
Abstract
Microtubule inhibiting agents (MIAs) characteristically induce phosphorylation of the major anti-apoptotic Bcl-2 family members Mcl-1, Bcl-2 and Bcl-xL, and although this leads to Mcl-1 degradation, the role of Bcl-2/Bcl-xL phosphorylation in mitotic death has remained controversial. This is in part due to variation in MIA sensitivity among cancer cell lines, the dependency of cell fate on drug concentration and uncertainty about the modes of cell death occurring, thus making comparisons of published reports difficult. To circumvent problems associated with MIAs, we used siRNA knockdown of the anaphase-promoting complex activator, Cdc20, as a defined molecular system to investigate the role, specifically in mitotic death, of individual anti-apoptotic Bcl-2 proteins and their phosphorylated forms. We show that Cdc20 knockdown in HeLa cells induces mitotic arrest and subsequent mitotic death. Knockdown of Cdc20 in HeLa cells stably overexpressing untagged wild-type Bcl-2, Bcl-xL or Mcl-1 promoted phosphorylation of the overexpressed proteins in parallel with their endogenous counterparts. Overexpression of Bcl-2 or Bcl-xL blocked mitotic death induced by Cdc20 knockdown; phospho-defective mutants were more protective than wild-type proteins, and phospho-mimic Bcl-xL was unable to block mitotic death. Overexpressed Mcl-1 failed to protect from Cdc20 siRNA-mediated death, as the overexpressed protein was susceptible to degradation similar to endogenous Mcl-1. These results provide compelling evidence that phosphorylation of anti-apoptotic Bcl-2 proteins has a critical role in regulation of mitotic death. These findings make an important contribution toward our understanding of the molecular mechanisms of action of MIAs, which is critical for their rational use clinically.
Similar content being viewed by others


Main
Microtubule inhibiting agents (MIAs) act by suppressing the dynamic instability of microtubules, and cells treated with MIAs do not proceed through mitosis because of sustained activation of the spindle checkpoint leading to prolonged mitotic arrest and subsequent apoptosis.1, 2 Despite the widespread use of MIAs in cancer therapy and the development of new antimitotic agents,3 the molecular mechanisms linking mitotic arrest and apoptosis are not clearly established. Elucidation of these apoptotic pathways is pivotal in understanding the molecular basis of sensitivity and resistance to these agents, and for their informed clinical use.3 Although MIAs promote a wide diversity of signaling events, one of the most widespread and common characteristics is the phosphorylation of anti-apoptotic Bcl-2 protein family members. Thus Bcl-2, Bcl-xL and Mcl-1 have all been shown to undergo phosphorylation in a wide variety of cell types in response to these agents.4, 5, 6, 7, 8, 9, 10, 11, 12, 13 Extensive phosphorylation of Mcl-1 by cyclin-dependent kinase 1 (Cdk1) and other kinases during mitotic arrest results in Mcl-1 degradation5, 6, 7 and release of bound pro-apoptotic Bcl-2 proteins such as Bak.7 Phosphorylation of Bcl-2 and Bcl-xL during mitotic arrest, on the other hand, does not affect protein expression levels8 but does appear to weaken interactions with pro-apoptotic Bcl-2 proteins such as Bax.9, 10 Recent evidence suggests that Cdk1 is the primary kinase responsible,11 although other kinases such as JNK have also been implicated.12, 13
Data suggesting that Bcl-2/Bcl-xL/Mcl-1 phosphorylation during mitotic arrest disables their anti-apoptotic function has been based largely on the use of mutants lacking major phospho-acceptor sites expressed in cells subsequently treated with MIAs.5, 6, 10, 12, 13 However, many of these studies have been conducted using tagged proteins, and it is well established that tags can interfere with protein function including that of Bcl-2 proteins.14, 15, 16 In addition, different cell types differ markedly in their sensitivity to MIAs, and the fate of cells treated with MIAs is highly dependent on drug concentration. For example, in breast cancer cells lines, low concentrations of docetaxel induced aberrant mitosis and subsequent necrosis, whereas higher concentrations induced mitotic arrest and apoptosis.17 We have found that DLD-1 colon carcinoma cells undergo largely mitotic slippage and die in interphase at low concentrations of paclitaxel, but tend to die in mitosis at higher concentrations.18 Furthermore, MIAs also target interphase microtubules,19 and the mechanisms of cell death during interphase are likely very different from those occurring during mitotic arrest where transcription and translation are repressed. Indeed, high levels of phosphorylation of anti-apoptotic Bcl-2 proteins are only observed during mitotic arrest and likely have a key role in mitotic death but not interphase death. Overall, the variation in drug sensitivity among cell lines, the dependency of cell fate on drug concentration and the lack of data on the modes of cell death occurring make it difficult to compare results from independent studies examining Bcl-2 protein phospho-mutants or to draw firm conclusions regarding the role of Bcl-2 protein phosphorylation. Finally, studies examining the role of anti-apoptotic Bcl-2 protein phosphorylation have typically focused on a single member of the family, and recent observations indicate that they can compensate for each other’s function. For example, cells that show resistance to Bcl-2 inhibitors express higher levels of Mcl-1,20, 21 and conversely, resistance to Mcl-1 inhibitors has been shown to be due to high levels of Bcl-xL expression.22
Based on these considerations, and in order to circumvent problems associated with the use of MIAs, we sought a defined molecular approach to induce mitotic arrest and mitotic death, where anti-apoptotic Bcl-2 protein phosphorylation is most relevant functionally. It is well established that the spindle assembly checkpoint prevents cells from entering anaphase by inhibiting activation of the anaphase-promoting complex (APC/C) through sequestration of its co-activator, Cdc20.23 Thus, knockdown of Cdc20 should prevent anaphase-promoting complex (APC/C) activation and induce mitotic arrest, and this was tested and verified in a recent study in which several cancer cell lines, including those resistant to MIAs, were shown to undergo mitotic arrest and apoptotic death after knockdown of Cdc20.24 In this study, we applied this approach to HeLa cells and then systematically examined the role of individual anti-apoptotic Bcl-2 proteins and their phospho-mutant forms, using untagged full-length proteins that closely mimicked the properties of their endogenous counterparts. The results show that untagged wild-type Bcl-2, Bcl-xL or Mcl-1 individually blocked mitotic death induced by Cdc20 knockdown, that phospho-defective Bcl-2 or Bcl-xL were more effective than wild-type proteins and that phospho-mimic Bcl-xL was unable to block mitotic death. These results provide conclusive evidence that phosphorylation of anti-apoptotic Bcl-2 proteins has a key role in regulation of mitotic death.
