
Abstract
Autophagy (macroautophagy) is an evolutionarily conserved lysosomal degradation process, in which a cell degrades long-lived proteins and damaged organelles. Recently, accumulating evidence has revealed the core molecular machinery of autophagy in carcinogenesis; however, the intricate relationship between autophagy and cancer continue to remain an enigma. Why does autophagy have either pro-survival (oncogenic) or pro-death (tumor suppressive) role at different cancer stages, including cancer stem cell, initiation and progression, invasion and metastasis, as well as dormancy? How does autophagy modulate a series of oncogenic and/or tumor suppressive pathways, implicated in microRNA (miRNA) involvement? Whether would targeting the oncogenic and tumor suppressive autophagic network be a novel strategy for drug discovery? To address these problems, we focus on summarizing the dynamic oncogenic and tumor suppressive roles of autophagy and their relevant small-molecule drugs, which would provide a new clue to elucidate the oncosuppressive (survival or death) autophagic network as a potential therapeutic target.
Similar content being viewed by others


Facts
-
Autophagy, an evolutionarily conserved lysosomal degradation process, is well-suited to have the role of the Roman God Janus in sealing the fate of cancer cells: survival or death.
-
Autophagy has either a pro-survival (oncogenic) or pro-death (tumor suppressive) role at different cancer stages, including cancer stem cell (CSC), initiation and progression, invasion and metastasis, and dormancy.
-
Autophagy may modulate a series of oncogenic and tumor suppressive pathways in cancer, implicated in microRNA (miRNA) involvement.
-
Targeting the oncogenic and tumor suppressive autophagic network would be a new promising therapeutic strategy.
Open Questions
-
Why does autophagy have either pro-survival (oncogenic) or pro-death (tumor suppressive) role at specific cancer stage, including CSC, initiation and progression, invasion and metastasis and dormancy?
-
How does autophagy modulate a series of oncogenic and/or tumor suppressive signaling pathways, implicated in miRNA involvement?
-
Whether would targeting the oncogenic and tumor suppressive autophagic network be a novel cancer therapeutic strategy?
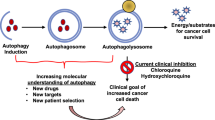
Autophagy, a term from the Greek words ‘auto’ (self) and ‘phagy’ (to eat), is an evolutionarily conserved, multistep lysosomal degradation process for the clearance of damaged or superfluous proteins and organelles.1 Macroautophagy (autophagy) is a pivotal mechanism for catabolic regulation that involves delivering the cytoplasmic cargo sequestered inside double-membrane vesicles to the lysosome and is highly regulated by a limited number of autophagy-related (ATG) genes.2
The complete autophagic flow is a highly regulated process that can generally be divided into the following five stages: induction, vesicle nucleation, vesicle elongation and completion, docking and fusion, and degradation and recycling.3 Induction of autophagy is initiated by the ULK complex, which contains the mammalian ATG1 homologs ULK1 or ULK2, ATG13, focal adhesion kinase family interacting protein of 200 kDa (FIP200) and ATG101 (Figure 1a).4 Then vesicle nucleation occurs, in which proteins and lipids are recruited for construction of the autophagosomal membrane. This process is initiated by activation of the class III PI3K/Beclin-1 complex.5 Numerous binding partners of this complex function as either positive or negative regulators, including Bax-interacting factor-1 (Bif-1), ATG14L, UV irradiation resistance-associated gene (UVRAG), activating molecule in Beclin-1-regulated autophagy protein 1 (Ambra1), Rubicon and Bcl-2/Bcl-XL (Figure 1b).6 Ubiquitin-like protein conjugation is required for vesicle elongation and autophagosome completion, mediated by ATG3, ATG5, ATG7, LC3 and so on, to fully encapsulate the cytosolic cargo (Figure 1c).7 Recruitment of cargo from the cytoplasm requires autophagy receptors such as p62. Finally, the process of docking and fusion is required for the disassembly of ATG protein complexes from matured autophagosomes, and regulated by ATG2, ATG9 and ATG18.8 Fusion of an autophagosome with endolysosomal compartments leads to the breakdown of cargoes by acidic hydrolases, resulting in degradation and recycling (Figure 1d).

The autophagic process involved in miRNAs. The autophagic process can generally be divided into the following five stages: induction, vesicle nucleation, vesicle elongation and completion, docking and fusion as well as degradation and recycling. MiRNAs, mainly playing the negatively regulators, can impede different steps of the autophagy process. (a) Autophagy induction (miR-18a, miR-101, miR-106a and miR-885-3p); (b) vesicle nucleation (miR-30a, miR-376b, miR-519a, miR-374a and miR-630); (c) vesicle elongation and completion (miR-519a, miR-30a, miR-181a, miR-374a, miR-885-3p, miR-101, miR-376b, miR-199a-5p, miR-375, miR-204 and miR-630); (d) docking and fusion (miR-34)
ATG family is delineated as physiological mechanisms of autophagy, with numerous links to cancer.9 Autophagy can exert cytoprotective effects, as autophagy keeps cells alive during nutrient and growth factor deprivation conditions due to the ability of autophagy to recycle nutrients, maintain cellular energy homeostasis and degrade toxic cytoplasmic constituents.10 The autophagy-mediated elimination of altered cytosolic constituents, including aggregated proteins or damaged organelles, can protect cells from further damage.11 Conversely, if the cellular stress leads to continuous or excessively induced autophagy, cell death would ensue. Under this circumstance, autophagy has a death-promoting role as type II programmed cell death (type II PCD), compared with apoptosis (type I PCD).12 On the basis of biochemical and functional considerations, the term autophagic cell death is used to indicate a death instance mediated by autophagy, suppressed by the inhibition of autophagy by chemicals and/or genetic means.13 From a purely morphological perspective, autophagic cell death occurs in the absence of chromatin condensation but is accompanied by large-scale autophagic vacuolization of the cytoplasm, but the term autophagic cell death may be highly prone to misinterpretation and thus being used with caution.14
However, these paradoxical studies are often confusing, depending on different cell types, autophagy seems to have either an oncogenic or tumor suppressive role for the regulation of core pathways, thereby sealing the distinctive fate of cancer cells.15 Thus, this review summarizes the Janus role of autophagy at different cancer cell stages, oncogenic and tumor suppressive autophagic pathways involved in miRNAs, as well as relevant autophagy-modulated drugs in cancer therapy.
