino-acid signal sequence, N-domain, P-domain, C-domain, and a C-terminal KDEL ER retrieval signal (not shown). Repeats A (amino-acid sequence PXXIXDPDAXKPEDWDE) and B (amino-acid sequence GXWXPPXIXNPXYX) are located in the P-domain. The N- and P-domains of calreticulin are responsible for the chaperone function of the protein. The C-terminal C-domain contains a large number of negatively charged amino acids and is involved in high-capacity Ca2+ storage. In addition, the P-domain is responsible for binding to ERp57. Calreticulin contains three redox-sensitive cysteine residues (C106, C137, and C163), which are important for calreticulin function. (b) A summary of the mechanism of calreticulin exposure
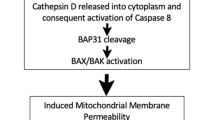
Calreticulin is exposed on the outer leaflet of cells during the early phase of cell death following treatment with anthracyclins. The formation of surface-exposed calreticulin (ecto-CRT) is an active process and precedes phosphatidylserine exposure and plasma membrane permeabilization. Ecto-CRT initiates ICD and facilitates the engulfment of dying tumor cells by DCs, which increases cancer immunogenicity.19 The mechanism for the translocation of ecto-CRT is variable and depends on ICD inducers, although the ER stress pathway has a central role. The anthracycline-stimulated translocation of ecto-CRT has been deconvoluted into three sequentially activated but functionally distinguishable signaling modules: (1) an ER stress module, (2) an apoptotic module, and (3) a translocation module. ER stress is characterized by the overgeneration of ROS, increased cytoplasmic Ca2+ concentrations, and the activation of EIF2AK3/PERK-mediated phosphorylation of eIF2a. The subsequent apoptosis induction involves caspase-8, Bax and Bak, and the ER protein Bap31. Finally, the generation of ecto-CRT is dependent on an ER-to-Golgi secretory pathway or a PI3K-dependent exocytosis pathway (Figure 5b). Moreover, interaction between calreticulin and ERp57 is responsible for their cotranslocation to the cell surface during ICD. Conversely, the pathway-mediated Hyp-PDT-induced calreticulin exposure only requires PERK and Bax/Bak, but it is independent on eIF2a, caspase-8, and ERp57.22 In addition, the calreticulin receptor CD91 is required for engulfment of dying tumor or nontumor cells by immature DCs or macrophages, respectively. These studies suggest that calreticulin exposure by preapoptotic cells is a complex and multiple step process.
HMGB1
HMGB1 was discovered in 1973 and is the most abundant non-histone nuclear protein (Figure 6a). The function of HMGB1 is dependent on its subcellular localization. In the nucleus, HMGB1 as a DNA-binding and -bending protein supports chromatin structure and regulates DNA repair and genomic stability. It is also a cell surface membrane-expressed protein on activated platelets and early neurons involved in migration and outgrowth. HMGB1 can be actively secreted from immune cells or passively released from dying, dead, or injured cells, and mediates sterile and infectious inflammation responses by binding to multiple cell surface receptors. The immune response mediated by HMGB1 lies not simply in the release and receptors of HMGB1 but rather also in the redox state of released HMGB1.27 In addition, extracellular HMGB1 acts to form heterocomplexes with other molecules to initiate a synergistic immune response.