Results
Knockdown of Cdc20 leads to mitotic arrest
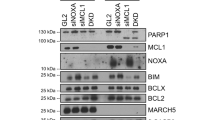
We first tested knockdown of Cdc20 as a molecular approach to induce mitotic arrest. Knockdown was performed in HeLa cells using siRNA specific to duplex 1 of Cdc20. Because mitotically arrested cells typically roundup and become less adherent, cells affected by siRNA treatment were enriched by gently shaking the plate to dislodge loosely adherent cells, which were pooled and analyzed separately from the remaining adherent cells. Non-adherent cells showed extensive knockdown of Cdc20, to below detectable levels, when compared with the adherent population that retained detectable Cdc20 expression (Figure 1a, lanes 4 and 5). Loss of Cdc20 was accompanied by increased levels of cyclin B1 and phospho-histone H3, which is similar to that observed after treatment with the microtubule inhibitor vinblastine (Figure 1a, lane 2), and control non-coding siRNA was without effect. Propidium iodide staining and flow cytometry indicated a significant increase in the percentage of cells with 4N DNA content after Cdc20 siRNA treatment, up to 75% of the population at 48 h, compared with control transfected cells (Figure 1b). These results taken together demonstrate that Cdc20 knockdown leads to mitotic arrest in HeLa cells.

Knockdown of Cdc20 cells leads to mitotic arrest, phosphorylation of Bcl-2/Bcl-xL, phosphorylation and degradation of Mcl-1, and Cdk1 activation. HeLa cells were either untreated (lane 1), treated with 30 nM vinblastine for 24 h (lane 2), transfected with negative control siRNA (lane 3) or siRNA targeting Cdc20 for 48 h (lanes 4 and 5). After Cdc20 siRNA transfection, remaining adherent (lane 4) or non-adherent (lane 5) cells were isolated separately by mitotic shake-off, as described in Materials and Methods. (a) Whole-cell extracts were prepared and immunoblotted for the indicated proteins. P-Histone 3 was detected using a phospho-specific antibody. Phosphorylation (P) of Bcl-2 or Bcl-xL was evidenced by mobility shift as indicated. GAPDH was used as a loading control. (b) Cells were subjected to propidium iodide staining and FACS analysis to determine the proportion of cells with 4N DNA content. The non-adherent population of cells was used after Cdc20 knockdown. Results given are mean±S.D. (n=3). (c) Cell extracts were prepared 48 h post transfection with control or Cdc20 siRNA, the latter prepared from the non-adherent cell population, and subjected to Cdk1 assay with H1 histone as substrate, as described in Materials and Methods. Values shown have been corrected for background kinase activity (no substrate) and represent mean±S.D. (n=3)
Knockdown of Cdc20 induces anti-apoptotic Bcl-2 phosphorylation and Cdk1 activation
Analysis of Bcl-2, Bcl-xL and Mcl-1 indicated that knockdown of Cdc20 resulted in phosphorylation of Bcl-2 and Bcl-xL as evidenced by robust mobility shifts observed in the immunoblots (Figure 1a). In addition, Cdc20 knockdown resulted in loss of expression of Mcl-1, which has previously been shown to occur during mitotic arrest by phosphorylation-triggered ubiqitination and subsequent degradation.5, 6, 7 Importantly, the phosphorylation of Bcl-2/Bcl-xL and degradation of Mcl-1 observed in response to Cdc20 knockdown mirrored those induced by vinblastine (Figure 1a, compare lanes 2 and 5). Because Cdk1/cyclin B1 has been implicated as a primary kinase involved in phosphorylation of Bcl-2/Bcl-xL/Mcl-1,5, 6, 7, 11 Cdk1 kinase assays were conducted, as described in Materials and Methods. Histone H1 was used as the substrate, as we have previously established that the major H1 kinase activity in mitotic extracts of HeLa cells is due to Cdk1/cyclin B1.11 Cdk1 activity was significantly increased at 48 h in cells that received siRNA to Cdc20 when compared with negative control siRNA (Figure 1c). We have previously reported that mitotic death induced by paclitaxel is strongly associated with elevated Cdk1 activity and Cdk1-mediated phosphorylation of Bcl-2/Bcl-xL and phosphorylation/degradation of Mcl-1.18 The results shown in Figure 1 confirm that Cdc20 knockdown induces the characteristic mitotic death signature associated with MIAs, and as such represents a robust molecular approach valuable in the analysis of Cdk1 pro-apoptotic signaling.
Knockdown of Cdc20 induces apoptosis
Our previous studies showed that when HeLa cells are treated with MIAs, they undergo prolonged mitotic arrest and subsequently die because of apoptosis.8 To determine whether knockdown of Cdc20 induced apoptosis with similar kinetics, a cell death ELISA assay was performed that quantitatively measures DNA fragmentation, as described in Materials and Methods. Although at 24 h, only low levels of apoptosis were observed under each condition, at 48 and 72 h post transfection, siRNA to Cdc20 strongly induced apoptosis when compared with control siRNA (Figure 2). These results show that apoptosis induced by Cdc20 knockdown initiates only after prolonged mitotic arrest, with kinetics similar to that observed with MIAs, and further validate the use of Cdc20 knockdown as a molecular mimic for MIA action.