Autophagy and Cancer Cell
Autophagy in CSC
CSCs have the ability to self-renew as well as to cause heterogeneous lineages of cancer cells, and these tumor-forming cells can originate from the stem, progenitor or differentiated cells.16 Autophagy has an essential role in the development of drug resistance, self-renewal, differentiation and tumorigenic potentials of CSCs. CD133+ liver CSCs (LCSCs) can be significantly enriched with higher autophagy levels after hypoxia and nutrient starvation (H/S) in hepatocellular carcinoma (HCC) Huh-7 cells; thereby autophagy may have an essential role in LCSC maintenance.17 The conversion of microtubule-associated protein LC3-I to LC3-II as well as increased accumulations of ATG7 and Beclin-1 are observed in pancreatic CSCs treated with Rottlerin (ROT), a widely used protein kinase C-delta (PKC-δ) inhibitor. Additionally, the gene silencing of ATG7 and Beclin-1, or co-treatment of the autophagosome inhibitor, 3-methyladenine (3-MA), can inhibit ROT-induced autophagic cell death.18 Moreover, breast cancer cells with low metastatic potentials can be induced into a reversible state of dormancy by farnesyl transferase inhibitors (FTIs), with expressions of ATG5, ATG12 and LC3B in these dormant stem cell-like breast cancer cells. The c-jun NH2 terminal kinase (JNK)-mediated autophagic pathway can also be upregulated in breast CSCs with the periods of FTI-induced dormancy.19 Knockdown of damage-regulated autophagy modulator 1 (DRAM1) may decrease p62 localization to autophagosome and its autophagy-mediated degradation, suggesting a crucial role of DRAM1 in p62-mediated autophagy. The DRAM1 expression has been correlated with the activation of mitogen-activated protein kinases (MAPKs) and the mesenchymal marker c-MET in glioblastoma stem cells.20 And, irradiation of CD133+ glioma stem cells (GSCs) can induce autophagy in a short time and then autophagy slightly decreases the viability of the cells. The gamma-radiation may also induce a large degree of autophagy in the CD133+ GSCs, and the CD133+ cells expresses higher levels of the ATG proteins such as ATG5, LC3 and ATG12 (Figure 2a).21

Autophagy and cancer cell. (a) Oncogenic autophagic pathways at CSC stage (e.g., PI3KCI-Akt-mTORC1 and Bcl-2/Bcl-XL); (b) oncosuppressive autophagic pathways at cancer initiation and progression stages (e.g., PI3KCI-Akt-mTORC1, cytoplasm p53, FIP200, NF-κB, Ras and BCR-ABL; nucleus p53, Beclin-1, UVRAG, Bif-1 and FoxO); (c) oncosuppressive autophagic pathways at cancer invasion and metastasis stages (e.g., HMGB1, MAPKs and PAK); (d) oncogenic autophagic pathways at cancer dormancy stage (e.g., PI3KCI and ARHI)
Autophagy in cancer initiation and progression
Cancer initiation is defined as ‘a process in which normal cells are changed so that they are able to form cancers’, whereas cancer progression is characterized by increased growth speed and invasiveness of the tumor cells.22 Multiple ATG members and their key regulators have been known to have crucial roles in the formation of the autophagosome and autophagic regulation, which are closely linked to cancer initiation and progression. The best known modulator of autophagy is mammalian target of rapamycin (mTOR), which forms two complexes, mTORC1 and mTORC2.23 FIP200, as a component of ULK1/ATG1-mATG13-FIP200-ATG101, can regulate different intracellular signaling pathways by interacting with mTORC1 and p53. Unlike mTORC1, Forkhead box O protein (FoxO), a group of transcription factors that is required for autophagy, can be inhibited by the PI3KCI/Akt pathway, which may indicate the suppressive effect of FoxO in tumorigenesis.24 In addition, BCR-ABL, which can cause over-activation of several intracellular signaling pathways causing hyper-proliferation and insensitivity to growth factor withdrawal, is another key oncogenic factor involved in mTORC1 regulation.25 Besides, oncogenic Ras can induce autophagy, which is associated with Beclin-1/ATG6, whereas the co-expression of Bcl-2 family members can inhibit Beclin-1.26 Ras-driven tumor cells have metabolic abnormalities that require high levels of autophagy for cell growth and survival. Beclin-1 can be overexpressed in breast cancer cells, thereby increasing autophagy and decreasing the growth of tumorigenicity, indicating that Beclin-1 may function as a tumor suppressor.6 During the p53 response to DNA damage, several genes are transcriptionally activated, which include cell death genes involved in autophagy.27 DRAM1 and the p53 upregulated modulator of apoptosis (PUMA) have been shown to be required for p53-induced autophagy. PUMA may target autophagy, whereas p53 is found to induce DRAM1-dependent autophagy in response to mitochondrial dysfunction (Figure 2b).28
Autophagy in cancer invasion and metastasis
Cancer invasion and metastasis can be promoted by proteins that stimulate tumor cell attachment to host cellular or extracellular matrix (ECM) determinants, tumor cell proteolysis of host barriers.29 Reduced levels of oxygen and nutrients and malfunction of ECM and endoplasmic reticulum (ER) stress are critical parameters modulating the tumor microenvironment. Thus, abnormality in the tumor microenvironment can induce autophagy to aid the maintenance of cancer cell viability and promote cancer cell metastasis under these stressful conditions.30 Moreover, autophagy can either promote or impede metastasis via complicated processes, and the initial step requires signals within the tumor microenvironment. Autophagy directly mediates tumor-associated inflammation by regulating immune-modulatory factors such as high-mobility group box protein1 (HMGB1), which is an extracellular signal in tumor metastasis.31 HMGB1, as a novel Beclin-1-binding protein, has a key role in neutralizing and eliminating reactive reactive oxygen species (ROS) under oxidative stress, which can induce autophagy through promotion of ATG4C.32 When released, HMGB1 may prevent metastasis, acting as an antitumor immune response. Statins can inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase. Activation of the AMP-activated protein kinase (AMPK)-mTOR signaling pathway may be a major mechanism of statin-induced autophagy, as epleting cellular geranylgeranyl diphosphate activates AMPK and inactivates mTOR, thereby leading to autophagic responses (Figure 2c).33
Autophagy in cancer dormancy
