
Abstract
The failure of adult hippocampal neurogenesis is increasingly considered as an important factor in the pathological correlates for memory decline in Alzheimer’s disease (AD). Loss of adult-born neurons and abnormalities of neural stem/progenitor cells (NSPCs) within the dentate gyrus (DG) of adult hippocampus might contribute to this process. In this study, we showed that amyloid-β1–42 (Aβ42) oligomer triggers senescent phenotype of NSPCs in vitro. Oligomerized Aβ42 induced the production of senescence-associated biomarkers p16 and senescence-associated β-galactosidase (SA-β-gal) in adult mouse hippocampal NSPCs, as well as inhibited cells proliferation and differentiation. In the DG of amyloid precursor protein/presenilin1 (APP/PS1) transgenic mice, the number of senescent NSPCs was significantly increased and senescence-associated protein p16 was upregulated. Formylpeptide receptor 2 (FPR2), one of Aβ42 functional receptors, may be involved in NSPCs senescence. The FPR2 antagonist WRW4 significantly inhibited NSPCs senescence induced by Aβ42. In addition, the activation of p38 mitogen-activated protein kinase (MAPK) in response to the accumulation of reactive oxygen species (ROS) was involved in NSPCs senescence induced by Aβ42. WRW4 inhibited the accumulation of ROS and the activation of p38 MAPK in NSPCs. Our data suggest that Aβ42 accelerates NSPCs senescence via FPR2-dependent activation of its downstream ROS-p38 MAPK signaling, which limits the function of NSPCs and contributes to failure of neurogenesis. This is the first demonstration of NSPCs senescence response to Aβ42.
Similar content being viewed by others


Main
In the mammalian brain, neurogenesis persists throughout adulthood in the subgranular zone (SGZ) of the dentate gyrus (DG) of the hippocampus and in the subventricular zone (SVZ) of the lateral ventricles.1 In the SGZ, adult neurogenesis involves activation of quiescent neural stem/progenitor cells (NSPCs) to yield proliferative progenitors, migration of neuroblasts and ultimately differentiation into granule cells that synaptically integrate into the existing neural network. Each of these stages is regulated by microenvironmental cues. Both the number of NSPCs and neurogenesis decline within SGZ has been involved in cognitive impairments linked with ageing and neurodegenerative diseases, and it was suggested to have an important role in Alzheimer’s disease (AD).2
AD is an age-related, progressive and irreversible neurodegenerative disease. The disease is characterized by extracellular deposition of β-amyloid (Aβ) peptides and intracellular hyperphosphorylation of tau protein, which result in progressive loss of memory and other cognitive functions.3 Over the past decade, researchers and clinicians have not made substantial progress in treating AD, due to the complexity of the disease.4 Recently, strategy for stem cell therapy has received considerable attention as an alternative treatment for neurodegenerative diseases. Accumulating studies showed that transplantation of NSPCs can reduce neuronal loss, improve cognition and memory in animal models of AD.5, 6, 7 However, up to now, stem cell therapy obtained limited benefits in AD, because hostile microenvironments of AD brain may impair the function of NSPCs, as it contains Aβ.
Considerable evidence, obtained from studies of patient, cell culture and animals models, suggests that Aβ is the key trigger of AD pathogenesis, and oligomeric amyloid-β1–42 (Aβ42) is thought to be the most toxic form of Aβ.3 Early works showed that Aβ impairs proliferation and neuronal differentiation of NSPCs.8, 9 Overproduction and consequent aggregation of Aβ in amyloid precursor protein/presenilin1 (APP/PS1) transgenic mice severely reduced the number of adult-generated hippocampal neurons.10 Some studies also showed that Aβ was directly associated with the reduction in hippocampal neurogenesis in AD animal models.8, 10, 11, 12 Hippocampal neurogenesis decline in neurogenic capacity has been suggested to underlie cognitive impairments that accompany senescence.13, 14 However, little is currently known about how Aβ affects hippocampal neurogenesis. Recently, Aβ has been described to be involved in senescent responses of neurons and astrocytes.15, 16 In addition, Aβ peptides caused endothelial cells and retinal pigment epithelial cells to enter senescence.17, 18 However, these responses have yet to be determined in NSPCs.
In this study, we showed that Aβ42 oligomer can promote the senescence of cultured adult mouse hippocampal NSPCs. Compared with wild-type mice, more senescent NSPCs were observed in the DG of APP/PS1 transgenic mice. Furthermore, we provided evidence suggesting that Aβ42 induces NSPCs senescence via the activation of formylpeptide receptor 2 (FPR2) and its downstream reactive oxygen species p38 mitogen-activated protein kinase (ROS-p38 MAPK) signaling pathways. These results therefore represent, to the best of our knowledge, the first study of the relationship between Aβ42 and NSPCs senescence. This finding may provide us new insights into the potential therapy of stem cell for neurodegeneration including AD.
Results
Aβ42 inhibits NSPCs proliferation and high-dose also induces apoptosis
To begin our study of the effects of Aβ42 on the adult hippocampal neurogenesis, we first isolated NSPCs from the hippocampus of adult mouse brain. NSPCs form clonal spheres called neurospheres, which are composed of NSPCs and more committed neural progenitors.19 Immunostaining experiments revealed that NSPCs markers Nestin and Sox2 were expressed on our cultured neurospheres or dissociated NSPCs (data not shown).