HMGB1 structure and release. (a) Linear representation of HMGB1 domains is shown. The protein contains two homologous DNA-binding domains (box A and box B) and a negatively charged C-terminal domain. Residues 150–183 are responsible for binding to RAGE, whereas residues 89–108 and residues 7–74 are responsible for binding to TLR4 and p53 transactivation domains, respectively. Two nuclear localization signals (NLS1 and NLS2) and two nuclear emigration signals (NES1 and NES2; not shown) control the nuclear transport of HMGB1. In addition, HMGB1 contains three redox-sensitive cysteine residues (C23, C45, and C106), which are important for HMGB1 activity. (b) A summary of the mechanisms of HMGB1 release
Cells can release HMGB1 actively or passively by several identified mechanisms (Figure 6b). There are two steps for HMGB1 secretion from activated immune cells: nuclear protein exporter exportin 1 (CRM1) mediates HMGB1 nuclear export and secretory lysosome-mediated HMGB1 exocytosis. HMGB1 is the prototypic DAMP released by necrotic cells in a PARP-1-dependent way. Both apoptotic and autophagic cancer cells also contribute to HMGB1 release, promoted by caspase3/7 activation and ATG5, respectively. More recently, PKR-mediated inflammasome activation was found to be an essential regulator of HMGB1 release during pyroptosis.28 In contrast to the inflammatory response initiated by necrotic cells, ROS produced by and HMGB1 released from apoptotic cells promote tolerance in DCs.14 Interestingly, macrophage engulfment of apoptotic cells induces HMGB1 release and an inflammatory response.29 These observations revealed phagocytosis-associated crosstalk between macrophages and apoptotic cells; therefore, the concept that apoptosis is noninflammatory is not always correct. More recently, it was discovered that oxidized mitochondrial DNA released from apoptotic cells promotes inflammasome activation.30 HMGB1-containing nucleosomes from apoptotic cells induce both inflammation and immune activation.31 Thus, HMGB1 both on its own as well as in its relationships with apoptosis and immunity are complex.
HMGB1 dysfunction is associated with each of the hallmarks of cancer.32 Serum HMGB1 and HMGB1 expression in cancer tissue are generally increased in the setting of cancer. HMGB1 release promotes tumor growth, proliferation, and metastasis owing to its cytokine, chemokine, and growth factor activity. However, HMGB1 also functions as a breast cancer suppressor by binding to retinoblastoma, a tumor suppressor protein.33 A dynamic relationship between intracellular and extracellular pools of HMGB1 in tumorigenesis remains to be fully investigated.
HMGB1 has positive and negative influences on tumor immunity.34 On one hand, HMGB1 suppresses lymph node DC survival, diminishes naturally acquired CD8+ T-cell-dependent antitumor immunity, and promotes membrane lymphotoxin-α1β2 expression on tumor-activated T cells and subsequent accumulation of tumor-associated macrophages. This in turn promotes tumor growth and metastasis. On the other hand, HMGB1 can trigger protective DC-based anti-neoplastic T-cell responses during ICD. Blocking the HMGB1/TLR4 pathway inhibits dying tumor cell-mediated anticancer immune responses upon chemotherapy. Surprisingly, tumor-associated DC expression of the receptor TIM-3 inhibits antitumor efficacy of DNA vaccines and chemotherapy by binding to HMGB1.35 Besides immune cells, cancer cells also express HMGB1 receptors and have varied roles in cancer formation and treatment. We found that loss of the receptor for advanced glycation end products (RAGE) increases chemotherapy sensitization and inhibits pancreatic carcinogenesis with a significant diminution of myeloid-derived suppressor cells.36, 37, 38 HMGB1 released from necrotic cancer cells treated with chemotherapy enhances regrowth and metastasis of the remaining cancer cells through RAGE activation39 and RAGE-promoted bioenergetics.40 These studies suggest that the function of extracellular HMGB1 in tumor immunity might be receptor dependent.
HSPs
The highly conserved HSPs are constitutively expressed and function as molecular chaperones, which facilitate the synthesis and folding of proteins. Increased expression of HSPs protects the cell mainly by stabilizing unfolded proteins, inducing proteasomal degradation, and preventing apoptosis. In addition, cell stress and injury-mediated release of HSPs results in pro-inflammatory cytokine/chemokine release and activation/maturation of antigen-presenting cells (APCs) to produce a robust innate immune response.41 However, controversy still exists as to whether HSPs themselves are DAMPs.