Knockdown of Cdc20 leads to apoptosis following prolonged mitotic arrest. HeLa cells were transfected with siRNA to Cdc20 or negative control siRNA for 24, 48 or 72 h. Cell extracts were prepared and subjected to an apoptotic cell death ELISA, as described in Materials and Methods. The non-adherent population of cells was used after Cdc20 knockdown. Results given are mean±S.D. (n=3)
Tagged Bcl-2 proteins behave aberrantly
In order to test the hypothesis that phosphorylation of Bcl-2 proteins is a key step in initiating apoptosis following mitotic arrest, HeLa cells were initially transiently co-transfected with Cdc20 siRNA and plasmids encoding full-length wild-type HA-tagged Bcl-2 or HA-tagged Bcl-xL. Cells were also co-transfected with Cdc20 siRNA and mutant plasmids encoding untagged AAA-Bcl-2 (with mutations T69A, S70A and S87A) or HA-tagged S62A-Bcl-xL, both of which are predicted to be phospho-defective based on identified sites of phosphorylation.10, 12 Analysis by immunoblotting revealed, however, that neither wild-type HA-Bcl-2 nor HA-Bcl-xL underwent a phosphorylation-dependent mobility shift in cells co-transfected with Cdc20 siRNA (Figure 3a). Indeed, although AAA-Bcl-2 failed to shift in response to Cdc20 siRNA treatment as expected, wild-type HA-Bcl-2 failed to shift also (note that AAA-Bcl-2 migrates faster than HA-Bcl-2 due to the lack of a tag). Similarly, the gel migration of wild-type HA-Bcl-xL was identical without or with Cdc20 siRNA treatment, and identical to that of S62A-HA-Bcl-xL (Figure 3a). To further analyze the phosphorylation status of the tagged proteins during mitotic arrest, cells were transiently transfected with plasmids encoding wild-type HA-Bcl-xL, S62A-HA-Bcl-xL, wild-type HA-Bcl-2 or AAA-Bcl-2, and then treated with vinblastine. We again observed a lack of mobility shift in wild-type HA-Bcl-xL (Figure 3b, lanes 1 and 2) or wild-type HA-Bcl-2 (Figure 3b, lanes 5 and 6) in response to vinblastine. However, in cells overexpressing HA-Bcl-2, vinblastine treatment clearly caused a mobility shift in endogenous Bcl-xL (Figure 3b, lanes 5 and 6). Likewise, in cells overexpressing HA-Bcl-xL, vinblastine-induced phosphorylation of endogenous Bcl-2; this is shown in Figure 3b, right panel, where lanes 1 and 2 have been subjected to overexposure to visualize endogenous Bcl-2. Vinblastine also failed to induce phosphorylation of HA-Bcl-xL when stably expressed (data not shown). Thus, conditions that induced phosphorylation of the endogenous proteins failed to promote phosphorylation of the exogenously expressed protein. To better understand the basis of these observations, the subcellular locations of stably overexpressed HA-Bcl-xL and stably overexpressed untagged Bcl-xL were compared with that of the endogenous protein using immunofluorescence microscopy (Figure 3c). In the upper panels of Figure 3c, it is evident that the green signal of endogenous Bcl-xL merged only in part with the red mitochondrial signal, and that a greater proportion of Bcl-xL was extra-mitochondrial, most likely cytosolic. Overexpressed untagged Bcl-xL exhibited an essentially identical pattern of distribution (Figure 3c, middle panels). However, HA-tagged Bcl-xL differed markedly, being localized almost exclusively to the mitochondria with little evidence of cytosolic expression (Figure 3c, lower panels). Taken together, these results suggested that tagged Bcl-2 proteins may exhibit aberrant behavior due to mislocalization.

HA-tagged Bcl-2/Bcl-xL do not undergo phosphorylation following mitotic arrest. (a) HeLa cells were co-transfected with siRNA targeting Cdc20 or negative control siRNA and plasmids encoding either wild-type (Wt) HA-Bcl-xL, HA-S62A-Bcl-xL, Wt HA-Bcl-2 or AAA-Bcl-2. Cells were harvested 48 h post transfection, and whole-cell extracts were immunoblotted for Bcl-2 or Bcl-xL. The non-adherent population of cells was used after Cdc20 knockdown. (b) HeLa cells were transfected with plasmids encoding either Wt HA-Bcl-2, Wt HA-Bcl-xL, HA-S62A-Bcl-xL, AAA-Bcl-2 or empty vector, followed by treatment with 30 nM vinblastine for 24 h. Whole-cell extracts were immunoblotted for Bcl-2 or Bcl-xL and GAPDH was used as a loading control. The right panel shows an overexposed blot of lanes 1 and 2 detecting endogenous Bcl-2. (c) Aberrant subcellular localization of HA-Bcl-xL. Control HeLa cells (endogenous) or cells stably overexpressing untagged Bcl-xL or HA-Bcl-xL were grown on glass coverslips, fixed, permeabilized and probed for expression of Bcl-xL using a Bcl-xL specific primary antibody and a FITC-conjugated secondary antibody (green). Mitotracker Red was used to visualize the mitochondria (red), and the right panel shows the merged signals
Untagged Bcl-2 proteins undergo phosphorylation like their endogenous counterparts
Based on the results discussed above, HeLa cells stably overexpressing untagged Bcl-2, Bcl-xL or Mcl-1 were prepared, and then transfected with control or Cdc20 siRNA. As shown in Figure 4a, wild-type overexpressed Bcl-2 underwent phosphorylation in response to Cdc20 knockdown, with several bands evident decremented in gel mobility (lane 4), much like the endogenous protein (Figure 1a, and note that the light exposure conditions needed to visualize overexpressed Bcl-2 in Figure 4a precluded detection of the endogenous protein). In contrast, AAA-Bcl-2 failed to undergo phosphorylation (lane 6). Similarly, wild-type overexpressed Bcl-xL underwent phosphorylation in response to Cdc20 knockdown, although it was incomplete (Figure 4b, upper blot, lane 4), whereas S62A-Bcl-xL failed to undergo a mobility shift (lane 6). To confirm these results, a phospho-specific antibody for Bcl-xL that recognizes the Serine-62 phosphorylation site was used. Phospho-Bcl-xL was robustly expressed in response to Cdc20 knockdown in cells overexpressing wild-type Bcl-xL, and only a