Cancer dormancy is a poorly understood phase of cancer progression and only recently have its underlying molecular mechanisms started to be revealed.34 With increasing scrutiny on how fundamental cellular stress-response pathways impact survival and expansion of dormant tumor cells, autophagy has emerged as an attractive target against dormant tumor cells. The aplasia Ras homolog member I (ARHI) is an imprinted tumor suppressor gene, which is associated with decreased progression-free survival. It has been demonstrated that re-expression of ARHI can inhibit cancer growth, initiate autophagy and induce tumor dormancy.35 ARHI localizes with LC3 and is associated with an increased expression of ATG4C, indicating its regulatory role in autophagy by direct physical interaction. Additionally, ARHI-induced autophagy is well-suited to suppress intracellular PI3KCI signaling, and the expression of ARHI can lead to a marked inhibition of PI3KCI-Akt-mTORC1 and MAPK signaling. When the level of ARHI remains at normal autophagic activity, tumor dormancy is likely to occur, as removed tumors can recover their abilities that are consistent with the autophagic process.36 The inhibition of ARHI-induced autophagy can dramatically reduce the regrowth of xenografted tumors upon the reduction of ARHI levels, suggesting that autophagy may contribute to the survival of dormant cells; therefore, ARHI can induce autophagic cell death as well as promoting tumor dormancy in the presence of factors that promote survival in the tumor microenvironment.36 p27Kip1, a cyclin-dependent kinase inhibitor involved in G0/G1 cell cycle arrest, is identified as a downstream target of the LKB1-AMPK pathway, inducing autophagy and facilitating cell survival (Figure 2d).37
Oncosuppressive Autophagic Pathways in Cancer
Oncogenic function of autophagy
Oncogenic autophagic pathways involving PI3KCI, Akt, mTORC1, Ras, BCR-ABL, STAT, Bcl-2 and Bcl-XL can have their key roles in cancer cell survival.38, 39 Of note, mTORC1 is the major inhibitory signal that shuts off autophagy in the presence of growth factors and abundant nutrients.40 PI3KCI/Akt signaling molecules can link receptor tyrosine kinases to mTORC1 activation, thereby repressing autophagy in response to insulin-like and other growth factor signals.41 The known mTORC1 targets in autophagy are the autophagic regulator kinase ULK1 and its accessory protein ATG13, in which mTORC1-mediated phosphorylation causes suppression of autophagy.42 In addition, Akt is a central node in a complex cascade of signaling with a feedback loop that influences the regulation of this kinase, whereas PI3KCI can antagonize autophagy through the activation of the Akt pathway.43 Other antiapoptotic Bcl-2 subfamilies, such as Bcl-2 and Bcl-XL, can also mediate autophagic pathways in cancer. Bcl-2 can block the interaction between tumor suppressive Beclin-1 and PI3KCIII/Vps34; thereby, downregulating autophagic activity.44 Although the precise mechanism of how Bcl-2 blocks Beclin-1 and PI3KCIII/Vps34 interaction remains unclear, the binding of Bcl-2 to Beclin-1 seems to be constitutive and its detachment from Beclin-1 is of great importance in autophagic induction. Moreover, oncogenic BCR-ABL can stimulate the transcription of mTORC1 via PI3KCI-Akt-FoxO signaling in chronic myeloid leukemia (CML).45 Ras, another oncoprotein, can induce autophagic cell death in glioma and gastric cancer cells, and thus inducing autophagic death that may constitute another level of control against cancer formation.46 In addition, hypoxic regions of tumors are protected from cytotoxic T-lymphocytes (CTL), dependent on the phosphorylation of STAT3 and autophagic degradation of p62 allows phospho-STAT3 to accumulate promoting resistance to CTL (Figure 3).47

Oncogenic and tumor suppressive autophagic pathways involved in miRNAs. Autophagic pathways include several oncogenic pathways, such as PI3KCI/Akt/mTORC1 signaling pathway, Ras-Raf-MAPK cascade and BCR-ABL as well as other tumor suppressive pathways, for example, Beclin-1 complexes and p53 signaling. Moreover, some oncogenic miRNAs such as miR-18a, miR-106a, miR-181a, miR-199a-5p, miR-221/222 and miR-376b, as well as tumor suppressive miRNAs including miR-9*, miR-30a, miR-34a, miR-101, miR-130a, miR-204 and miR-375 may have their key roles in the regulation of the aforementioned autophagic pathways in cancer
Tumor suppressive function of autophagy
Tumor suppressive autophagic pathways contain some key regulators, most notably Beclin-1, UVRAG, Bif-1, ATG5, p53 and FoxO1. Beclin-1 can positively regulate autophagy by combining with PI3KCIII/Vps34, and other positive and negative cofactors such as Vps15, ATG14L/Barkor, UVRAG, Bif-1, Rubicon, Ambra1, HMGB1, Survivin, Akt and Bcl-2/Bcl-XL to form the Beclin-1 interactome.48 Beclin-1 is mapped to a tumor susceptibility locus that is monoallelically deleted in a high percentage of human breast, ovarian and prostate cancers, and the ectopic Beclin-1 expression may reduce cancer cell proliferation in vitro and decrease tumorigenic potential in vivo, indicating a role for autophagy in tumor suppression.49 In addition to Beclin-1, the genetic alteration of other cofactors has been found in various types of cancer. These include nonsense mutations in UVRAG in colon cancer cells and gastric carcinomas, as well as the downregulation of Bif-1, another positive regulator of autophagy in colon adenocarcinomas.50 Intriguingly, some antiapoptotic Bcl-2 family members, such as Bcl-2 and Bcl-XL, contain four Bcl-2 homology domains and inhibit autophagy via interacting with Beclin-1. Bcl-2 can block Beclin-1 interaction with PI3KCIII, decrease PI3KCIII activity and downregulate autophagy through either the disassociation of Beclin-1/PI3KCIII or inhibition of Beclin-1 activity.51 In addition, the deletion of ATG5, an essential autophagy gene, has been in natural killer cell malignancies. Importantly, programmed cell death protein 4 (PDCD4), a suppressor of gene transcription and translation, has a crucial inhibitory role in several types of human tumors, can inhibit the expression of an essential autophagy-related gene, ATG5 and the formation of an ATG12-ATG5 complex.52 Moreover, p53, which interacts with some pro-apoptotic Bcl-2 subfamily members, has a controversial influence on autophagy, depending on its different subcellular localizations. In the nucleus, p53 abets autophagy mainly through interacting with its targets, DRAM and sestrin 1/2. DRAM, a p53 target gene which can encode a lysosomal protein that induces autophagy, is found to be an effector of p53-mediated cell death, as it illustrates the direct link between p53 and autophagy.53 Sestrin-1 and sestrin-2 are two targets of DRAM, whose expressions are usually induced upon DNA damage and oxidative stresses, thus being considered to be p53-mediated tumor expression through an mTORC1 signaling.54 Cytoplasm p53 can inhibit autophagy without the assistance of its role as a transcriptional factor. In addition, p53 can activate pro-apoptotic members of Bcl-2 family such as Bax, Noxa and PUMA, which are involved in the permeabilization of the outer mitochondrial membrane.55 Moreover, the acetylated tumor suppressive FoxO1 can bind to ATG7, which is an important regulator in autophagosome expansion, and the FoxO1/ATG7 complex may impact autophagy in human colon cancer HCT116 cells or in HeLa cells.56 X-box binding protein 1u (XBP1u) has a critical role in FoxO1 degradation by recruiting FoxO1 to the 20S proteasome. Moreover, the phosphorylation of XBP1u by MAPK1/3 has been found to be essential for the enhancement of the interaction between XBP1u and FoxO1 (Figure 3).57
Oncogenic and Tumor Suppressive miRNAs in Autophagy
Oncogenic miRNAs in autophagic pathways
