
Abstract
Wild-type p53-induced phosphatase 1 (Wip1) is a p53-inducible serine/threonine phosphatase that switches off DNA damage checkpoint responses by the dephosphorylation of certain proteins (i.e. p38 mitogen-activated protein kinase, p53, checkpoint kinase 1, checkpoint kinase 2, and uracil DNA glycosylase) involved in DNA repair and the cell cycle checkpoint. Emerging data indicate that Wip1 is amplified or overexpressed in various human tumors, and its detection implies a poor prognosis. In this study, we show that Wip1 interacts with and dephosphorylates BAX to suppress BAX-mediated apoptosis in response to γ-irradiation in prostate cancer cells. Radiation-resistant LNCaP cells showed dramatic increases in Wip1 levels and impaired BAX movement to the mitochondria after γ-irradiation, and these effects were reverted by a Wip1 inhibitor. These results show that Wip1 directly interacts with and dephosphorylates BAX. Dephosphorylation occurs at threonines 172, 174 and 186, and BAX proteins with mutations at these sites fail to translocate efficiently to the mitochondria following cellular γ-irradiation. Overexpression of Wip1 and BAX, but not phosphatase-dead Wip1, in BAX-deficient cells strongly reduces apoptosis. Our results suggest that BAX dephosphorylation of Wip1 phosphatase is an important regulator of resistance to anticancer therapy. This study is the first to report the downregulation of BAX activity by a protein phosphatase.
Similar content being viewed by others


Main
Prostate cancer is the most common form of cancer in men and the second leading cause of cancer deaths in North American men.1 Radiotherapy is an important treatment option for prostate cancer, either as a single modality or when combined with prostatectomy and hormonal therapy, although little is known about the treatment-related genetic changes and radiocurability of these tumors.2 The identification of novel genetic biomarkers, able to predict clinical responsiveness, would facilitate the selection of patients likely to respond to treatment.
Wild-type p53-induced protein phosphatase 1 [(Wip1 or protein phosphatase, Mg2+/Mn2+ dependent, 1D (PPM1D)] is a member of the protein phosphatase type 2C (PP2C) and p53 target gene family.3, 4 Wip1 is activated by p38 mitogen-activated protein kinase (MAPK) and the p53 pathway in response to various stresses, including UV, γ-radiation, and alkylating agents.5, 6, 7 Activated Wip1 directly dephosphorylates checkpoint kinase 1 (Chk1), checkpoint kinase 2 (Chk2), p38 MAPK, uracil DNA glycosylase (UNG), ataxia–telangiectasia-mutated (ATM) kinase, mdm2, γ-H2AX, and p53,8, 9, 10, 11, 12 suggesting its role as a homeostatic regulator that reverses protein kinase cascades such that damaged cells are induced to re-enter the normal cell cycle after completing DNA repair.13, 14, 15 Moreover, the Wip1 gene is frequently amplified/overexpressed in human cancers, including breast cancers, ovarian clear-cell adenocarcinomas, pancreatic neuroendocrine tumors, neuroblastomas, and pancreatic cancers, all of which rarely carry a p53 mutation.16, 17, 18
Ionizing radiation induces single- or double-stranded DNA breaks and initiates the DNA damage response (DDR). The latter is initially recognized by multiple DDR factors, such as MDC1, 53BP1, BRCA1, and TRF2, as well as MRE11-RAD50-NBS1 (MRN) complexes. Thereafter, ATM-Chk1/Chk2-p53 kinase pathways are activated, followed by the induction of G1–S and G2–M checkpoint activities and, finally, Wip1.19, 20 A recent study reported that Wip1 protects normal cells from hyperactivation during DNA repair activity, thereby reducing toxicity in response to chemotherapeutic agents.21, 22 The precise role of overexpressed Wip1 in the response to radiotherapy is still unclear. In fact, two opposing responses could be proposed, one involving resistance and the other hypersensitivity to apoptosis in malignant cancers through premature dephosphorylation/inactivation of DNA repair genes or by the total depletion of these signaling responses even before activation.
BAX is a 21-kDa protein that belongs to the Bcl-2 family and promotes apoptosis. Although mainly localized in the cytoplasm, BAX translocates to the mitochondria in response to stress stimuli.23 Both the prosurvival protein kinase AKT and protein kinase Cζ (PKCζ) phosphorylate BAX at Ser184, which suppresses its proapoptotic activity.24, 25 By contrast, the phosphorylation of either Thr167 by p38 MAPK and c-Jun NH2-terminal kinase (JNK) or Ser163 by glycogen synthase kinase-3β (GSK-3β) enhances the apoptotic activity of BAX.26, 27
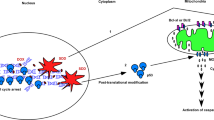
Here we show that Wip1 induction reduces the activities of the stress-activated kinases ataxia–telangiectasia and Rad3-related (ATR), mitogen-activated protein kinase kinase 4 (MKK4), p38 MAPK, and JNK, decreases apoptotic death following cell exposure to ionizing radiation, and directly dephosphorylates BAX at threonines 172, 174, and 186, all of which inhibit BAX translocation and BAX-related apoptosis. This study is the first to report the regulation of BAX activity by a protein phosphatase.
Results
Wip1 phosphatase expression represses apoptotic cell death in response to γ-irradiation
Considering that Wip1 functions as a silencer of checkpoint kinases, including ATM, ATR, and γ-H2AX,11, 13, 19 we hypothesized that a high level of Wip1 expression modulates the radiosensitivity of prostate cancer cells. We therefore examined three prostate cancer cell lines differing in their p53 status: PC3 (null for p53), DU145 (expressing partially functional P223L and non-functional V274F p53 mutant), and LNCaP (expressing wild-type p53). All three cell lines were established from metastatic prostate cancers and are among the most radioresistant human tumor cells.28 Analyses of the level of Wip1 mRNA levels by quantitative real-time reverse transcription PCR (qRT-PCR) after exposure of the cells for 6 h to 10 or 20 Gy of ionizing radiation (IR) showed that they were greatly elevated in LNCaP cells, but only slightly induced in PC3 and DU145 cells (Figure 1a). PC3 and LNCaP cells showed concordant increases in Wip1 protein expression in response to IR (Figure 1b). Based on these results, LNCaP cells were chosen for further investigation.