We then tested the effect of Aβ42 on cell viability. Cells were treated with Aβ42 (2.5, 5 or 10 μM) for 1, 3 and 5 days. Then, cell viability was determined by CCK-8 assay. We found that at 3 days, the significant inhibitory effect of Aβ42 was detectable only at 10 μM. However, at 5 days, Aβ42 significantly inhibited the viability of NSPCs in concentration-dependent manners (Figure 1a), which led us to choose 5 days of incubation to detect the following experiments. These data indicate that Aβ42 decreases NSPCs viability in dose-dependent and time-dependent manners.

Aβ42 inhibits NSPCs proliferation and high-dose induces NSPCs apoptosis. (a) Cell viability assay (CCK-8) showed that Aβ42 suppressed NSPCs viability in dose- and time- dependent manners. The number of newly formed neurospheres (b) and the size of newly formed neurospheres (c) were reduced significantly in Aβ42-treated NSPCs. (d) NSPCs were treated with the indicated concentrations of Aβ42 for 5 days, apoptotic cells were analyzed with Annexin V-FITC kit by flow cytometry. The effects of Aβ42 (0 or 10 μM) on the NSPCs apoptosis are shown. (e) The percentage of apoptotic cells was calculated by Annexin V+/PI− and Annexin V+/PI+ together. The mean±S.D. of three independent experiments is shown. *P<0.05, **P<0.01 compared with control
To investigate the effect of Aβ42 on the biology of NSPCs, we used the neurospheres proliferation assay to detect the number and diameter of neurospheres. The number of neurospheres reflects self-renewing activity of NSPCs, and the diameter of neurospheres reflects proliferative capacity within each neurosphere. The number of Aβ42-treated neurospheres was decreased dose-dependently compared with control (Figure 1b). The maximal inhibition was observed at 10 μM Aβ42. This result was consistent with that of CCK-8 assay. The dose of Aβ42 (10 μM) was applied for the following studies. Furthermore, the decrease of neurosphere diameter was also found in Aβ42-treated cells. However, there was no significant difference among these treatment conditions (Figure 1c).
To identify whether the proliferation-inhibitory effect of Aβ42 was due to NSPCs apoptosis, we further examined the effect of Aβ42 on apoptosis. The data showed that incubation with low-dose Aβ42 (2.5 or 5 μM) did not alter the percentage of apoptotic cell (Figure 1e). However, high-dose Aβ42 (10 μM) increased the percentage of apoptotic cell by 1.3-fold in comparison with control (Figures 1d and e). Taken together, these data indicate that low-dose Aβ42 inhibits NSPCs proliferation and resists to apoptosis, whereas, in addition to inhibition proliferation, high-dose Aβ42 also induces NSPCs apoptosis.
Aβ42 treatment results in morphological and differentiated changes in NSPCs
Morphologically, in suspension culture conditions, NSPCs appeared as cell clusters with a smooth and well-defined border. However, NSPCs treated with 10 μM Aβ42 displayed smaller in size and less in number than control, and usually had an uneven border (Figure 2a). In monolayer cultivation conditions, untreated cells grew in clusters with tight cell-cell contacts. In contrast, Aβ42-treated cells exhibited obvious reduction in total cell number, which was accompanied by a unique flattened and enlarged morphology, and tended to grow disjointed (Figure 2b). These morphological changes in Aβ42-induced NSPCs confirm to cellular senescence phenotype.

Aβ42 alters morphology and differentiation capacity of NSPCs. Either in the absence or presence of 10 μM Aβ42 for 5 days, the morphology of neurospheres (a) or monolayer cells (b) is observed. (c) Either in the absence or presence of 10 μM Aβ42, NSPCs were differentiation for 5 days, and then were immunostained for neurons (red, β-III-tubulin) and astrocytes (green, GFAP). (d) Untreated and 10 μM Aβ42-treated NSPCs were dissociated to single cells and then at the same density to be grown onto glass coverslips coated with PDL in differentiation condition for 5 days, and stained for lineage markers as above. Quantification analyzed the percentage of β-III-tubulin or GFAP-positive cells. The mean±S.D. of three independent experiments is shown. *P<0.05, **P<0.01 compared with control. Scale bars=100 μm
Multipotency is an important characteristic of NSPCs, which can differentiate into the neuron, astrocyte or oligodendrocyte. As shown in Figure 2c, in normal conditions, NSPCs rapidly spread out from the center to the periphery in the differentiation media and were mainly consisted of both β-III-tubulin-positive and GFAP-positive cells. In Aβ42-treated conditions, NSPCs began to differentiate much later (data not shown), displayed sphere morphology with little process extension and reduced expressions of neural phenotype β-III-tubulin and GFAP. To rule out the possibility that smaller neurosphere has lower differentiation capacity than bigger one, untreated and Aβ42-treated NSPCs were dissociated to single cells and then grown onto glass coverslips coated with poly-D-lysine hydrobromide (PDL) at the same cell density in differentiation media. After 5 days of differentiation, we stained the cells for lineage markers. Quantitative analysis showed that there were decrease of β-III-tubulin-positive cells (1.2-fold) and GFAP-positive cells (3.1-fold) (Figure 2d). These results suggest that Aβ42 suppresses NSPCs differentiation capacity, which leads to neurogenesis impairment.
Aβ42 induces NSPCs senescence in vitro
One possible explanation to Aβ42 suppresses neurogenesis could be that Aβ42 induces NSPCs senescence. To test this hypothesis, we assessed the number of senescence-associated β-galactosidase (SA-β-gal)-positive cells, because of SA-β-gal staining as a marker of cellular senescence.20 Our results showed that the number of SA-β-gal-positive cells was significantly increased by Aβ42 (5 or 10 μM) compared with that in the control (Figures 3a and b). These data suggest that Aβ42 results in NSPCs senescence.