miRNAs, non-coding RNA molecules with 18–25 nucleotides in length, are estimated to regulate about 30% human gene expression at both the post-transcriptional and the translational levels.58 MiR-18a potentially regulates ataxia telangiectasia mutated (ATM) that can upregulate autophagic process, and the impact of miR-18a on autophagy and ATM expression has recently been revealed in HCT116 colon cancer cells. Overexpression of miR-18a in HCT116 cells is found to enhance autophagy and ionize radiation-induced autophagy, as well as leading to the increase of ATM and suppression of mTORC1 activity. Therefore, the fact that miR-18a can regulate autophagy and ATM gene expression in colon cancer cells reveals a novel function for miR-18a in a critical cellular event and on a crucial gene with significant impacts in cancer development, progression and treatment.59 The overexpression of miR-181a can result in the attenuation of starvation-induced and rapamycin-induced autophagy in MCF-7, Huh-7 and K562 cells. Indeed, the cellular level of ATG5 can decrease upon miR-181a overexpression and increase following some antagomirs, thereby revealing that ATG5 is identified as a miR-181a target.60 In addition, the level of miR-199a-5p is found to be significantly reduced in HCC patients treated with cisplatin-based chemotherapy, as the forced expression of miR-199a-5p can promote cisplatin-induced inhibition of cell proliferation. In addition, cisplatin treatment can activate autophagy in Huh-7 and HepG2 cells. Further, the downregulation of miR-199a-5p can enhance autophagy activation by targeting ATG7.61 MiR-221/222 may inhibit the cell cycle inhibitor p27Kip1, a downstream modulator of PI3KCI/Akt, leading to autophagic cell death in HER2/neu-positive primary human breast carcinoma MCF-7 cells, and the ectopic expression of miR-221/222 is found to render the parental MCF-7 cells resistant to tamoxifen. Thus, a relationship has been established between miR-221/222 expression and HER2/neu overexpression in breast cancers that are generally resistant to tamoxifen therapy.62 Both ATG4C and Beclin-1 have been identified as cellular targets of miR-376b. Upon the overexpression of the miRNA, mRNA and protein levels of ATG4C and Beclin-1 decrease in MCF-7 and Huh-7 cells. Antagomir-mediated inactivation of the endogenous miR-376b can lead to an increase in levels of ATG4C and Beclin-1 (Figure 3).63
Tumor suppressive miRNAs in autophagic pathways
ATG7 is a potential target for miR-17, and this tumor suppressive miRNA can negatively regulate ATG7 expression, resulting in a modulation of the autophagic status in T98G glioblastoma cells. Interestingly, anti-miR-17 administration can activate autophagy through autophagosome formation, which is resulted by LC3B and ATG7 protein expression increase in living cells.64 Overexpression of miR-23b inhibits radiation-induced autophagy, whereas an inhibitor of miR-23b promotes autophagy in pancreatic cancer cells. In addition, overexpression of miR-23b can sensitize pancreatic cancer cells to radiation. The target of miR-23b, ATG12, has been found to be overexpressed in radioresistant cells; levels of ATG12 correlate with the occurrence of autophagy.65 Autophagic activity in cancer cells may also increase after cis-dichloro-diamine platinum (cis-DDP) or Taxol treatment, as indicated by the enhanced expression of Beclin-1. The forced expression of miR-30a significantly reduces Beclin-1 and the autophagy activity of tumor cells induced by cis-DDP. Thus, miR-30a can sensitize tumor cells to cis-DDP via reducing Beclin-1-mediated autophagy and that increasing miR-30a level in tumor cells represents a novel approach to enhance the efficacy of chemotherapy during cancer treatment.66 The tumor suppressive miR-101 can act as a potent inhibitor of basal autophagy, and three novel miR-101 targets such as Stathmin 1 (STMN1), Ras-related protein Rab-5A (RAB5A) and ATG4D have been identified.67 STMN1 overexpression will partially rescue cells from miR-101-mediated inhibition of autophagy, indicating a functional importance of this target. In an in vivo tumor setting, progressive loss of miR-101 can contribute to elevated levels of autophagy in cancer cells, enabling long-term tumor cell survival by allowing them to cope with metabolic stress and promoting eventual regrowth following treatment.68 MiR-130a can directly regulate ATG2B, the downregulation of whose expression can be transfected with miR-130a in chronic lymphocytic leukemia (CLL). ATG2B can then interact with ATG2A and WDR45, and miR-130a is involved in autophagy and cell survival in CLL cells by regulating maturation.69 Additionally, the role of tumor suppressive miR-204 in autophagy regulation was initially recognized in cardiomyocytes and further confirmed in renal cell carcinoma (RCC) via LC3B.70 The overexpression of miR-204 can arrest subcutaneous tumor growth relative to a control miRNA with a mutated seed sequence and are rescued upon re-expression of LC3B lacking the 3′-untranslated region (3'UTR). Thus, a negative correlation between LC3B and miR-204 is shown in RCC. Interestingly, the regulation of miR-204 in autophagy and cytotoxity occurs only when the von Hippel-Lindau tumor-suppressor gene (VHL) is absent.71 MiR-375 has a predominantly inhibitory role in autophagy activation by attenuating the protective role of autophagy by targeting ATG7, ATG4D, STMN1 and RAB5A in HCC. Moreover, miR-375, normally downregulated in HCC when exogenously expressed, can inhibit autophagy in response to hypoxia by targeting ATG7, reducing the conversion of LC3-I to LC3-II. In mice, xenograft tumors that express miR-375 have fewer autophagic cells, larger areas of necrosis and grow more slowly than tumors from HCC cells that express lower levels of miR-375 (Figure 3).72
Autophagic Network as a Cancer Therapeutic Target
Autophagy-modulated drugs targeting oncogenic pathways
Rapamycin, a well-known inducer of mTORC1-dependent autophagy, is used as an antitumor agent in malignant glioma and breast cancer cells via binding to mTORC1 by forming a complex with FKBP12, thus preventing interaction between mTORC1 and its kinase substrates.73 Because of significant therapeutic effects of rapamycin, its synthetic analogues including temsirolimus (CCI-779), everolimus (RAD001) and deforolimus (AP23573) are developed.74 Temsirolimus can exert an antitumor effect by inducing autophagy in malignant glioma and breast cancer cells or by causing the downregulation of p21 to induce autophagy in mantle-cell lymphoma cells.75 Additionally, everolimus can induce autophagy by enhancing the antitumor effect of the oncolytic adenovirus Delta-24-RGD in ovarian cancer cells.76 Some autophagy activators such as perhexiline, niclosamide, amiodarone and ROT have also been identified to promote autophagy by inhibiting the function of mTORC1 in cancers under nutrient-rich conditions without blocking mTORC2.77 Sorafenib, a multiple tyrosine kinase inhibitor, can induce the accumulation of autophagosome, thereby inhibiting the mTORC1 pathway in HCC.78 The autophagic inducer, AZD8055, an ATP-competitive inhibitor of mTOR, can potently block phosphorylation of mTORC1 substrates as well as Akt in order to suppress tumor growth in mice by inducing autophagy.79 In lung cancer cells, sulindac sulfide amide (SSA), a N,N-dimethylethyl amine derivative of sulindac sulfide, can display potent tumor cell growth-inhibitory activity by inhibiting Akt/mTOR signaling.80 And, tricribine can induce autophagy through dephosphorylation of Akt and inhibition of mTORC1 in T-cell acute lymphoblastic leukemia.81 In human prostate cancer cells, phenethyl isothiocyanate (PEITC) can suppress the phosphorylation of both Akt and mTOR, which are implicated in regulation of autophagy.82 Tetrahydrocannabinol (THC) induces cancer cell death through activation of autophagy via enhancing the ER stress that activates autophagy via inhibition of Akt and mTORC1.83 Moreover, the treatment of cancer cells with the ER stress-inducing drug nelfinavir can result in the expression of endogenous mTOR inhibitor sestrin-2, and transient overexpression of ectopic sestrin-2 may lead to mTOR inhibition and autophagy, confirming a link among ER stress, sestrin-2 up-regulation and mTOR inhibition.84 The upregulation of sestrin-2 can also occur in cells treated with the proteasome inhibitor bortezomib.84