minimal level of phospho-Bcl-xL was detected in the S62A mutant that can be attributed to the endogenous protein (Figure 4b, middle blots). Finally, as shown in Figure 5c, endogenous Mcl-1 underwent phosphorylation and degradation in response to Cdc20 knockdown, as already illustrated in Figure 1a. In cells stably overexpressing wild-type untagged Mcl-1, the exogenous protein exhibited a similar fate (Figure 4c). To more closely examine the kinetics of Mcl-1 degradation in relation to initiation of apoptosis following mitotic arrest, control cells and cells stably overexpressing Mcl-1 were synchronized using a double thymidine block followed by treatment with the microtubule inhibitor vinblastine. Cells were harvested 10–36 h post release and Mcl-1 expression was examined together with Poly(ADP-ribose) polymerase (PARP) cleavage as an indicator of caspase activation and initiation of apoptosis. The kinetics of degradation of Mcl-1 in cells treated with vinblastine was similar for both the endogenous and overexpressed protein, though the latter remained detectable at later time points (Figure 4d). PARP cleavage was delayed by about 4 h in cells overexpressing Mcl-1 in concert with the persistence of Mcl-1 expression (Figure 4d). Although numerous sites of phosphorylation have been identified in Mcl-1 during mitotic arrest,5, 6 there exists many additional sites that remain to be identified,25 thus, a phosphorylation/degradation-resistant mutant of Mcl-1 is not currently available.

Untagged anti-apoptotic Bcl-2 proteins phosphorylate normally in response to Cdc20 knockdown. HeLa cells stably overexpressing Bcl-2, AAA-Bcl-2, Bcl-xL, SA62-Bcl-xL or Mcl-1 were transfected with siRNA targeting Cdc20 or negative control siRNA. Non-adherent cells were collected and harvested 48 h post transfection, and cell extracts prepared and immunoblotted for Bcl-2 (a), Bcl-xL (b) or Mcl-1 (c). Phosphorylation was detected by mobility shift (P, phosphorylated) for Bcl-2 in a and Bcl-xL in b; phospho-Bcl-xL was also detected in b using a Ser62-phospho-specific antibody. GAPDH was used as a loading control. (d) Degradation kinetics of Mcl-1 in response to vinblastine. Control HeLa cells or cells stably overexpressing Mcl-1 were synchronized using a double thymidine block, treated with vinblastine (30 nM) 1 h post release and harvested at the indicated times intervals post release. Cell extracts were subjected to immunoblot analysis for Mcl-1 and PARP, with intact PARP and its cleavage product indicated. GAPDH was used as a loading control

Expression of anti-apoptotic Bcl-2 proteins or their phospho-defective mutants confers resistance to cell death induced by Cdc20 knockdown. HeLa cells stably expressing plasmids encoding Bcl-2, AAA-Bcl-2, Bcl-xL, SA62-Bcl-xL or Mcl-1 were transfected with siRNA targeting Cdc20 or negative control siRNA. (a) Non-adherent cells were collected 48 h post transfection, and cell death was measured by the percentage of cells that uptake propidium iodide. Results are given are mean±S.D. (n=3). (b) Non-adherent cells were collected and harvested 48 h post transfection. Caspase-3 activity was measured in whole-cell extracts as described in Materials and Methods (mean±S.D., n=3). Significantly different from HeLa: *P-value <0.05; ***P-value <0.001
Phosphorylation negatively regulates the ability of anti-apoptotic Bcl-2 proteins to confer resistance to mitotic death
We next tested the hypothesis that anti-apoptotic Bcl-2 proteins are key substrates for pro-death Cdk1 signaling, and that phosphorylation is a key event in their inactivation, by examining cell death induced by Cdc20 knockdown and vinblastine in cells stably expressing untagged wild-type or phospho-defective mutants. Cell death was first analyzed by propidium iodide permeability as an indicator of outer membrane integrity. Cells overexpressing Bcl-2 or Bcl-xL showed significant protection from Cdc20 siRNA-mediated death, and importantly, cells expressing phospho-defective AAA-Bcl-2 or SA62-Bcl-xL provided greater protection (Figure 5a). To further test the role of phosphorylation, Cdc20 was knocked down in cells stably overexpressing phospho-mimic S62D-Bcl-xL, which would not be predicted to protect cells from death due to its inability to bind and sequester pro-apoptotic Bax.10 Consistent with phosphorylation acting to disable anti-apoptotic function, phospho-mimic S62D-Bcl-xL was ineffective (Figure 5a). Overexpression of Mcl-1 failed to protect against Cdc20 siRNA-mediated death (Figure 5a), and this was not unexpected, as the overexpressed protein is subject to degradation (Figures 4c and d). Caspase-3 activity was next assessed as an independent and more specific indicator of apoptotic death. Upon knockdown of Cdc20, HeLa cells showed a fivefold increase in caspase-3 activity compared with cells transfected with control siRNA (Figure 5b). When Cdc20 was knocked down in cells stably expressing wild-type Bcl-2 or Bcl-xL, a decrease in caspase-3 activation was observed relative to HeLa cells. AAA-Bcl-2 showed greater protection than wild-type Bcl-2, consistent with phosphorylation acting to disable Bcl-2 function. However, both wild-type Bcl-xL and SA62-Bcl-xL completely protected the cells from Cdc20 siRNA-mediated caspase-3 activation (Figure 5b). The apparent absence of a difference between wild-type and phospho-defective Bcl-xL could be the result of incomplete phosphorylation of wild-type Bcl-xL under these conditions (Figure 4b). To test this directly, Cdc20 was knocked down in cells stably overexpressing phospho-mimic S62D-Bcl-xL, and much of the protection afforded by Bcl-xL or S62A-Bcl-xL was reversed (Figure 5b). Cells overexpressing Mcl-1 showed a significant but only modest decrease of caspase-3 activation (Figure 5b), which may reflect partial protection resulting from residual Mcl-1 expression (Figure 4d).