The expression of HSPs is increased in various human tumors (especially those of epithelial origin or gliomas) and their expression often correlates with increased cell proliferation, lymph node metastases, poor response to chemotherapy, and poor prognosis. Thus, knockdown or knockout of HSPs increase anticancer drug-induced apoptosis and inhibit tumor grow. Apart from their intracellular location, several HSPs such as HSP90 and HSP70 have been found on the plasma membrane of tumor cells and in the extracellular milieu during cell death. HSP90 as well as HSP70 peptide complexes are potent stimulators of the adaptive immune system.42 HSP-chaperoned peptides are taken up by professional and nonprofessional APCs via receptor-mediated endocytosis and thus become cross-presented as classical antigens for CD8-positive cytotoxic T cells on MHC class I molecules.41, 43 TLR4 is the major receptor that recognizes HSP exposed by tumor cells, which facilitates intracellular antigen processing and presentation. Other receptors such as TLR2, CD40, CD91, CCR5, and members of the scavenger receptor family also mediate binding and uptake of HSPs into APCs. Surface-exposed HSP70/HSP90 with calreticulin can mediate T-cell-based antitumor immunity and contribute to IDC.21, 22 The mechanism of export and uptake of HSPs between immunogenic tumor cells with immature DCs remains to be elucidated.
ATP
Intracellular ATP is the major cellular energy currency involved in many cellular functions. Extracellular ATP can propagate signals via P2 receptors including P2Y and P2X that are essential for adhesion, proliferation, differentiation, mobility, immunity, and inflammation.44 Extracellular ATP contributes to inflammation and immunity, primarily by activation of the NALP3 inflammasome pathway. Extracellular ATP contributes to tumorigenesis in multiple ways, including serving as a growth factor for stem cells and a Ca2+ regulator for tumor invasion and metastasis. However, higher concentrations of extracellular ATP can also induce apoptosis and/or necrosis in cancer cells. Extracellular ATP can act as a ‘find me’ signal when it accumulates in the extracellular medium during apoptosis and promote P2Y2-dependent recruitment of phagocytes.45 Although the mechanisms responsible for ATP release during cell death have not been defined, recent studies suggest that pannexin 1 channels, individual ATGs (ATG5, ATG7, and beclin-1), PERK-based trafficking, and PI3K-dependent exocytosis regulate ATP release during cell death.
Extracellular ATP released from cells undergoing ICD activates purinergic P2X7 receptors on DCs.23 This activates the NALP3-ASC-inflammasome and drives the secretion of IL-1β, which is required for polarization of IFN-γ-producing CD8+ T cells and for the immune response to tumor cells.23 HMGB123 or calreticulin46 could synergize with ATP to induce IL-1β release by DCs. As a negative feedback mechanism, however, extracellular ATP could also be converted to immunosuppressive adenosine following hydrolysis of ATP by the action of surface expressed ectonucleotidases, such as CD39 and CD73.
Autophagy, Immunity, and Tumor Cell Death
Autophagy helps clear apoptotic cells during embryogenesis by the generation of energy-dependent engulfment signals including ‘eat me’ and ‘find me’ signals. In addition, LC3-associated phagocytosis (LAP) is also involved in macrophage-mediated clearance of necrotic, apoptotic, and necroptotic cells and the subsequent immune silence by inhibition of IL-1β release.47, 48 LAP is a recently described form of phagocytosis in which the activation of PI3K leads to direct recruitment of autophagy factors ATG5, ATG7, and LC3 to phagosomes.47, 48 Interplay between autophagy, LAP, and phagocytosis regulates the clearance of dying cells.49
There is a critical role for autophagy in tumor immunology and immunotherapy.50 Autophagy is upregulated at the immunological synapse during DC–T-cell contact.51 Suppression of autophagy in DCs results in hyperstable contacts between DCs and CD4+ T cells and increases T-cell activation.51 Autophagy contributes to CD4+ T-cell survival by degradation of pro-apoptotic proteins.52 DCs use autophagy to promote cross-presentation of tumor antigens on MHC class I complexes for cytotoxic T-lymphocyte activation and to facilitate antigen expression on MHC class II molecules for T-helper cell activation. Other studies show that autophagy within tumor cells favors cross-priming of tumor-associated antigen-specific CD8+ T cells.53, 54 Autophagosomes from cancer cells are a source of specific tumor-associated antigens, and autophagosome-enriched and autophagosome-based vaccines have potent antitumor efficacy. These findings suggest that autophagy contributes to APC-mediated antitumor immunity. In addition, autophagy contributes to IFNγ/STAT1-mediated antimetastatic immunotherapy. Autophagy also has an immunosuppressive effect in cancer therapy. Hypoxia-induced autophagy increases STAT3 phosphorylation, which impairs CTL-mediated tumor cell lysis in lung cancer cells.55 Thus, autophagy has a dual role in tumor immunity and has in some instances a paradoxical influence, requiring further elucidation.