A new class of PI3K inhibitors with multiple-target abilities has been uncovered. Perifosine can exhibit the antitumor activity in various cancers and this activity is partly associated with its ability to inhibit mTORC1 through PI3KCI-Akt signaling.85 The imidazo[4,5-c]quinoline derivative NVP-BEZ235 inhibits PI3K and mTOR activities by binding to the ATP-binding cleft of both enzymes, thereby leading to the induction of autophagy in glioma cells.86 PI103, a potent inhibitor of PI3KCI, Akt and mTORC1 can lead to autophagy enhancement and inhibit the proliferation and invasion of a wide variety of cancer cells.87 Acting as a central nervous system stimulant, caffeine is known to inhibit the kinase activity of some proteins, mainly dependent on the PI3KCI-Akt-mTORC1 signaling pathway.88 Two downstream kinases of PI3KCI, Akt and mTORC1, can be suppressed by resveratrol, resulting in an increase in both GFP-LC3 puncta and LC3-II levels.89, 90 Dexamethasone is another potent synthetic member of the glucocorticoid family, which may inhibit Akt phosphorylation and PI3KCI-mTORC1 in acute lymphoblastic leukemia (ALL).91 Ophiopogonin B (OP-B), a bioactive component of Radix Ophiopogon Japonicus, is a prospective inhibitor of PI3K/Akt in non-small cell lung cancer (NSCLC) cells.92
Imatinib (Gleevec) is a powerful drug in the treatment of CLL and other malignancies that can inhibit the chronic myelogenous leukemia-specific tyrosine kinase BCR-ABL by binding to its ATP-binding site.93 Dasatinib, another tyrosine kinase inhibitor of BCR-ABL, can enhance the therapeutic effect of chemotherapy by inducing autophagic cell death in glioma cells.94 By preventing the tyrosine kinase activity of EGFR, two tyrosine kinase inhibitors including gefitinib and erlotinib have been developed for the treatment of NSCLC. Both gefitinib and erlotinib can activate autophagy to arrest tumor growth both in vitro and in vivo.95 Lapatinib is another tyrosine kinase inhibitor that can target EGFR for the treatment of various solid tumors, including breast, head, colon, prostate and stomach cancers (Supplementary Table S1).96
Autophagy-modulated drugs targeting tumor suppressive pathways
Tamoxifen, a well-recognized antitumor drug for breast cancer treatment, can increase the level of Beclin-1 to stimulate autophagy.97 The BH3 mimetic ABT-737 can specifically decrease the interaction between Bcl-2 and Bcl-XL with the BH3 motif of Beclin-1 and then stimulate the Beclin-1-dependent activation of PI3KCIII.98 As a chemotherapeutic vitamin D analog, EB1089 may trigger and induce Beclin-1-dependent autophagy in MCF-7 cells.99 Spautin-1 promotes the degradation of PI3KCIII/Vps34 complexes by inhibiting two ubiquitin-specific peptidases, USP10 and USP13, as well as targeting the Beclin-1 subunit of Vps34 complexes.100 Xestosponging B can disrupt Beclin-1 through an indirect link established by Bcl-2.101 RAD001, known as an mTORC1 inhibitor, can increase Beclin-1 expression to induce autophagy in leukemia.102 In addition, L-Securinine has an antitumor effect against colon cancer SW480 cell by inducing autophagy in SW480 cell in vitro, which is related with the upregulation of Beclin-1.103 Moreover, chloroquine, 3-MA, wortmannin and LY294002 have been widely used as autophagic inhibitors.104, 105 Wortamannin is a microbial secondary metabolite that can irreversibly inhibit PI3KCIII through covalent binding, whereas LY294002 is a morpholine derivate of quercetin binding to PI3KCIII, which is less potent than wortmannin (Supplementary Table S1).104, 105
Chloroquine and hydroxychloroquine can inhibit autophagy induced by p53-controlled apoptosis in Myc-driven lymphoma and augmented the anticancer activity of cyclophosphamide.106 3-MA can cause autophagic/lysosomal protein degradation, and thus inhibiting PI3KCIII, whereas paclitaxel can increase p53 expression before the regulation of LC3B, thus inhibiting autophagic induction.107 Another attractive strategy is the combined inhibition of autophagy and the proteasome-two processes which are thought to be partly complementary. Proteasome inhibition induces autophagy and autophagy inhibition leads to the accumulation of poly-ubiquitinated proteins.108 Sulphathiazole and sulphacetamide can overexpress critical genes involved in autophagy such as p53 and DRAM in T-47D breast cancer cells (Supplementary Table S1).109
Conclusions
As an evolutionary conserved lysosomal degradation process, autophagy may play the Janus role by regulating some oncogenes (e.g., PI3KCI, Akt, mTORC1, Ras, Raf and BCR-ABL) or other tumor suppressors (e.g., Beclin-1, p53, FoxO1 and BNIP-3), which is implicated in autophagic relevant pathways to jointly seal the fate of cancer cell. Intriguingly, several miRNAs can modulate some oncogenic and tumor suppressive pathways, thus having their negatively regulatory roles in the autophagic process (Figure 4a).

The balance between oncogenic and tumor suppressive autophagy in drug discovery. (a) Conflicting effects of autophagy on cancer: (a) when there are more oncogenes and fewer tumor suppressors, cytoprotective autophagy may occur and thus leading to cancer cell survival; (b) when there are more tumor suppressors and fewer oncogenes, autophagic death may occur and thus culminating in cancer cell demise; (b) oncogenic and tumor suppressive autophagic pathways as drug targets: (a) mTORC1/mTORC2; (b) PI3KCI/Akt; (c) Ras/Raf/MAPKs; (d) BCR-ABL (indicated in red); (a) PI3KCIII/Beclin-1complex; (b) p53; (c) FoxO1; (d) BNIP-3 (indicated in green)
However, these oncogenic and tumor suppressive autophagic pathways involved in miRNA regulation could be integrated into the ‘dynamic’ autophagy network, as they have distinctive roles in survival and death at each cancer stage, such as CSC stage (i.e., PI3KCI/Akt/mTORC1 and Bcl-2/Bcl-XL), initiation and progression (i.e., PI3KCI-Akt-mTORC1, cytoplasmic p53, FIP200, Ras and BCR-ABL; nucleus p53, Beclin-1, UVRAG, Bif-1 and FoxO), invasion and metastasis (i.e., HMGB1, MAPKs and PAK) and dormancy (i.e., ARHI, PI3KCI-Akt-mTORC1 and MAPKs). Hitherto, accumulating evidence demonstrates that some oncogenic and tumor suppressive miRNAs may regulate the autophagic pathways involved in mTORC1 cascade, Beclin-1, p53 and other autophagic components. Thus, unraveling the roles of miRNAs would be crucial for understanding of the importance of miRNA regulation of autophagy in cancer (Figure 4b).