To further demonstrate the importance of Bcl-2 protein phosphorylation in regulation of mitotic death, cells stably expressing wild-type or phospho-mutant proteins were treated with vinblastine, and caspase-3 activity was assessed. The data are presented in Figure 6, and it is evident that the same general trends apply, with the phospho-defective Bcl-2/Bcl-xL mutants affording greater protection than their wild-type counterparts, phospho-mimic Bcl-xL being relatively ineffective and Mcl-1 being modestly protective at 24 h but unable to sustain protection at 48 h, consistent with its degradation kinetics (Figure 4d).

Expression of anti-apoptotic Bcl-2 family proteins or their phospho-defective mutants inhibits caspase-3 activation in response to vinblastine. Control HeLa cells or cells stably overexpressing the indicated Bcl-2 family protein were untreated or treated with 30 nM vinblastine for 24 or 48 h, and cell extracts were subjected to caspase-3 assay, as described in Materials and Methods. Results given are mean±S.D. (n=3)
To ensure that the results obtained reflected the properties of the expressed proteins and not the characteristics of the specific stable cell line clones chosen, additional clones were examined (Supplementary Figure S1). Expression levels of the overexpressed Bcl-2 proteins in representative clones are shown in Supplementary Figure S1A. The clones indicated were selected for further study and subjected to a cell death assay after vinblastine treatment (Supplementary Figure S1B). Although some variation between clones of the same type was observed, for example, the three overexpressing Bcl-2, different clones of each type behaved similarly, and the same overall trends were found as for the single clones examined earlier. Thus, clonal cell lines expressing wild-type Bcl-2 or Bcl-xL protected against vinblastine-induced cell death, those expressing phospho-defective Bcl-2/Bcl-xL afforded greater protection than their wild-type counterparts, those expressing phospho-mimic Bcl-xL were consistently less protective and clones expressing Mcl-1 were consistently non-protective.
Discussion
We show here that Cdc20 knockdown induced mitotic arrest and mitotic death in HeLa cells, and that overexpression of untagged wild-type Bcl-2 or Bcl-xL blocked mitotic death induced by Cdc20 knockdown. A key role for phosphorylation was demonstrated by showing that phospho-defective Bcl-2 or Bcl-xL were more effective than wild-type proteins, and that phospho-mimic Bcl-xL was much less effective in blocking mitotic death. Importantly, the same trends were observed for two different inducers of mitotic death (Cdc20 knockdown and treatment with vinblastine), for two independent cell death assays and for several different clonal cell lines of each type. Previous studies have led to conflicting reports, with phosphorylation of Bcl-2, for example, being described as disabling Bcl-2 anti-apoptotic activity on the one hand12 or enhancing Bcl-2 anti-apoptotic activity on the other hand.26 Our data are highly consistent with phosphorylation acting to antagonize anti-apoptotic Bcl-2 protein function and thus serve to clarify this uncertainty. A recent study indicated that a key role of Bcl-xL is to retrotranslocate Bax from the mitochondria to the cytosol.27 The observation that Bcl-xL localizes to both the mitochondria and cytosol (Figure 3c) is consistent with such a role, and it is tempting to speculate that because phosphorylation disrupts Bcl-xL:Bax interaction, as we showed previously,10 it would also reduce retrotranslocation and allow Bax to accumulate at the mitochondria. Consistent with this fact, we previously reported that vinblastine promotes translocation of Bax from the cytosol to the mitochondria.28 Further work is needed to determine whether phosphorylation alters the binding affinity of Bcl-2 or Bcl-xL to pro-apoptotic Bcl-2 proteins such as Bax, or affects the rate of Bcl-xL-mediated Bax retrotranslocation.
Data from several other lines of investigation support the conclusion that phosphorylation inactivates anti-apoptotic Bcl-2 proteins during mitotic arrest and as such is a key regulatory feature of mitotic death. For example, DLD-1 colon cancer cells, which have a relatively weak spindle checkpoint and are slippage-prone, only transiently phosphorylate Bcl-xL/Mcl-1 in response to Taxol, and after slippage, survive or die in subsequent interphase.18 Moreover, if slippage-resistant HT29 colon cancer cells, which normally die in mitosis when treated with Taxol, are exposed to Cdk1 inhibitors during mitotic arrest, they survive concomitantly with reversal of Bcl-xL/Mcl-1 phosphorylation.18 In addition to the conclusion derived from the use of phospho-mutants described here, other evidence supports the idea that phosphorylation disables anti-apoptotic activity. For example, if Bcl-2 or Bcl-xL are knocked down or cells are treated with the Bcl-2/Bcl-xL inhibitor ABT-263, the level of cell death induced by Cdc20 knockdown is unaltered (unpublished observations). As knockdown or chemical inhibition was without additional effect, this suggests that Bcl-2/Bcl-xL phosphorylation induced by Cdc20 knockdown strongly inhibits their anti-apoptotic activity. It is noteworthy that overexpression of wild-type Bcl-2 or Bcl-xL, despite retaining the relevant sites for phosphorylation, blocked to some degree Cdc20-mediated death (Figures 5 and 6), and this may seem counter to the hypothesis that phosphorylation completely disables anti-apoptotic activity. However, exogenously overexpressed proteins may overwhelm the phosphorylation machinery, and phosphorylation may be incomplete, as was most clearly observed for overexpressed Bcl-xL (Figure 4b).