Aβ42 promotes NSPCs senescence. (a) NSPCs were treated with Aβ42 (0, 2.5, 5 or 10 μM) for 5 days, and then detected by SA-β-gal staining. The senescent NSPCs were observed by blue cells staining visualized under an inverted microscope. Pictures were acquired and analyzed using the NIS Nikon software. (b) The percentage of SA-β-gal-positive cells was quantified by counting 102 cells on six separate fields for each condition. (c) Immunoblot for levels of p16 and pRb expressions in NSPCs treated with different concentrations of Aβ42. (d) Quantification by densitometric scanning was presented, and results were normalized to the untreated condition. The mean±S.D. of three independent experiments is shown. *P<0.05, **P<0.01 compared with untreated NSPCs. Scale bar=100 μm
To further confirm that Aβ42 induces NSPCs senescence, we also monitored the expressions of prosenescence molecules p16 and pRb. The p16-Rb axis maintains the growth arrest, which is known to induce cellular senescence.21 An inverse relationship was observed between p16 and pRb in that as p16 expression increased, pRb levels decreased correspondingly in Aβ42-treated NSPCs compared with the control (Figures 3c and d). These effects exhibit a concentration-dependent manner; therefore, these results further indicate that Aβ42 accelerates NSPCs senescence.
The number of senescent NSPCs has a significant increase in APP/PS1 transgenic mice
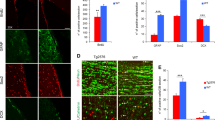
Having established that Aβ can induce NSPCs senescence in vitro, we examined the brain tissue from a transgenic mouse model of AD. Transgenic mouse that carries an APP andPS1 mutated gene shows AD-like pathology and memory impairment, and is useful for studying AD and testing possible treatments.22, 23, 24 It had been reported that Aβ is deposited in the DG of the hippocampus of APP/PS1 transgenic mice at 9 months and impaired adult hippocampus neurogenesis.11 Therefore, 9-month-old APP/PS1 transgenic mice and age-matched wild-type control allowed us to test whether Aβ was associated with NSPCs senescence in vivo. As confirmed by Aβ immunohistochemistry, only APP/PS1 mice harbored Aβ deposits in the cerebral cortex and hippocampus, including the DG, whereas no such was found in the wild-type mice (Figure 4a). Interestingly, we also found that compared with wild-type control, the number of SA-β-gal-positive cells was increased in the DG of APP/PS1 mice (1.7-fold) (Figure 4b). Furthermore, brain sections were screened for p16-positive NSPCs using double immunofluorescence for cellular senescent biomarker p16 and NSPCs marker Nestin (Figure 4c). APP/PS1 transgenic mice exhibited a 1.3-fold increase in the number of p16-positive NSPCs within the DG compared with wild-type counterparts (Figure 4d). These results indicate that the number of senescent NSPCs is increased in APP/PS1 transgenic mice.

The number of senescent NSPCs is increased in the DG of APP/PS1 transgenic mice. (a) Brain slices of APP/PS1 transgenic mice and wild-type mice at 9 months were immunofluorescence staining with Aβ antibody (red). Aβ was specific to the cerebral cortex and hippocampus of APP/PS1 transgenic mice. DIC microphotographs were taken at the same location. (b) Brain slices as above were stained with SA-β-gal labeling assay. The senescent cells in the DG were detected by blue cells staining visualized under an inverted microscope. Pictures were acquired and analyzed using stereological methods. (c) Brain slices as above were double immunofluorescence staining with Nestin (green) and p16 (red), and nuclei were counterstained with DAPI (blue). (d) The number of p16-positive NSPCs in the DG was quantified using stereological methods. *P<0.05 compared with wild-type mice. Scale bars (a)=250 μm. Scale bars (b and c)=100 μm
FPR2 involves in Aβ42-induced NSPCs senescence
Recent evidence suggested that FPR2, a G protein-coupled receptor (GPCR), mediates a range of inflammatory responses to Aβ42 in AD.25 To gain insight into the molecular mechanisms by which Aβ42 induced NSPCs senescence, we investigated whether FPR2 is involved in this process. First, brain sections of 9-month-old APP/PS1 transgenic mice and age-corresponding wild-type mice were stained with FPR2 antibody. We found that FPR2 is expressed in the DG of the hippocampus with higher (3.4-fold) expression in transgenic mice (Figure 5a). DG niches contain NSPCs, committed progenitor and differentiated progeny. To confirm that FPR2 is expressed in NSPCs, dissociated NSPCs were immunostained with FPR2 antibody. As shown in Figure 5b, FPR2 is expressed in purified NSPCs in culture. To quantify the levels of FPR2 in NSPCs affected by Aβ42, NSPCs were treated with 10 μM Aβ42 for 5 days. Western blotting experiments confirmed that Aβ42 induced FPR2 protein expression in NSPCs (Figure 5c).