The best hope for cancer therapeutics may lie in discovering candidate small-molecule drugs targeting oncogenic or tumor suppressive autophagic pathways and even the entire autophagic network (multiple-target strategy), rather than the individual gene or protein (single target). The idea of multitarget attacks is not new, as the first formal advocate of the multitarget concept might be the military strategist Carl von Clausewitz, who argued that the strategy should aim at the enemy’s forces, resources, and their will to fight, instead of simultaneously striving for a successful single battle.110 On the basis of this point, elucidating survival or death mechanisms of autophagy would be a promising avenue for discovering more novel drugs targeting the oncogenic and tumor suppressive autophagic network.
Abbreviations
- HMG-CoA:
-
3-hydroxy-3-methylglutaryl coenzyme A
- 3-MA:
-
3-methyladenine
- 3' UTR:
-
3′-untranslated region
- Ambra1:
-
activating molecule in Beclin-1-regulated autophagy protein 1
- AMPK:
-
AMP-activated protein kinase
- ARHI:
-
aplasia Ras homolog member I
- ATG:
-
autophagy-related
- ATM:
-
ataxia telangiectasia mutated
- Bif-1:
-
Bax-interacting factor-1
- CMA:
-
chaperone-mediated autophagy
- cis-DDP:
-
cis-dichloro-diamine platinum
- CLL:
-
chronic lymphocytic leukemia
- CML:
-
chronic myeloid leukemia
- CSC:
-
cancer stem cell
- DRAM1:
-
damage-regulated autophagy modulator 1
- CTL:
-
cytotoxic T-lymphocytes
- ECM:
-
extracellular matrix
- ER:
-
endoplasmic reticulum
- FIP200:
-
focal adhesion kinase family interacting protein of 200 kDa
- FoxO:
-
Forkhead box O protein
- FTI:
-
farnesyl transferase inhibitor
- GSC:
-
glioma stem cell
- HCC:
-
hepatocellular carcinoma
- HMGB1:
-
high-mobility group box protein1
- JNK:
-
c-jun NH2 terminal kinase
- LCSC:
-
liver cancer stem cell
- MAPK:
-
mitogen-activated protein kinase
- miRNA:
-
microRNA
- mTOR:
-
mammalian target of rapamycin
- NSCLC:
-
non-small cell lung cancer
- OP-B:
-
Ophiopogonin B
- PCD:
-
programmed cell death
- PDCD4:
-
programmed cell death protein 4
- PEITC:
-
phenethyl isothiocyanate
- PKC-δ:
-
protein kinase C-delta
- PUMA:
-
the p53 upregulated modulator of apoptosis
- RAB5A:
-
Ras-related protein Rab-5A
- ROS:
-
reactive oxygen species
- ROT:
-
Rottlerin
- RCC:
-
renal cell carcinoma
- SSA:
-
sulindac sulfide amide
- STMN1:
-
Stathmin 1
- THC:
-
Tetrahydrocannabinol
- UVRAG:
-
UV irradiation resistance-associated gene
- VHL:
-
von Hippel-Lindau tumor-suppressor gene
- XBP1u:
-
X-box binding protein 1u
References
Choi AM, Ryter SW, Levine B . Autophagy in human health and disease. N Engl J Med 2013; 368: 651–662.
Yang Z, Klionsky DJ . Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12: 814–822.
Oczypok EA, Oury TD, Chu CT . It's a cell-eat-cell world: autophagy and phagocytosis. Am J Pathol 2013; 182: 612–622.
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20: 1992–2003.
Yang Z, Klionsky DJ . Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22: 124–131.
Fu LL, Cheng Y, Liu B . Beclin-1: Autophagic regulator and therapeutic target in cancer. Int J Biochem Cell Biol 2013; 45: 921–924.
Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC . Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010; 12: 747–757.
Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17: 654–666.
Meschini S, Condello M, Lista P, Arancia G . Autophagy: molecular mechanisms and their implications for anticancer therapies. Curr Cancer Drug Targets 2011; 11: 357–379.
Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MC et al. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta 2009; 1793: 1524–1532.
Kroemer G, Levine B . Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9: 1004–1010.
Maiese K, Chong ZZ, Shang YC, Wang S . Targeting disease through novel pathways of apoptosis and autophagy. Expert Opin Ther Targets 2012; 16: 1203–1214.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19: 107–120.
Clarke PG, Puyal J . Autophagic cell death exists. Autophagy 2012; 8: 867–869.
Levine B, Kroemer G . Autophagy in the pathogenesis of disease. Cell 2008; 132: 27–42.
Sanchez CG, Penfornis P, Oskowitz AZ, Boonjindasup AG, Cai DZ, Dhule SS et al. Activation of autophagy in mesenchymal stem cells provides tumor stromal support. Carcinogenesis 2011; 32: 964–972.
Song YJ, Zhang SS, Guo XL, Sun K, Han ZP, Li R et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett 2013; 339: 70–81.
Singh BN, Kumar D, Shankar S, Srivastava RK . Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochem Pharmacol 2012; 84: 1154–1163.
Chaterjee M, van Golen KL . Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Res 2011; 2011: 362938.
Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013; 32: 699–712.
Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer 2009; 125: 717–722.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Zhou J, Tan SH, Codogno P, Shen HM . Dual suppressive effect of MTORC1 on autophagy: Tame the dragon by shackling both the head and the tail. Autophagy 2013; 9: 803–805.
Zhang Y, Gan B, Liu D, Paik JH . FoxO family members in cancer. Cancer Biol Ther 2011; 12: 253–259.
Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL et al. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011; 30: 1855–1867.
Elgendy M, Sheridan C, Brumatti G, Martin SJ . Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell 2011; 42: 23–35.
Xie X, Le L, Fan Y, Lv L, Zhang J . Autophagy is induced through the ROS-TP53-DRAM1 pathway in response to mitochondrial protein synthesis inhibition. Autophagy 2013; 8: 1071–1084.
Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 2013; 27: 1016–1031.
Beleva E, Grudeva-Popova J . From Virchow's triad to metastasis: circulating hemostatic factors as predictors of risk for metastasis in solid tumors. J BUON 2013; 18: 25–33.
Avivar-Valderas A, Bobrovnikova-Marjon E, Alan Diehl J, Bardeesy N, Debnath J, Aguirre-Ghiso JA . Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 2013; 32: 4932–4940.
Kenific CM, Thorburn A, Debnath J . Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol 2010; 22: 241–245.
Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal 2012; 16: 1264–1284.
Zhang J, Yang Z, Xie L, Xu L, Xu D, Statins Liu X . Autophagy and cancer metastasis. Int J Biochem Cell Biol 2013; 45: 745–752.