Overexpression of Mcl-1 afforded only minor protection against mitotic death (Figures 5, 6 and Supplementary Figure S1), likely because the overexpressed protein was degraded with kinetics similar to, though slightly delayed from, endogenous Mcl-1 (Figure 4d). Thus, the processing machinery responsible for endogenous Mcl-1 degradation appears sufficiently active to handle excess protein. Numerous sites of phosphorylation have been identified in Mcl-1,29 and major sites of mitotic phosphorylation have been reported as Ser6430 and Thr92.5 However, we have found that a Mcl-1 mutant with alanine replacements of five putative major sites (Ser64, Thr92, Ser121, Ser159 and Thr163)29 undergoes phosphorylation and degradation in HeLa cells treated with vinblastine, and 2D gel analysis of wild-type protein reveals up to eight distinct phosphorylated Mcl-1 species.25 Similarly, paclitaxel induced a complex pattern of Mcl-1 phosphorylation in Burkitt lymphoma cells, as determined by thermolysin digestion and 2D-phosphopeptide mapping.31 Thus, there appears to be many additional phosphorylation sites in Mcl-1 associated with mitotic arrest yet to be identified, and a phosphorylation/degradation-resistant Mcl-1 mutant remains to be described.
Different cells lines depend on different members of the anti-apototic Bcl-2 subfamily for survival. Many leukemia cell lines are Bcl-2 dependent, for example,32 and we have found that HeLa cells are strictly dependent on Mcl-1 (JM Eichhorn et al., manuscript in preparation). Despite this dependency, different anti-apoptotic Bcl-2 proteins expressed in a cell have overlapping and compensatory functions. This is best illustrated by studies showing that sensitivity to Bcl-2 or Mcl-1 inhibitors is dependent to a large degree on the relative expression level of the non-targeted protein. For example, high levels of Mcl-1 confers resistance to the Bcl-2/Bcl-xL inhibitor ABT-263;20, 21 sensitivity to Bcl-2/Bcl-xL inhibitors can be restored under conditions in which Mcl-1 expression is downregulated;33, 34 and cells that exhibit resistance to Mcl-1 inhibitors show high levels of Bcl-xL expression.22 These compensatory mechanisms likely underlie the observation that, in response to inducers of mitotic arrest, Bcl-2, Bcl-xL and Mcl-1 become phosphorylated simultaneously in a highly coordinated fashion.7, 8, 11 In untreated HeLa cells, one of the functions of the major survival protein Mcl-1 is to sequester Bak, and during mitotic arrest, the phosphorylation and degradation of Mcl-1 release bound Bak.7 Released Bak could potentially be sequestered by Bcl-2 or Bcl-xL if they remained unphosphorylated, but by coordinating Bcl-2 and Bcl-xL phosphorylation with Mcl-1 phosphorylation, the capacity to sequester freed Bak through its interaction with Bcl-2 or Bcl-xL is restricted. In Mcl-1-dependent HeLa cells, the actual loss of Mcl-1 expression by phosphorylation/degradation is coordinated with the functional loss of Bcl-2 and Bcl-xL by phosphorylative modification, severely diminishing anti-apoptotic capacity and promoting apoptosis. As mitotic death is uniquely mediated by elimination of anti-apoptotic Bcl-2 protein function,18 the existence of these compensatory mechanisms makes it imperative to coordinate the inactivation of the dominant family member with inactivation of the other members, and this is achieved through a conserved pathway leading to their inactivation by phosphorylation.
Materials and Methods
Materials
Antibodies against Cdc20 (4823), Bcl-xL (2762), P-Histone 3 (9701) and GAPDH (2118) were purchased from Cell Signaling (Beverly, MA, USA); antibodies against Cyclin B1 (sc-245), Bcl-2 (sc-509) and Mcl-1 (M22) were purchased from Santa Cruz (Santa Cruz, CA, USA); antibody for PARP was purchased from BD Biosciences (San Jose, CA, USA); antibody specific for phospho-Ser-62-Bcl-xL was purchased from Abcam (Cambridge, MA, USA); FITC-conjugated anti-rabbit IgG secondary antibody for immunofluorescence was purchased from Jackson ImmunoResearch (West Grove, PA, USA). Vinblastine was purchased from Sigma-Aldrich (St Louis, MO, USA) and thymidine was purchased from EMD Biosciences (Gibbstown, NJ, USA). Plasmids encoding Bcl-2 (Plasmid 8768, Stanley Korsmeyer, Yamamoto 1999), Bcl-2 (AAA) (Plasmid 13340, Stanley Korsmeyer, Yamamoto, 1999), Bcl-xL (Plasmid 8749, Stanley Korsmeyer, Chao 1995), and Mcl-1 (Plasmid 21605, Ulrich Maurer, Maurer 2006) were purchased from Addgene (Cambridge, MA, USA)); Bcl-xL (S62A) and Bcl-xL (S62D) were generated using Stratagene QuickChange site-directed mutagenesis kit from Agilent Technologies (Santa Clara, CA, USA). For immunofluorescence, Mitotracker Red and ProLong Gold antifade reagent with DAPI were purchased from Invitrogen (Carlsbad, CA, USA). Cell death analysis was measured using Cell Death Detection ELISA from Roche (Penzberg, Germany), Caspase-3 activity Kit from Millipore (APT165; Billerica, MA, USA), and PI/RNAse staining buffer from BD Biosciences.
Cell culture, preparation of whole-cell extracts and immunoblotting
HeLa human cervical carcinoma cell line was maintained in monolayer culture at 37 °C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 50 units/ml penicillin and 50 μg/ml streptomycin. Cells were synchronized at the G1/S boundary by double thymidine block as described previously.8 Whole-cell extracts for kinase assays and immunoblot analysis were prepared as described previously,11, 35 and protein concentration was determined using the Bradford method.