Chemotherapy-induced autophagy promotes ICD and antitumor immune responses by regulation of ATP release56 or mannose 6-phosphate receptor accumulation on the cell membrane from dying cells.24 Interestingly, autophagy is not required for calreticulin exposure and HMGB1 release by cancer cells upon ICD,56 although other studies show that autophagy is a critical regulator of HMGB1 release during starvation, infection, and chemotherapy. These findings indicate that autophagy has a selective role in regulation of ICD as well as HMGB1 release.
Strange Attractors: Interplay between DAMPs and Autophagy
HMGB1, one of the best characterized DAMP, has important nuclear, cytosolic, and extracellular roles in the regulation of autophagy (Figure 7). Cytoplasmic HMGB1 induces autophagy by binding to beclin-1 and subsequent activation of the autophagy-initiating beclin-1–PI3KC3 complex.57 p53 and ULK1 have opposing roles in regulation of the HMGB1–beclin-1 complex formation in cancer cells.58, 59 Nuclear HMGB1 modulates the expression of HSPB1/HSP27 and dynamic intracellular trafficking during autophagy and mitophagy.60 The Pink1/Parkin pathway has been implicated in mitophagy and mitochondrial dysfunction in humans, which is also required for HSPB1/HMGB1-mediated mitophagy.60 These findings reveal a novel pathway coupling autophagy and cellular energy metabolism. Extracellular HMGB1 induces autophagy, and this role is dependent on its redox state and RAGE expression.61 HMGB1- and RAGE-mediated autophagy promote chemoresistance in cancer cells, including colon cancer, pancreatic cancer, osteosarcoma, leukemia, gastric cancer, and ovarian cancer.38, 58, 59, 62 Moreover, RAGE modulates crosstalk between two pro-survival pathways (STAT3 and autophagy) in pancreatic ductal adenocarcinoma tumor cells, and contributes to early pancreatic intraepithelial neoplasia formation.37 Whether the HMGB1–RAGE-mediated autophagy pathway is involved in other settings, including ICD, remains unknown.

The interplay between HMGB1 and autophagy. HMGB1 has important nuclear, cytosolic, and extracellular roles in the regulation of autophagy. Autophagy may also have a central role in the regulation of cellular traffic, secretion, and degradation of HMGB1. Although the connection between HMGB1 and autophagy is well recognized, the fully explicated molecular basis for it remains elusive
Autophagy may have a central role in regulation of the cellular traffic, secretion, and degradation of HMGB1 (Figure 7). Autophagy not only regulates passive HMGB1 release from dying cells but also regulates active HMGB1 secretion from immune and cancer cells by ROS-dependent signals. Intriguingly, it was recently shown that induction of autophagy contributes to HMGB1 release during formation of IL-1β-mediated neutrophil extracellular traps,63, 64 and with treatment with several anti-inflammatory drugs (for example, Tanshinone IIA sodium sulfonate and green tea)-induced HMGB1 degradation.65, 66
The complex interplay between DAMPs other than HMGB1 (for example, S100, DNA, and ATP) and autophagy as ‘strange attractors’ has been demonstrated in several recent studies.67 For example, autophagy-deficient macrophages accumulate dysfunctional mitochondria, which produce higher mitochondrial ROS, translocate more mitochondrial DNA, and activate caspase-1 more robustly than normal macrophages.68 Thus, preservation of mitochondrial integrity by autophagy proteins is important in regulating mitochondrial DAMP-mediated NLRP3 inflammasome activation. In addition to the previously known role of LAP in acceleration of phagosome maturation,47 LAP is required for type I interferon secretion following TLR9 stimulation by DNA-immune complexes.69 ATP-induced autophagy contributes to phagocytosis and destruction of invading microbes,70 whereas S100A8/A9-induced autophagy has a key role in the removal of damaged mitochondria in the setting of apoptosis.71 These findings suggest a link between DAMP and autophagic clearance during infection and cell death.