Sosa MS, Bragado P, Debnath J, Aguirre-Ghiso JA . Regulation of tumor cell dormancy by tissue microenvironments and autophagy. Adv Exp Med Biol 2013; 734: 73–89.
Lu Z, Bast RC Jr . The tumor suppressor gene ARHI (DIRAS3) inhibits ovarian cancer cell migration through multiple mechanisms. Cell Adh Migr 2013; 7: 232–236.
Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest 2008; 118: 3917–3929.
Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 2007; 9: 218–224.
Liu JJ, Lin M, Yu JY, Liu B, Bao JK . Targeting apoptotic and autophagic pathways for cancer therapeutics. Cancer Lett 2011; 300: 105–114.
Rubinsztein DC, Codogno P, Levine B . Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11: 709–730.
Jewell JL, Russell RC, Guan KL . Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol 2013; 14: 133–139.
Morselli E, Galluzzi L, Kepp O, Mariño G, Michaud M, Vitale I et al. Oncosuppressive functions of autophagy. Antioxid Redox Signal 2011; 14: 2251–2269.
Vadlakonda L, Pasupuleti M, Pallu R . Role of PI3K-AKT-mTOR and Wnt Signaling Pathways in Transition of G1-S Phase of Cell Cycle in Cancer Cells. Front Oncol 2013; 3: 85.
Beauchamp EM, Platanias LC . The evolution of the TOR pathway and its role in cancer. Oncogene 2012; 32: 3923–3932.
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15: 741–750.
Calabretta B, Salomoni P . Suppression of autophagy by BCR/ABL. Front Biosci (Schol Ed) 2012; 4: 453–460.
Wu SY, Lan SH, Cheng DE, Chen WK, Shen CH, Lee YR et al. Ras-related tumorigenesis is suppressed by BNIP3-mediated autophagy through inhibition of cell proliferation. Neoplasia 2011; 13: 1171–1182.
Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res 2011; 71: 5976–5986.
Huang W, Choi W, Hu W, Mi N, Guo Q, Ma M et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res 2012; 22: 473–489.
Zhang X, Chen LX, Ouyang L, Cheng Y, Liu B . Plant natural compounds: targeting pathways of autophagy as anti-cancer therapeutic agents. Cell Prolif 2012; 45: 466–476.
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif 2012; 45: 487–498.
Kang R, Zeh HJ, Lotze MT, Tang D . The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18: 571–580.
Song X, Zhang X, Wang X, Zhu F, Guo C, Wang Q . Tumor suppressor gene PDCD4 negatively regulates autophagy by inhibiting the expression of autophagy-related gene ATG5. Autophagy 2013; 9: 743–755.
Napoli M, Flores ER . The family that eats together stays together: new p53 family transcriptional targets in autophagy. Genes Dev 2013; 27: 971–974.
Morselli E, Galluzzi L, Kroemer G . Mechanisms of p53-mediated mitochondrial membrane permeabilization. Cell Res 2008; 18: 708–710.
Brady CA, Attardi LD . p53 at a glance. J Cell Sci 2010; 123: 2527–2532.
Medema RH, Jäättelä M . Cytosolic FoxO1: alive and killing. Nat Cell Biol 2010; 12: 642–643.
Zhao Y, Li X, Cai MY, Ma K, Yang J, Zhou J et al. XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell Res 2013; 23: 491–507.
Frankel LB, Lund AH . MicroRNA regulation of autophagy. Carcinogenesis 2012; 33: 2018–2025.
Qased AB, Yi H, Liang N, Ma S, Qiao S, Liu X . MicroRNA-18a upregulates autophagy and ataxia telangiectasia mutated gene expression in HCT116 colon cancer cells. Mol Med Rep 2013; 7: 559–564.
Tekirdag KA, Korkmaz G, Ozturk DG, Agami R, Gozuacik D . MIR181A regulates starvation- and rapamycin-induced autophagy through targeting of ATG5. Autophagy 2013; 9: 374–385.
Xu N, Zhang J, Shen C, Luo Y, Xia L, Xue F et al. Cisplatin-induced downregulation of miR-199a-5p increases drug resistance by activating autophagy in HCC cell. Biochem Biophys Res Commun 2012; 423: 826–831.
Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem 2008; 283: 29897–29903.
Korkmaz G, le Sage C, Tekirdag KA, Agami R, Gozuacik D . miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy 2012; 8: 165–176.
Comincini S, Allavena G, Palumbo S, Morini M, Durando F, Angeletti F et al. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol Ther 2013; 14: 574–586.
Wang P, Zhang J, Zhang L, Zhu Z, Fan J, Chen L et al. MicroRNA 23b Regulates autophagy Associated with radioresistance of Pancreatic Cancer Cells. Gastroenterology 2013 doi:10.1053/j.gastro.2013.07.048.
Zou Z, Wu L, Ding H, Wang Y, Zhang Y, Chen X et al. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J Biol Chem 2012; 287: 4148–4156.
Xu Y, An Y, Wang Y, Zhang C, Zhang H, Huang C et al. miR-101 inhibits autophagy and enhances cisplatin-induced apoptosis in hepatocellular carcinoma cells. Oncol Rep 2013; 29: 2019–2024.
Frankel LB, Wen J, Lees M, Høyer-Hansen M, Farkas T, Krogh A et al. microRNA-101 is a potent inhibitor of autophagy. EMBO J 2011; 30: 4628–4641.
Kovaleva V, Mora R, Park YJ, Plass C, Chiramel AI, Bartenschlager R et al. miRNA-130a targets ATG2B and DICER1 to inhibit autophagy and trigger killing of chronic lymphocytic leukemia cells. Cancer Res 2012; 72: 1763–1772.
Mikhaylova O, Stratton Y, Hall D, Kellner E, Ehmer B, Drew AF et al. VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 2012; 21: 532–546.
Xiao J, Zhu X, He B, Zhang Y, Kang B, Wang Z et al. MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. J Biomed Sci 2011; 18: 35.
Chang Y, Yan W, He X, Zhang L, Li C, Huang H et al. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology 2012; 1: 177–187.
Benjamin D, Colombi M, Moroni C, Hall MN . Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discovery 2011; 10: 868–880.
Martin LA, André F, Campone M, Bachelot T, Jerusalem G . mTOR inhibitors in advanced breast cancer: Ready for prime time? Cancer Treat Rev 2013; 39: 742–752.
Chen T, Shen L, Yu J, Wan H, Guo A, Chen J et al. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell 2011; 10: 908–911.
Liu Q, Thoreen C, Wang J, Sabatini D, Gray NS . mTOR mediated anti-cancer drug discovery. Drug Discov Today Ther Strateg 2009; 6: 47–55.
Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG et al. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One 2009; 4: e7124.
Shimizu S, Takehara T, Hikita H, Kodama T, Tsunematsu H, Miyagi T et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer 2012; 131: 548–557.
Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 2010; 70: 288–298.
Gurpinar E, Grizzle WE, Shacka JJ, Mader BJ, Li N, Piazza NA et al. A novel sulindac derivative inhibits lung adenocarcinoma cell growth through suppression of Akt/mTOR signaling and induction of autophagy. Mol Cancer Ther 2013; 12: 663–674.