Generation of cell lines stably expressing Bcl-2 proteins
Cells stably overexpressing human untagged Bcl-2, AAA-Bcl-2 (S69A, S70A and S87A), Bcl-xL, SA62-Bcl-xL or Mcl-1 were generated by transfecting HeLa cells, at ∼70% confluence, using Lipofectamine 2000 in serum-free Opti-MEM (Invitrogen). After 6 h, the transfection medium was replaced with DMEM containing 10% fetal bovine serum. After an additional 24 h, G418 was added to the medium to a final concentration of 1 mg/ml, and the cells were maintained for 2 weeks. Drug-resistant colonies were selected, expanded and maintained in growth medium containing 0.4 mg/ml G418. Clones were screened for overexpression of the transfected Bcl-2 family protein by immunoblotting using specific antibodies. HeLa subline (KB-3) cells stably expressing HA-tagged Bcl-xL were described previously.10
Transfections
Depletion of Cdc20 was performed using Ambion Silencer Select siRNA against Cdc20 (s2748) at a final concentration of 50 nM with Silencer Select negative control siRNA (4390844) (50 nM) as control. A mitotic shake-off technique was used to collect cells arrested in mitosis. Briefly, mitotic cells were detached into the media by gentle agitation of the plate and the remaining adherent cells were collected separately after trypsinization. Unless otherwise indicated, all experiments performed after Cdc20 knockdown used the non-adherent population of cells. Transient transfections of HA-tagged Bcl-2 family proteins were performed using 0.5 μg plasmid DNA with 10 μl Lipofectamine 2000 reagent. Fresh media containing serum was added 6 h post transfection. Both plasmid DNA and siRNA transfections were performed using Lipofectamine 2000 (Invitrogen) in Opti-MEM according to manufacturer’s instructions.
Cell cycle analysis
After transfection with Cdc20 siRNA, HeLa cells were collected by mitotic shake off and resuspended in DMEM at a concentration of 106 cells per ml. Cell cycle analysis was performed using propidium iodide staining and flow cytometry according to the manufacturer’s instructions (BD Pharmingen, San Diego, CA, USA) by the UAMS Flow Cytometry Core Facility using a FACSCalibur (Becton Dickinson, Mountain View, CA, USA). The data were analyzed using the ModFit DNA analysis program (Verity Software House, Topsham, ME, USA).
Cdk1 kinase assay
Cdk1 activity was measured as described previously.11 Briefly, whole-cell extracts containing 5 μg protein were incubated with H1 histone (5 μg) in a reaction mixture containing 25 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 5 mM DTT, 1 μ M ATP and 1 μCi [γ32P]ATP at 30 °C for 20 min. The reaction was stopped by the addition of 20 mM EDTA, acidified by the addition of 15% (v/v) acetic acid to and spotted onto 2-cm P81 phosphocellulose filter discs (Fisher, Waltham, MA, USA) for the measurement of 32P incorporation by scintillation counting. Background activity, obtained in reactions incubated in the absence of substrate, was subtracted.
Caspase-3 and apoptosis assays
Caspase-3 activity was measured using a Caspase-3 Colorimetric Assay Kit (Millipore) according to manufacturer’s instructions. Briefly, 100 μg whole-cell extract was incubated with caspase-3 substrate (15 μg) at 37 °C for 3 h followed by quantification of the cleaved substrate by measuring absorbance at 490 nm. Values were corrected for background absorbance obtained in assays conducted in the absence of sample. Control assays were performed in the presence of the caspase-3 inhibitor Ac-DEVD-CHO to account for non-specific substrate cleavage. PARP cleavage into characteristic fragments of 116 and 89 kDa was used as a qualitative measure of apoptosis. Cell permeability to propidium iodide was used to monitor cell membrane integrity as an indicator of apoptosis. Propidium iodide staining was done according to manufacturer’s instructions (BD Pharmingen) and analysis of propidium iodide incorporation was performed by the UAMS Flow Cytometry Core Facility. Apoptosis was also determined using a Cell Death ELISA assay according to manufacturer’s instructions (Roche Applied Science).
Immunofluorescence microscopy
Cells were grown on glass coverslips followed by fixation with 4% formaldehyde and permeabilization with 0.2% Triton X-100, and then blocked with 1% bovine serum albumin. Cells were probed for expression of Bcl-xL using a specific primary antibody (1 : 100) and a FITC-conjugated secondary antibody (1 : 100). Cells were co-stained with Mitotracker Red and visualized using an Olympus FluoView FV1000 confocal microscope (Olympus, Center Valley, PA, USA).
Abbreviations
- MIA:
-
microtubule inhibiting agents
- Cdk1:
-
cyclin-dependent kinase 1
- APC/C:
-
anaphase promoting complex
- PARP:
-
poly(ADP-ribose) polymerase
References
Jordan MA, Wilson L . Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004; 4: 253–265.
Mollinedo F, Gajate C . Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 2003; 8: 413–450.
Jackson JR, Patrick DR, Dar MM, Huang PS . Targeted anti-mitotic therapies: can we improve on tubulin agents? Nat Rev Cancer 2007; 7: 107–117.
Ruvolo PP, Deng X, May WS . Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 2001; 15: 515–522.
Harley ME, Allan LA, Sanderson HS, Clarke PR . Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J 2010; 29: 2407–2420.
Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011; 471: 110–114.
Chu R, Terrano DT, Chambers TC . Cdk1/cyclin B plays a key role in mitotic arrest-induced apoptosis by phosphorylation of Mcl-1, promoting its degradation and freeing Bak from sequestration. Biochem Pharmacol 2012; 83: 199–206.
Du L, Lyle CS, Chambers TC . Characterization of vinblastine-induced Bcl-xL and Bcl-2 phosphorylation: evidence for a novel protein kinase and a coordinated phosphorylation/dephosphorylation cycle associated with apoptosis induction. Oncogene 2005; 24: 107–117.