Aβ42 induces NSPCs senescence via FPR2. (a) Brain slices were stained with antibody against FPR2 (red) to identify its expression in the DG of APP/PS1 transgenic mice and wild-type mice aged 9 months. The number of FPR2-positive cells in the DG was quantified using stereological methods. (b) NSPCs were performed by immunofluorescence staining with antibody against FPR2 (red) to identify its expression and nuclei were counterstained with DAPI (blue). (c) NSPCs were treated with 10 μM Aβ42 for 5 days, and cell lysates were subjected to western blot probed with FPR2 antibody. GAPDH was selected as an inner standard. Quantification by densitometric scanning was presented. Results were normalized to the untreated condition. (d) NSPCs were preincubated with 10 μM WRW4 for 1 h followed with 10 μM Aβ42 treatment. The senescent NSPCs were determined by SA-β-gal staining and the percentage of SA-β-gal-positive cells was statistically analyzed as above. (e) Cell lysates collected from NSPCs treated with 10 μM Aβ42 for indicated time with or without preincubation of 10 μM WRW4 were subjected to western blot using antibody against p16 and pRb. GAPDH was selected as an inner standard. Quantification by densitometric scanning was presented. Results were normalized to the untreated condition. The mean±S.D. of three independent experiments is shown. *P<0.05, **P<0.01 compared with wild-type mice or compared with untreated NSPCs; #P<0.05 compared with Aβ42-treated NSPCs. Scale bars=100 μm
To determine the role of FPR2 on Aβ42-induced NSPCs senescence, NSPCs were pretreated with 10 μM WRW4 (a selective and potent antagonist of FPR2) for 1 h before exposure to 10 μM Aβ42, and then senescence-associated indexes were detected 5 days later. We found that WRW4 significantly decreased the percentage of SA-β-gal-positive cells in NSPCs during Aβ42 incubation (Figure 5d). This was paralleled by an increase of pRb (2.1-fold) and a decrease of p16 (1.5-fold) (Figure 5e). Thus, FPR2 antagonist WRW4 ameliorates Aβ42-induced NSPCs senescent phenotype. Together, these results suggest that FPR2 involves in Aβ42-induced NSPCs senescence.
Elevated intracellular ROS levels and activated p38 MAPK are associated with Aβ42-induced NSPCs senescence
The ROS-p38 MAPK pathway has been shown to induce cellular senescence.26 Furthermore, Aβ increases intracellular ROS production by acting at FPR2 in the brain.27 This led us to hypothesize that the ROS-p38 MAPK pathway might contribute to the Aβ42-mediated induction of NSPCs senescence. To test the hypothesis, NSPCs were pretreated with 10 μM WRW4, 1 mM antioxidant N-acetylcysteine (NAC) or 10 μM p38 MAPK inhibitor SB203580 for 1 h before exposure to 10 μM Aβ42. During the 5-day experimental period, Aβ42 obviously increased intracellular ROS production, which was significantly inhibited by WRW4 or NAC (Figure 6a). Western blot showed that WRW4 or NAC also obviously suppress phosphorylation of p38 MAPK induced by Aβ42 (Figure 6b). Moreover, NAC or SB203580 significantly decreased the number of SA-β-gal-positive cells during Aβ42 insult (Figure 6c). And NAC reduced the number of senescent cells to the control level. This was also confirmed by the detection expressions of p16 and pRb proteins (Figure 6d). These findings strongly support that Aβ42 promotes NSPCs senescence through activation of FPR2 and its downstream ROS-p38 MAPK signaling pathway.

ROS-P38 MAPK signaling pathway is involved in the Aβ42-induced NSPCs senescence via FPR2. NSPCs were pretreated with 10 μM WRW4, 1 mM NAC or 10 μM SB203580 for 1 h before exposure to 10 μM Aβ42. After 5 days, the following were assessed: (a) Intracellular ROS levels in all groups were measured by DCFH-DA; (b) The expressions of p-p38 and p38 proteins in all groups were detected by immunoblot and results were normalized to the untreated condition. (c) The senescent NSPCs were observed by SA-β-gal staining in all groups and statistically analyzed as above. (d) The expressions of pRb and p16 proteins in all groups were determined by western blotting. GAPDH was selected as an inner standard. Quantification by densitometric scanning was presented. Results were normalized to the untreated condition. The mean±S.D. of three independent experiments is shown. *P<0.05, **P<0.01 compared with untreated NSPCs; #P<0.05, ##P<0.01 compared with Aβ42-treated NSPCs. Scale bar=100 μm
Discussion
In this study, we have demonstrated for the first time that Aβ42 induces NSPCs senescence in vitro, and the number of senescent NSPCs is increased in the DG of APP/PS1 double transgenic mice. Furthermore, results from our mechanistic studies suggest that Aβ42 induction of NSPCs senescence is mediated via the activation of FPR2 and its downstream ROS-p38 MAPK signaling pathway as summarized in Figure 7.

An integrated model depicting the mechanism underlying the Aβ42-induced NSPCs senescence via FPR2 in a ROS-P38 MAPK-dependent fashion
Several lines of evidence demonstrated that Aβ42 triggers senescent phenotype of NSPCs in vitro. Aβ42 significantly inhibited proliferation and differentiation capacities of NSPCs. Furthermore, Aβ42 (2.5 or 5 μM) also exhibited resistance to apoptosis. In contrast, Aβ42 (10 μM) significantly induced NSPCs apoptosis. Morphologically, Aβ42-treated neurospheres are smaller in size and less in number than untreated cells. Under the condition of monolayer cultivation, Aβ42-treated cells displayed a unique flattened and enlarged morphology. These features of Aβ42-treated NSPCs displayed typical cellular senescence phenotype. This notion is further supported by the observation that Aβ42 dose-dependently increased the number of SA-β-gal-positive cells and upregulated p16 and decreased pRb phosphorylation. p16 is cyclin-dependent kinase inhibitor, which can maintain downstream Rb protein in a hypophosphorylated and active state.20 The p16-Rb axis maintains the growth arrest, which is known to induce cellular senescence.21 Our results support the view that expression of p16 increases in NSPCs of the aged mouse brain, which limits NSPCs proliferation and inhibits neurogenesis.28 All of these results indicate that Aβ42 directly alters the phenotypical features of NSPCs and accelerates NSPCs senescence.