Wip1 expression is increased after γ-radiation in prostate cancer cell lines. (a and b) Cells were stimulated by ionizing radiation. At the indicated time points, the levels of Wip1 transcripts and proteins were determined by qRT-PCR (a) and immunoblot analysis (b). The mean of three experiments is shown in each column; bars correspond to the S.D. Asterisks (*) indicate P<0.05, according to a two-tailed Student’s t-test
Wip1 depletion reverses the radiation resistance of LNCaP prostate cancer cells
First, the sensitivity of LNCaP cells to 10 Gy IR was investigated. At 3 days after IR exposure ∼5% of the cells had died compared with ∼22% of radiosensitive HeLa cells used as a positive control, according to the analysis of annexin V-FITC- and propidium iodide-positive subpopulations (Figure 2a). Wip1 depletion has been shown to activate the p38 MAPK-p53 and MKK-JNK-c-Jun pathways, as well as apoptotic death in response to DNA damage in both mouse embryonic fibroblasts and Wip1 overexpressing tumor cells.29 To examine the radiosensitization effect of Wip1 inhibition, LNCaP cells were pretreated with 50 nM of Wip1 siRNA and then subjected to IR. Wip1 depletion by pretreatment with Wip1 siRNA resulted in a significant increase in apoptotic cells (41%) compared with control siRNA-treated cells (11%) (Figure 2b). Next, the effects of Wip1 knockdown on apoptosis were assessed by assaying caspase-3 activity in LNCaP cells. The transfection of Wip1 siRNA resulted in a twofold increase in caspase-3 activity compared with control siRNA (Figure 2c). These data suggested that, in LNCaP cells, Wip1 negatively regulates cell death in response to IR.