Conclusions and Future Challenges
Dysfunction of cell death participates in the pathogenesis of many diseases, including cancer and autoimmunity. Three distinct types of cell death can be distinguished by morphological criteria, namely, apoptosis, necrosis, and autophagic cell death. However, their function is significantly different following cellular stress. In general, induction of apoptosis and necrosis is accompanied by protective autophagy, and induction of autophagy promotes cell survival under stressful conditions. As we have reviewed, the study of dying cell clearance and DAMP release has a key role in the underlying mechanisms of immune response to cell death. Phagocytosis, autophagy, and LAP contribute to the clearance of dying cells through phagocytic cells from the innate immune system. DAMPs are released by dying cells, including apoptotic, necrotic, and autophagic cells, which alert the host to cell death and trigger an inflammatory and immune response. Of note, the interplay between autophagy and DAMPs regulates cell death and shapes the immune response to dying cells, including ICD and TCD. Despite efforts to understand the basis of ICD/TCD, the precise mechanism of ICD/TCD and their roles in disease remains stubbornly unclear. In addition, new insights into the mechanistic basis of autophagy-mediated stress responses and DAMP-mediated immune responses will open new perspectives for the development of molecular targeted treatment approaches and thus have great potential in drug discovery.
Abbreviations
- DAMPs:
-
damage-associated molecular patterns
- ICD:
-
immunogenic tumor cell death
- TCD:
-
tolerogenic cell death
- PDT:
-
photodynamic treatment
- PAMPs:
-
pathogen-associated molecular patterns
- TLRs:
-
Toll-like receptors
- RAGE:
-
the receptor for advanced glycation end products
- HMGB1:
-
high mobility group box 1
- Hyp-PDT:
-
hypericin-based photodynamic therapy
- ecto-CRT:
-
surface-exposed calreticulin
- LAP:
-
LC3-associated phagocytosis
- PI3KC3:
-
class III phosphatidylinositol 3 kinase
References
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
White E . Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12: 401–410.
Green DR, Ferguson T, Zitvogel L, Kroemer G . Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009; 9: 353–363.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19: 107–120.
Chen L, Park SM, Tumanov AV, Hau A, Sawada K, Feig C et al. CD95 promotes tumour growth. Nature 2010; 465: 492–496.
Labi V, Erlacher M, Krumschnabel G, Manzl C, Tzankov A, Pinon J et al. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev 2010; 24: 1602–1607.
Michalak EM, Vandenberg CJ, Delbridge AR, Wu L, Scott CL, Adams JM et al. Apoptosis-promoted tumorigenesis: gamma-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev 2010; 24: 1608–1613.
Tang D, Lotze MT, Kang R, Zeh HJ . Apoptosis promotes early tumorigenesis. Oncogene 2011; 30: 1851–1854.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G . Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11: 700–714.
Klionsky DJ, Emr SD . Autophagy as a regulated pathway of cellular degradation. Science 2000; 290: 1717–1721.
Kang R, Zeh HJ, Lotze MT, Tang D . The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18: 571–580.
Rock KL, Kono H . The inflammatory response to cell death. Annu Rev Pathol 2008; 3: 99–126.
Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S et al. Differential antigen processing by dendritic cell subsets in vivo. Science 2007; 315: 107–111.
Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA . Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008; 29: 21–32.
Zitvogel L, Kepp O, Kroemer G . Decoding cell death signals in inflammation and immunity. Cell 2010; 140: 798–804.
Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT . PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev 2012; 249: 158–175.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L . Immunogenic cell death in cancer therapy. Annu Rev Immunol 2012; 31: 51–72.
Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P . Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012; 12: 860–875.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13: 54–61.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007; 13: 1050–1059.
Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV . Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood 2007; 109: 4839–4845.
Garg AD, Krysko DV, Vandenabeele P, Agostinis P . Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother 2012; 61: 215–221.
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 2009; 15: 1170–1178.
Ramakrishnan R, Huang C, Cho HI, Lloyd M, Johnson J, Ren X et al. Autophagy induced by conventional chemotherapy mediates tumor cell sensitivity to immunotherapy. Cancer Res 2012; 72: 5483–5493.
Ciampricotti M, Hau CS, Doornebal CW, Jonkers J, de Visser KE . Chemotherapy response of spontaneous mammary tumors is independent of the adaptive immune system. Nat Med 2012; 18: 344–346 author reply 346.
Krause KH, Michalak M . Calreticulin. Cell 1997; 88: 439–443.
Tang D, Billiar TA, Lotze MT . A Janus tale of two active HMGB1 redox states. Mol Med 2012; 18: 1360–1362.
Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012; 488: 670–674.
Qin S, Wang H, Yuan R, Li H, Ochani M, Ochani K et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med 2006; 203: 1637–1642.
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012; 36: 401–414.
Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med 2008; 205: 3007–3018.
Tang D, Kang R, Zeh HJ 3rd, Lotze MT . High-mobility group box 1 and cancer. Biochim Biophys Acta 2010; 1799: 131–140.
Jiao Y, Wang HC, Fan SJ . Growth suppression and radiosensitivity increase by HMGB1 in breast cancer. Acta Pharmacol Sin 2007; 28: 1957–1967.
Kang R, Zhang Q, Zeh HJ, Lotze M, Tang D . HMGB1 in cancer: good, bad, or both? Clin Cancer Res 2013; 19: 4046–4057.
Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol 2012; 13: 832–842.
Vernon PJ, Loux TJ, Schapiro NE, Kang R, Muthuswamy R, Kalinski P et al. The receptor for advanced glycation end products promotes pancreatic carcinogenesis and accumulation of myeloid-derived suppressor cells. J Immunol 2013; 190: 1372–1379.
Kang R, Loux T, Tang D, Schapiro NE, Vernon P, Livesey KM et al. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc Natl Acad Sci USA 2012; 109: 7031–7036.
Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ 2010; 17: 666–676.
Luo Y, Chihara Y, Fujimoto K, Sasahira T, Kuwada M, Fujiwara R et al. High mobility group box 1 released from necrotic cells enhances regrowth and metastasis of cancer cells that have survived chemotherapy. Eur J Cancer 2013; 49: 741–751.
Kang R, Tang D, Schapiro NE, Loux T, Livesey KM, Billiar TR et al. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 2013 e-pub ahead of print 14 January 2013; doi:10.1038/onc.2012.631.
Tsan MF, Gao B . Heat shock proteins and immune system. J Leukoc Biol 2009; 85: 905–910.
Suto R, Srivastava PK . A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science 1995; 269: 1585–1588.
Srivastava P . Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol 2002; 2: 185–194.
Corriden R, Insel PA . Basal release of ATP: an autocrine-paracrine mechanism for cell regulation. Sci Signal 2010; 3: re1.
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009; 461: 282–286.
Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. Embo J 2012; 31: 1062–1079.
Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450: 1253–1257.
Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci USA 2011; 108: 17396–17401.
Vernon PJ, Tang D . Eat-me: autophagy, phagocytosis, and reactive oxygen species signaling. Antioxid Redox Signal 2013; 18: 677–691.
Townsend KN, Hughson LR, Schlie K, Poon VI, Westerback A, Lum JJ . Autophagy inhibition in cancer therapy: metabolic considerations for antitumor immunity. Immunol Rev 2012; 249: 176–194.
Wildenberg ME, Vos AC, Wolfkamp SC, Duijvestein M, Verhaar AP, Te Velde AA et al. Autophagy attenuates the adaptive immune response by destabilizing the immunologic synapse. Gastroenterology 2012; 142: 1493–1503 e1496.