Interestingly, it has been reported that at the same concentration, Aβ42 oligomers lead to astrocyte senescence in vitro.16 Astrocytes, as a crucial component of the neurogenic niche, has an important role for adult hippocampal neurogenesis, and support neuronal homeostasis in NSPCs niche.29, 30 Therefore, senescent astrocytes may also indirectly induce NSPCs senescence, for example, through secreted interleukin-6 (IL-6).16
However, our results in vitro studies differ from some others showing that Aβ42 has neurogenic effect on NSPCs.31, 32, 33 The major difference is that we used NSPCs isolated from the hippocampus of adult mice, used much higher concentrations of Aβ42 and much shorter incubation time. Taken together, these results suggest that neurogenic effect of much lower concentrations of Aβ42 may be a compensatory response as one of the endogenous brain repair mechanism. With the accumulation of Aβ42, it drives cellular senescence, ultimately results in neurogenesis dysfunction.
Evidence coming mainly from the APP/PS1 double transgenic mouse model indicates that Aβ first appears between 4 and 5 months of age and worsen with age, and it is abundant in the hippocampus and cortex by 9 months of age.34 Consistent with these observations, we found that compared with age-corresponding wild-type mice, Aβ pathology was specific to APP/PS1 mice at 9 months. In addition, our in vivo data also showed that the number of SA-β-gal-positive cells and senescent NSPCs was increased in the DG of APP/PS1 transgenic mice. The animal model studies further suggest that Aβ possibly associates with adult hippocampal NSPCs senescence.
As a functional receptor of Aβ42, FPR2 has been suggested to participate in the pathogenesis of AD.25 Upon binding to FPR2, Aβ42 generates oxidative stress through activation of microglial cells and induces these cells migration response to Aβ42.27 FPR2 has also shown to be involved in the endocytosis process of Aβ42 and its cytotoxicity for neuronal cells.35 Recent findings demonstrated that FPR2 also regulates cell cycle by inducing p21 and p16 accumulation.25, 36 In the present paper, we have first observed that the expression of FPR2 was also present in the DG of the hippocampus and purified NSPCs. The expression of FPR2 was significantly increased in the APP/PS1 transgenic mice and Aβ42-treated NSPCs. FPR2 antagonist WRW4 inhibited the increased number of SA-β-gal-positive cells by Aβ42 and downregulated the expressions of senescent-associated proteins. Therefore, FPR2 antagonist WRW4 ameliorates Aβ42-induced NSPCs senescent phenotype. These results suggest that FPR2 might be a potential target for the regulation of neurogenesis in the process of AD.
Our study also provided evidence suggesting that Aβ42-induced NSPCs senescence via FPR2 is mediated through the ROS-p38 MAPK pathway. Activation of FPR2 initiates the activation of microglial cells and produces ROS.27 ROS, one of signal molecules, can regulate a variety of cellular functions, including NSPCs proliferation, differentiation and mobilization at physiological levels.37, 38, 39, 40 Abnormal increase of ROS production under pathological conditions can inhibit NSPCs self-renewal and adult neurogenesis.41 It has been reported that activation of the p38 MAPK pathway by elevated level of ROS results in defective function of NSPCs, which may leads to premature exhaustion of NSPCs and dysfunction of neurogenesis.42 Our data showed that Aβ42 increased intracellular ROS levels and p38 kinase phosphorylation, which can be significantly inhibited by WRW4. Antioxidant NAC or p38 MAPK inhibitor SB203580 significantly decreased the number of SA-β-gal-positive cells and downregulated p16 and upregulated pRb expressions. Therefore, these results further provide strong evidence that Aβ42 induces NSPCs senescence by the activation of FPR2 and its downstream ROS-p38 MAPK signaling pathway.
In conclusion, our study established for the first time a novel link between Aβ42 and NSPCs senescence. Besides inhibiting proliferation and differentiation capacities, Aβ42 induces senescent phenotype of NSPCs through activating FPR2 and its downstream ROS-p38 MAPK pathway. In addition, the number of senescent NSPCs increases in the animal model of AD. These results may be of significance for explanation of AD pathogenesis and also provide valuable information on the potential of stem cell therapy for neurodegeneration.
Materials and Methods
Animals
Eight-week-old male C57BL/6 mice were purchased from Slaccas Experimental Animal Company. Nine-month-old APP/PS1 transgenic mice and age-corresponding wild-type C57BL/6 mice were purchased from The Jackson Laboratory (B6C3-Tg (APPswe, PSEN1dE9) 85Dbo/J; stock no. 004462). The experimental protocol used in this study was approved by the Ethics Committee for Animal Experimentation and was conducted in strict accordance with the Guidelines for Animal Experimentation of Shanghai Jiao Tong University.
Mouse NSPCs cultures
NSPCs were isolated from the hippocampus of 8-week-old male C57BL/6 mice and were cultured as described previously in detail.43 Briefly, hippocampus tissues were dissociated in 0.1% Papain mixture (Worthington, Lakewood, NJ, USA). For neurospheres culture, cells were plated in serum-free neurosphere medium containing DMEM/F12, 2% B27 supplement, 20 ng/ml bFGF and 10 ng/ml EGF (Invitrogen, Carlsbad, CA, USA). For adherent monolayer cultivation, the methods have been previously described in detail.44 Cells were grown on dishes precoated with PDL and laminin (both from Sigma, St. Louis, MO, USA) containing the above mentioned medium. Cells were incubated at 37 °C in an atmosphere of 5% CO2.