Shitashige M, Toi M, Yano T, Shibata M, Matsuo Y, Shibasaki F . Dissociation of Bax from a Bcl-2/Bax heterodimer triggered by phosphorylation of serine 70 of Bcl-2. J Biochem 2001; 130: 741–748.
Upreti M, Galitovskaya EN, Chu R, Tackett AJ, Terrano DT, Granell S et al. Identification of the major phosphorylation site in Bcl-xL induced by microtubule inhibitors and analysis of its functional significance. J Biol Chem 2008; 283: 35517–35525.
Terrano DT, Upreti M, Chambers TC . Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol Cell Biol 2010; 30: 640–656.
Yamamoto K, Ichijo H, Korsmeyer SJ . BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol 1999; 19: 8469–8478.
Basu A, Haldar S . Identification of a novel Bcl-xL phosphorylation site regulating the sensitivity of taxol- or 2-methoxyestradiol-induced apoptosis. FEBS Lett 2003; 538: 41–47.
Skube SB, Chaverri JM, Goodson HV . Effect of GFP tags on the localization of EB1 and EB1 fragments in vivo. Cytoskeleton 2010; 67: 1–12.
Swulius MT, Jensen GJ . The helical MreB cytoskeleton in Escherichia coli MC1000/pLE7 is an artifact of the N-Terminal yellow fluorescent protein tag. J Bacteriol 2012; 194: 6382–6386.
Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW . Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol 2008; 6: e147.
Hernandez-Vargas H, Palacios J, Moreno-Bueno G . Molecular profiling of docetaxel cytotoxicity in breast cancer cells: uncoupling of aberrant mitosis and apoptosis. Oncogene 2007; 26: 2902–2913.
Sakurikar N, Eichhorn JM, Chambers TC . Cyclin-dependent kinase-1 (Cdk1)/cyclin B1 dictates cell fate after mitotic arrest via phosphoregulation of antiapoptotic Bcl-2 proteins. J Biol Chem 2012; 287: 39193–39204.
Komlodi-Pasztor E, Sackett DL, Fojo AT . Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res 2012; 18: 51–63.
van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10: 389–399.
Lin X, Morgan-Lappe S, Huang X, Li L, Zakula DM, Vernetti LA et al. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene 2007; 26: 3972–3979.
Wei G, Margolin AA, Haery L, Brown E, Cucolo L, Julian B et al. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell 2012; 21: 547–562.
Musacchio A, Salmon ED . The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 2007; 8: 379–393.
Huang HC, Shi J, Orth JD, Mitchison TJ . Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell 2009; 16: 347–358.
Chu R, Chambers TC . Unexpected complexity in Mcl-1 phosphorylation in response to microtubule inhibitors In: Proceedings of the 104th Annual Meeting of the American Association for Cancer Research, 6–10 April 2013, Washington, DC, 2013. Abstract no 1739.
Deng X, Gao F, Flagg T, May WS Jr . Mono- and multisite phosphorylation enhances Bcl2's antiapoptotic function and inhibition of cell cycle entry functions. Proc Natl Acad Sci USA 2004; 101: 153–158.
Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011; 145: 104–116.
Upreti M, Lyle CS, Skaug B, Du L, Chambers TC . Vinblastine-induced apoptosis is mediated by discrete alterations in subcellular location, oligomeric structure, and activation status of specific Bcl-2 family members. J Biol Chem 2006; 281: 15941–15950.
Thomas LW, Lam C, Edwards SW . Mcl-1; the molecular regulation of protein function. FEBS Lett 2010; 584: 2981–2989.
Kobayashi S, Lee SH, Meng XW, Mott JL, Bronk SF, Werneburg NW et al. Serine 64 phosphorylation enhances the antiapoptotic function of Mcl-1. J Biol Chem 2007; 282: 18407–18417.
Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW . MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 2004; 23: 5301–5315.
Vogler M, Dinsdale D, Dyer MJ, Cohen GM . Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ 2009; 16: 360–367.
Boiani M, Daniel C, Liu X, Hogarty MD, Marnett LJ . The stress protein BAG3 stabilizes Mcl-1 and promotes survival of cancer cells and resistance to ABT-737. J Biol Chem 2013; 288: 6980–6990.
Russo M, Spagnuolo C, Volpe S, Tedesco I, Bilotto S, Russo GL . ABT-737 resistance in B-cells isolated from chronic lymphocytic leukemia patients and leukemia cell lines is overcome by the pleiotropic kinase inhibitor quercetin through Mcl-1 down-regulation. Biochem Pharmacol 2013; 85: 927–936.
Fan M, Goodwin M, Vu T, Brantley-Finley C, Gaarde WA, Chambers TC . Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J Biol Chem 2000; 275: 29980–29985.
Acknowledgements
This work was supported by National Institutes of Health Grant CA-109821 from the National Cancer Institute to TCC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by H-U SImon
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Eichhorn, J., Sakurikar, N., Alford, S. et al. Critical role of anti-apoptotic Bcl-2 protein phosphorylation in mitotic death. Cell Death Dis 4, e834 (2013). https://doi.org/10.1038/cddis.2013.360
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.360
Keywords
This article is cited by
-
Alternative CDC20 translational isoforms tune mitotic arrest duration
Nature (2023)
-
Protein kinase RNA-activated controls mitotic progression and determines paclitaxel chemosensitivity through B-cell lymphoma 2 in ovarian cancer
Oncogene (2021)
-
The anaphase-promoting complex/cyclosome: a new promising target in diffuse large B-cell lymphoma and mantle cell lymphoma
British Journal of Cancer (2019)
-
Universal response in the RKO colon cancer cell line to distinct antimitotic therapies
Scientific Reports (2018)
-
The impact of phosphatases on proliferative and survival signaling in cancer
Cellular and Molecular Life Sciences (2018)