Evangelisti C, Ricci F, Tazzari P, Chiarini F, Battistelli M, Falcieri E et al. Preclinical testing of the Akt inhibitor triciribine in T-cell acute lymphoblastic leukemia. J Cell Physiol 2011; 226: 822–831.
Bommareddy A, Hahm ER, Xiao D, Powolny AA, Fisher AL, Jiang Y et al. ATG5 regulates phenethyl isothiocyanate-induced autophagic and apoptotic cell death in human prostate cancer cells. Cancer Res 2009; 69: 3704–3712.
Vara D, Morell C, Rodríguez-Henche N, Diaz-Laviada I . Involvement of PPARγ in the antitumoral action of cannabinoids on hepatocellular carcinoma. Cell Death Dis 2013; 4: e618.
Brüning A, Rahmeh M, Friese K . Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol Oncol 2013 pii S1574-7891: 00107–5.
Fu L, Kim YA, Wang X, Wu X, Yue P, Lonial S et al. Perifosine inhibits mammalian target of rapamycin signaling through facilitating degradation of major components in the mTOR axis and induces autophagy. Cancer Res 2009; 69: 8967–8976.
Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM et al. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther 2009; 8: 2204–2210.
Fan QW, Weiss WA . Inhibition of PI3K-Akt-mTOR signaling in glioblastoma by mTORC1/2 inhibitors. Methods Mol Biol 2012; 821: 349–359.
Saiki S, Sasazawa Y, Imamichi Y, Kawajiri S, Fujimaki T, Tanida I et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 2011; 7: 176–187.
Lin CJ, Lee CC, Shih YL, Lin TY, Wang SH, Lin YF et al. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic Biol Med 2012; 52: 377–391.
Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R . Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ 2008; 15: 1318–1329.
Laane E, Tamm KP, Buentke E, Ito K, Kharaziha P, Oscarsson J et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ 2009; 16: 1018–1029.
Chen M, Du Y, Qui M, Wang M, Chen K, Huang Z et al. Ophiopogonin B-induced autophagy in non-small cell lung cancer cells via inhibition of the PI3K/Akt signaling pathway. Oncol Rep 2013; 29: 430–436.
El-Metnawy WH, Mattar MM, El-Nahass YH, Samra MA, Abdelhamid HM, Abdlfattah RM et al. Predictive value of pretreatment BCR-ABL(IS) transcript level on response to imatinib therapy in Egyptian patients with Chronic Phase Chronic Myeloid Leukemia (CPCML). Int J Biomed Sci 2013; 9: 48–53.
Schafranek L, Leclercq TM, White DL, Hughes TP . Clarithromycin enhances dasatinib-induced cell death in chronic myeloid leukemia cells, by inhibition of late stage autophagy. Leuk Lymphoma 2013; 54: 198–201.
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One 2011; 6: e18691.
Huang HL, Chen YC, Huang YC, Yang KC, Hy Pan, Shih SP et al. Lapatinib induces autophagy, apoptosis and megakaryocytic differentiation in chronic myelogenous leukemia K562 cells. PLoS One 2011; 6: e29014.
John S, Nayvelt I, Hsu HC, Yang P, Liu W, Das GM et al. Regulation of estrogenic effects by beclin 1 in breast cancer cells. Cancer Res 2008; 68: 7855–7863.
Malik SA, Orhon I, Morselli E, Criollo A, Shen S, Mariño G et al. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene 2011; 30: 918–929.
Høyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jäättelä M . Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ 2005; 12: 1297–1309.
Wang H, Yu J, Li Y, Hao Y, Choi A, Ke H et al. Beclin 1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011; 147: 223–234.
Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L et al. The inositol 1,4,5- trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 2009; 16: 1006–1017.
Crazzolara R, Bradstock KF, Bendall LJ . RAD001 (Everolimus) induces autophagy in acute lymphoblastic leukemia. Autophagy 2009; 5: 727–728.
Xia YH, Cheng CR, Yao SY, Zhang Q, Wang Y, Ji ZN . L-securinine induced the human colon cancer SW480 cell autophagy and its molecular mechanism. Fitoterapia 2011; 82: 1258–1264.
Wu Y, Wang X, Guo H, Zhang B, Zhang XB, Shi ZJ et al. Synthesis and screening of 3-MA derivatives for autophagy inhibitors. Autophagy 2013; 9: 595–603.
Baek KH, Park J, Shin I . Autophagy-regulating small molecules and their therapeutic applications. Chem Soc Rev 2012; 41: 3245–3263.
Janku F, McConkey DJ, Hong DS, Kurzrock R . Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8: 528–539.
Hayashi S, Yamamoto A, You F, Yamashita K, Ikegame Y, Tawada M et al. The stent-eluting drugs sirolimus and paclitaxel suppress healing of the endothelium by induction of autophagy. Am J Pathol 2009; 175: 2226–2234.
Choudhury S, Kolukula VK, Preet A, Albanese C, Avantaggiati ML . Dissecting the pathways that destabilize mutant p53: the proteasome or autophagy? Cell Cycle 2013; 12: 1022–1029.
Mohammadpour R, Safarian S, Sheibani N, Norouzi S, Razazan A . Death inducing and cytoprotective autophagy in T-47D cells by two common antibacterial drugs: sulphathiazole and sulphacetamide. Cell Biol Int 2013; 24: 348–358.
Von Clausewitz C Vom Kriege (On War). Dümmlers Verlag, Berlin 1832.
Acknowledgements
We would like to dedicate this review to Professor Xinsheng Yao for celebrating his 80th birthday on 24 October 2013. This work was supported in part by grants from the National 973 Basic Research Program of China (Nos. 2013CB911300 and 2010CB529900), Department of Defense of USA (No. BC103654), Key Projects of the National Science and Technology Pillar Program (No. 2011BAZ02590) and National Natural Science Foundation of China (No. 81260628 and No. 81202403).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by GM Fimia
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Liu, B., Wen, X. & Cheng, Y. Survival or death: disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis 4, e892 (2013). https://doi.org/10.1038/cddis.2013.422
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.422
Keywords
This article is cited by
-
Leucine-rich repeat and sterile alpha motif containing 1 promotes the oncogenic growth of human hepatocellular carcinoma cells
Cancer Cell International (2019)
-
The interaction between SPARC and GRP78 interferes with ER stress signaling and potentiates apoptosis via PERK/eIF2α and IRE1α/XBP-1 in colorectal cancer
Cell Death & Disease (2019)
-
Glutathione S-transferases P1 protects breast cancer cell from adriamycin-induced cell death through promoting autophagy
Cell Death & Differentiation (2019)
-
Nitazoxanide, an antiprotozoal drug, inhibits late-stage autophagy and promotes ING1-induced cell cycle arrest in glioblastoma
Cell Death & Disease (2018)
-
Toxoplasma gondii GRA8 induces ATP5A1–SIRT3-mediated mitochondrial metabolic resuscitation: a potential therapy for sepsis
Experimental & Molecular Medicine (2018)