Oligomeric Aβ42 preparation
Oligomeric Aβ42 peptide was prepared as described previously.45 Aβ42 peptide (Invitrogen) was first dissolved to 1 mM in 100% hexafluoroisopropanol (HFIP), and then HFIP was removed under vacuum. The peptide was resuspended in dimethyl sulfoxide (DMSO) to 5 mM. F12 (without phenol red) culture medium (Invitrogen) was added to bring the peptide to a final concentration of 100 μM and incubated at 4 °C for 24 h. The solution was centrifuged at 13 000 r.p.m. for 20 min, and the supernatant was stored at −20 °C before use.
Cell viability assays
For cell viability assay, after trypsinizing neurospheres, single NSPCs were resuspended in proliferation medium. The cells were seeded into 96-well plates at a density of 102 cells per well. The compounds under study were added when the cells were seeded. After incubation for indicated time, cell viability was determined by a commercial Cell Counting Kit (CCK-8, Dojindo, Japan). CCK-8 reagent was added to each well and incubated at 37 °C for 1 h. Absorbance was measured at 450 nm in a spectrophotometer. Each experiment was performed in triplicate wells and repeated at least three times.
Cell proliferation assays
For neurosphere proliferation assay, single NSPCs were plated in 24-well plates at a density of 1800 cells per well. After incubation for indicated time, the numbers of newly formed neurospheres per well were counted using phase contrast microscopy, and the neurosphere sizes were estimated using a microimage analysis system from Olympus IX51 inverted microscope (Olympus, Tokyo, Japan).
Neurosphere differentiation assays
After the 7-day culture period described previously, neurospheres treated with Aβ42 (0 or 10 μM) were grown onto glass coverslips coated with PDL and laminin within 24-well tissue culture plates containing growth factor-free medium and 1% fetal bovine serum (FBS). Neurospheres were differentiated for 5 days before assessment by immunocytochemistry.
Immunocytochemistry
Animals received an overdose of sodium pentobarbital and were transcardially perfused with saline followed by ice-cold 4% paraformaldehyde (PFA) in 0.1 M KPBS. Brains were removed, postfixed overnight and placed in 20% sucrose in 0.1 M phosphate buffer for 24 h. Coronal sections (30 μm) were cut on a freezing microtome and stored in cryoprotective solution. Free-floating sections were preincubated in 0.25% Triton X-100 in KPBS containing 5% normal goat serum for 1 h. Sections were then incubated with primary antibodies overnight at 4 °C. Primary antibodies included the following: rabbit anti-Aβ (Cell Signaling Technology, Danvers, MA, USA; 1 : 250), mouse anti-Nestin (Cell Signaling Technology; 1 : 200), rabbit anti-p16 (Santa Cruz, Dallas, TX, USA; 1 : 200) and rabbit anti-FPR2 (Proteintech, Chicago, IL, USA; 1 : 50).
Cells grown on PDL/laminin-coated coverslips were fixed with 4% PFA for 30 min at room temperature (RT) and rinsed with PBS three times before being permeabilized in 1% Triton X-100 for 10 min. Nonspecific antibody binding sites were blocked by incubating with normal goat serum for 2 h at RT before labeling with primary antibodies overnight at 4 °C. Primary antibodies included the following: mouse anti-β-III-tubulin (Cell Signaling Technology; 1 : 50), rabbit anti-GFAP (Epitomics, Burlingame, CA, USA; 1 : 100) and rabbit anti-FPR2 (Proteintech; 1 : 50).
After washing three times in PBS for 5 min, sections were reacted for 1 h at 37 °C in the dark with second antibodies. The second antibodies included AlexaFluor 488 goat anti-mouse IgG and DyLight 594 goat anti-rabbit IgG (1 : 500). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). After final washing, the stained slides were mounted with Olympus DP control and DP software.
Determination of NSPCs apoptosis
For apoptosis analysis, cells were assessed with the Annexin V-FITC kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions. The percentage of early apoptotic cells (Annexin V+/propidium iodide−) and late-stage apoptotic cells (Annexin V+/propidium iodide+) together were considered total apoptotic cells. The apoptotic index was analyzed by flow cytometry.
Analysis of intracellular ROS
Intracellular ROS levels were monitored by using 2′,7′-dichlorofluorescin diacetate (DCFH-DA), which forms the fluorescent compound dichlorofluorescein on oxidation with ROS. After NSPCs were incubated with 10 μM DCFH-DA at 37 °C for 30 min, the fluorescence was monitored by scanning the whole well using a fluorescent plate reader at excitation and emission wavelengths of 485 and 525 nm.
SA-β-gal labeling assays
SA-β-gal labeling assays were used to identify senescent cells in tissues and NSPCs. In brief, frozen sections or cells were washed three times with PBS and fixed for 30 min at RT with fixative solution. After incubation with the staining solution overnight at 37 °C, slides were viewed with a compound microscope and scored for the SA-β-gal label as indicated by blue/green reactivation product over the cell soma. SA-β-gal-positive cells were quantified by counting six random fields per slides.