Kovacs JR, Li C, Yang Q, Li G, Garcia IG, Ju S et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ 2012; 19: 144–152.
Li Y, Wang LX, Pang P, Twitty C, Fox BA, Aung S et al. Cross-presentation of tumor associated antigens through tumor-derived autophagosomes. Autophagy 2009; 5: 576–577.
Li Y, Hahn T, Garrison K, Cui ZH, Thorburn A, Thorburn J et al. The vitamin E analogue alpha-TEA stimulates tumor autophagy and enhances antigen cross-presentation. Cancer Res 2012; 72: 3535–3545.
Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res 2011; 71: 5976–5986.
Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011; 334: 1573–1577.
Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P et al. Endogenous HMGB1 regulates autophagy. J Cell Biol 2010; 190: 881–892.
Livesey KM, Kang R, Vernon P, Buchser W, Loughran P, Watkins SC et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res 2012; 72: 1996–2005.
Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R et al. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res 2012; 72: 230–238.
Tang D, Kang R, Livesey KM, Kroemer G, Billiar TR, Van Houten B et al. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab 2011; 13: 701–711.
Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010; 29: 5299–5310.
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011; 25: 23–31.
Kambas K, Mitroulis I, Apostolidou E, Girod A, Chrysanthopoulou A, Pneumatikos I et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS One 2012; 7: e45427.
Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I et al. Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLoS One 2011; 6: e29318.
Zhang Y, Li W, Zhu S, Jundoria A, Li J, Yang H et al. Tanshinone IIA sodium sulfonate facilitates endocytic HMGB1 uptake. Biochem Pharmacol 2012; 84: 1492–1500.
Li W, Zhu S, Li J, Assa A, Jundoria A, Xu J et al. EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochem Pharmacol 2011; 81: 1152–1163.
Zhang Q, Kang R, Zeh HJ 3rd, Lotze MT, Tang D . DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy 2013; 9: 451–458.
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12: 222–230.
Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L et al. Noncanonical autophagy is required for type I interferon secretion in response to dna-immune complexes. Immunity 2012; 37: 986–997.
Biswas D, Qureshi OS, Lee WY, Croudace JE, Mura M, Lammas DA . ATP-induced autophagy is associated with rapid killing of intracellular mycobacteria within human monocytes/macrophages. BMC Immunol 2008; 9: 35.
Ghavami S, Eshragi M, Ande SR, Chazin WJ, Klonisch T, Halayko AJ et al. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res 2010; 20: 314–331.
Acknowledgements
We thank Christine Heiner (Department of Surgery, University of Pittsburgh) for her critical reading of the manuscript. This work was supported by the National Institutes of Health (NIH) (R01CA160417 to DT) and the American Association for Cancer Research (AACR) (13-20-25-TANG to DT). We apologize to the researchers who are not referenced because of space limitations.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Agostini
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Hou, W., Zhang, Q., Yan, Z. et al. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis 4, e966 (2013). https://doi.org/10.1038/cddis.2013.493
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.493
Keywords
This article is cited by
-
Repurposing thioridazine for inducing immunogenic cell death in colorectal cancer via eIF2α/ATF4/CHOP and secretory autophagy pathways
Cell Communication and Signaling (2023)
-
Cancer cells adapt FAM134B/BiP mediated ER-phagy to survive hypoxic stress
Cell Death & Disease (2022)
-
PLP2-derived peptide Rb4 triggers PARP-1-mediated necrotic death in murine melanoma cells
Scientific Reports (2022)
-
All-Trans Retinoic Acid–Preconditioned Mesenchymal Stem Cells Improve Motor Function and Alleviate Tissue Damage After Spinal Cord Injury by Inhibition of HMGB1/NF-κB/NLRP3 Pathway Through Autophagy Activation
Journal of Molecular Neuroscience (2022)
-
Targeting redox regulation and autophagy systems in cancer stem cells
Clinical and Experimental Medicine (2022)