Quantitative analysis of immunopositive cells densities in the DG
Quantification of Nestin+/P16+, FPR2+ or SA-β-gal+ cells in the DG were conducted using unbiased stereological methods, as described previously.10 Briefly, this analysis was quantified from a one-in-six series of peroxidase-labeled sections spaced at 240 μm spanning the full rostrocaudal extent of the hippocampus. The number of Nestin+/P16+,FPR2+ or SA-β-gal+ cells in the DG region were estimated using a computerized stereology system (Stereologer; Systems Planning and Analysis, Alexandria, VA, USA). Every 10th section was selected and generated 10–15 sections per reference space in a systematic-random manner. Immunopositive cells were counted using the optical-fractionator method with the dissector principle and unbiased counting rules. All measurements were made by an investigator, and the experimenter was blind to genotype of the mice during analysis.
Western blot
Cells were lysed with RIPA lysis buffer. Insoluble material was removed by centrifugation at 13 000 r.p.m. for 20 min at 4 °C. Total proteins from cell lysates were denatured at 100 °C for 5 min and 50 μg proteins were electrophoresed on 8–12% SDS-PAGE and transferred to a PVDF membrane. The membrane was blocked with 5% nonfat milk at RT for 1 h and incubated overnight with primary antibodies at 4 °C. The membrane was washed and then incubated with secondary antibody at RT for 1 h. The primary antibodies included polyclonal-pRb (Assaybiotech, Los Angeles, CA, USA; 1 : 500), polyclonal-p16 (Santa Cruz; 1 : 500), polyclonal-FPR2 (1 : 500), polyclonal-pp38 (Cell Signaling Technology; 1 : 1000), polyclonal-p38 (Cell Signaling Technology; 1 : 800) and monoclonal-GAPDH (Proteintech; 1 : 2000). Detection was carried out using an enhanced chemiluminescence detection kit (Pierce, Rockford, IL, USA). The optical density of each band was quantified using image analysis software.
Statistical analysis
All data are presented as mean±S.D. Quantitative data were analyzed by one-way analysis of variance (ANOVA), followed by the Student-Newman-Keuls test and the Dunnett’s test. Differences were considered significant when P<0.05.
Abbreviations
- Aβ42:
-
amyloid-β1–42
- NSPCs:
-
adult hippocampal neural stem/progenitor cells
- DG:
-
dentate gyrus
- SGZ:
-
subgranular zone
- SVZ:
-
subventricular zone
- AD:
-
Alzheimer’s disease
- FPR2:
-
formylpeptide receptor 2
- ROS:
-
reactive oxygen species
- p38 MAPK:
-
p38 mitogen-activated protein kinase
- SA-β-gal:
-
senescence-associated β-galactosidase
- NAC:
-
N-acetylcysteine
- SB:
-
SB203580
References
Taupin P . Adult neurogenesis, neural stem cells and Alzheimer's disease: developments, limitations, problems and promises. Curr Alzheimer Res 2009; 6: 461–470.
Mu Y, Gage FH . Adult hippocampal neurogenesis and its role in Alzheimer's disease. Mol Neurodegen 2011; 6: 85.
Querfurth HW, LaFerla FM . Alzheimer's disease. N Engl J Med 2010; 362: 329–344.
Huang Y, Mucke L . Alzheimer mechanisms and therapeutic strategies. Cell 2012; 148: 1204–1222.
Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Muller FJ, Loring JF et al. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci USA 2009; 106: 13594–13599.
Yamasaki TR, Blurton-Jones M, Morrissette DA, Kitazawa M, Oddo S, LaFerla FM . Neural stem cells improve memory in an inducible mouse model of neuronal loss. J Neurosci 2007; 27: 11925–11933.
Hampton DW, Webber DJ, Bilican B, Goedert M, Spillantini MG, Chandran S . Cell-mediated neuroprotection in a mouse model of human tauopathy. J Neurosci 2010; 30: 9973–9983.
Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP . Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem 2002; 83: 1509–1524.
Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP . Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer's disease. Neuromolecular Med 2002; 1: 125–135.
Verret L, Jankowsky JL, Xu GM, Borchelt DR, Rampon C . Alzheimer's-type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J Neuroscience 2007; 27: 6771–6780.
Zhang C, McNeil E, Dressler L, Siman R . Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer's disease. Exp Neurol 2007; 204: 77–87.
Rodriguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM et al. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS One 2008; 3: e2935.
Kempermann G, Kuhn HG, Gage FH . Experience-induced neurogenesis in the senescent dentate gyrus. J Neurosci 1998; 18: 3206–3212.
Kuhn HG, Dickinson-Anson H, Gage FH . Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci 1996; 16: 2027–2033.
Golde TE, Miller VM . Proteinopathy-induced neuronal senescence: a hypothesis for brain failure in Alzheimer's and other neurodegenerative diseases. Alzheimer's Res Ther 2009; 1: 5.
Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU et al. Astrocyte senescence as a component of Alzheimer's disease. PLoS One 2012; 7: e45069.
Donnini S, Solito R, Cetti E, Corti F, Giachetti A, Carra S et al. A peptides accelerate the senescence of endothelial cells in vitro and in vivo, impairing angiogenesis. FASEB J 2010; 24: 2385–2395.
Cao L, Wang H, Wang F, Xu D, Liu F, Liu C . Abeta-induced senescent retinal pigment epithelial cells create a proinflammatory microenvironment in AMD. Invest Ophthalmol Vis Sci 2013; 54: 3738–3750.
Reynolds BA, Weiss S . Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992; 255: 1707–1710.
Campisi J, d'Adda di Fagagna F . Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8: 729–740.
Takahashi A, Ohtani N, Yamakoshi K, Iida S-i, Tahara H, Nakayama K et al. Mitogenic signalling and the p16INK4a–Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol 2006; 8: 1291–1297.
Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995; 373: 523–527.
Arendash GW, King DL, Gordon MN, Morgan D, Hatcher JM, Hope CE et al. Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res 2001; 891: 42–53.
Guenette SY, Tanzi RE . Progress toward valid transgenic mouse models for Alzheimer's disease. Neurobiol Aging 1999; 20: 201–211.
Cattaneo F, Guerra G, Ammendola R . Expression and signaling of formyl-peptide receptors in the brain. Neurochem Res 2010; 35: 2018–2026.
Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 2006; 12: 446–451.
Tiffany HL, Lavigne MC, Cui YH, Wang JM, Leto TL, Gao JL et al. Amyloid-beta induces chemotaxis and oxidant stress by acting at formylpeptide receptor 2, a G protein-coupled receptor expressed in phagocytes and brain. J Biol Chem 2001; 276: 23645–23652.
Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006; 443: 448–452.
Song H, Stevens CF, Gage FH . Astroglia induce neurogenesis from adult neural stem cells. Nature 2002; 417: 39–44.
Ashton RS, Conway A, Pangarkar C, Bergen J, Lim K-I, Shah P et al. Astrocytes regulate adult hippocampal neurogenesis through ephrin-B signaling. Nat Neurosci 2012; 15: 1399–1406.
Heo C, Chang K-A, Choi HS, Kim H-S, Kim S, Liew H et al. Effects of the monomeric, oligomeric, and fibrillar Aβ42 peptides on the proliferation and differentiation of adult neural stem cells from subventricular zone. J Neurochem 2007; 102: 493–500.
Lopez-Toledano MA . Neurogenic effect of -amyloid peptide in the development of neural stem cells. J Neurosci 2004; 24: 5439–5444.
Sotthibundhu A, Li Q-X, Thangnipon W, Coulson EJ . Aβ1–42 stimulates adult SVZ neurogenesis through the p75 neurotrophin receptor. Neurobiol Aging 2009; 30: 1975–1985.
Jankowsky JL . Mutant presenilins specifically elevate the levels of the 42 residue -amyloid peptide in vivo: evidence for augmentation of a 42-specific secretase. Hum Mol Genetics 2003; 13: 159–170.
Chen K, Iribarren P, Huang J, Zhang L, Gong W, Cho EH et al. Induction of the formyl peptide receptor 2 in microglia by IFN-gamma and synergy with CD40 ligand. J Immunol 2007; 178: 1759–1766.
Mitchell D, Rodgers K, Hanly J, McMahon B, Brady HR, Martin F et al. Lipoxins inhibit Akt/PKB activation and cell cycle progression in human mesangial cells. Am J Pathol 2004; 164: 937–946.
Yoneyama M, Kawada K, Gotoh Y, Shiba T, Ogita K . Endogenous reactive oxygen species are essential for proliferation of neural stem/progenitor cells. Neurochem Int 2010; 56: 740–746.
Borchelt DR, Walton NM, Shin R, Tajinda K, Heusner CL, Kogan JH et al. Adult neurogenesis transiently generates oxidative stress. PLoS One 2012; 7: e35264.
Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2011; 8: 59–71.
Kennedy KAM, Sandiford SDE, Skerjanc IS, Li SSC . Reactive oxygen species and the neuronal fate. Cell Mol Life Sci 2011; 69: 215–221.
Taupin P . A dual activity of ROS and oxidative stress on adult neurogenesis and Alzheimer's disease. Cent Nerv Syst Agents Med Chem 2010; 10: 16–21.
Bose R, Moors M, Tofighi R, Cascante A, Hermanson O, Ceccatelli S . Glucocorticoids induce long-lasting effects in neural stem cells resulting in senescence-related alterations. Cell Death Dis 2010; 1: e92.
Brewer GJ, Torricelli JR . Isolation and culture of adult neurons and neurospheres. Nature Protocols 2007; 2: 1490–1498.
Kempermann G, Palmer T, Rünker AE, Kannan S, Claasen J-H, Babu H . A protocol for isolation and enriched monolayer cultivation of neural precursor cells from mouse dentate gyrus. Front Neurosci 2011; 5: 1–10.
Dahlgren KN . Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem 2002; 277: 32046–32053.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81270432 and 31171033) and the Polytechnic crossover Foundation of Shanghai Jiao Tong University (AE1700003).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
He, N., Jin, WL., Lok, KH. et al. Amyloid-β1–42 oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis 4, e924 (2013). https://doi.org/10.1038/cddis.2013.437
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.437
Keywords
This article is cited by
-
Applications of nanodiamonds in the diagnosis and treatment of neurological diseases
Journal of Nanoparticle Research (2022)
-
Functions of resolvin D1-ALX/FPR2 receptor interaction in the hemoglobin-induced microglial inflammatory response and neuronal injury
Journal of Neuroinflammation (2020)
-
Aβ40 Oligomers Promote Survival and Early Neuronal Differentiation of Dentate Gyrus-Isolated Precursor Cells Through Activation of the Akt Signaling Pathway
Neurotoxicity Research (2020)
-
Annexin A1-derived peptide Ac2-26 in a pilocarpine-induced status epilepticus model: anti-inflammatory and neuroprotective effects
Journal of Neuroinflammation (2019)
-
Amyloid β Peptide Compromises Neural Stem Cell Fate by Irreversibly Disturbing Mitochondrial Oxidative State and Blocking Mitochondrial Biogenesis and Dynamics
Molecular Neurobiology (2019)