Depletion of Wip1 enhances IR-induced apoptotic cell death. (a) HeLa or LNCaP cells were irradiated with 10 Gy. After 72 h, the cells were harvested and stained with propidium iodide (PI) and annexin V-fluorescein isothiocyanate (FITC). The proportions of apoptotic cells within the annexin V-FITC and PI-positive subpopulations were determined using fluorescence-activated cell sorting (FACS). (b) LNCaP cells were transfected with 50 nM of either control small interfering (si)RNA or Wip1 siRNA. At 48 h post-transfection, the cells were irradiated with 10 Gy. After 72 h, the cells were stained with PI and annexin V-FITC. Apoptotic cell death was determined by FACS. (c) LNCaP cells were transfected with 50 nM of either control siRNA or Wip1 siRNA for 36 h, and then irradiated with 10 Gy. After 40 h, the transfectants were assayed for caspase-3 activity. All assays were performed in triplicate. The data show the mean±S.D. (*P<0.05)
DDR kinases and Wip1 expression are inversely activated by radiation
We then examined whether Wip1 expression regulates stress-responsive signaling pathways. In control HeLa cells irradiated with 10 Gy, phosphorylation of JNK at Thr183 and Tyr185, c-JUN at Ser 73, and p38 MAPK at Thr180 and Tyr182 was significantly increased; the exception was the absence of ATR phosphorylation at Ser428. Interestingly, there was no increase in the cellular expression of Wip1 proteins despite the robust accumulation of its upstream regulator p53 (Figure 3a). In LNCaP cells, by contrast, large decreases in the amounts of phosphorylated ATR, MKK4, JNK, c-JUN, and p38 MAPK, and a concomitant increase in Wip1 protein were determined. These changes were accompanied by an initial increase in p53 followed by its decline (Figure 3b). A 1-h pretreatment of LNCaP cells with CCT007093 failed to alter the increased expression of Wip1 protein, whereas phosphorylated forms of ATR, MKK4, JNK, c-JUN, and p38 MAPK accumulated, as in HeLa cells (Figure 3c). These data suggested that once induced, Wip1 rapidly inactivates the JNK, p38 MAPK, and ATR pathways, thereby preventing a cytotoxic response after cellular exposure to IR.

IR-induced Wip1 expression is inversely related to the activation of DNA damage response kinases. (a and b) HeLa (a) or LNCaP (b) cells were irradiated with 10 Gy and harvested at the indicated time points thereafter. Extracts prepared from the cells were subjected to western blot analysis using the indicated antibodies. β-Actin served as a loading control. (c) LNCaP cells were incubated with 20 μM CCT007093 for 1 h before 10 Gy IR. The effects of the Wip1 inhibitor were evaluated as described in (b)
Wip1 impairs BAX mitochondrial translocation following cellular IR
Upon treatment with apoptotic stimuli, proapoptotic BAX protein is activated through its phosphorylation by JNK, p38 MAPK, or GSK-3β, resulting in its translocation to the outer mitochondrial membrane.26, 27 To monitor the localization of BAX, HeLa and LNCaP cells were irradiated with 10 Gy, and the endogenous BAX proteins were then stained. Confocal microscopy showed that in irradiated HeLa cells, BAX densely localized to the mitochondria, which stained positively with MitoTracker Red 580, whereas in irradiated LNCaP cells, diffuse BAX staining was observed (Figure 4a). To evaluate the mitochondrial localization of BAX proteins further, mitochondrial proteins were extracted and subjected to western blot analysis. The results showed that LNCaP cells displayed considerably less mitochondrion-associated BAX than HeLa cells. However, CCT007093 pretreatment reversely increased BAX protein levels in the mitochondria (Figure 4b). We then asked whether Wip1 expression affects the stress-induced conformational change of BAX. Accordingly, LNCaP cells were exposed to 10 Gy and then immunoprecipitated with 6A7 antibody, which reacts only with active BAX. As seen in Figure 4c, Wip1 inhibition was associated with a strong increase in the amount of the active form of BAX, consistent with the negative regulation by Wip1 of the activating conformational changes in BAX that occur upon cellular IR exposure.

Wip1 inhibits radiation-activated BAX mitochondrial translocation. (a) HeLa and LNCaP cells were irradiated (IR) with 10 Gy. After 48 h, the cells were fixed and double-fluorescence stained with MitoTracker (red) for the mitochondria and anti-BAX (green). 4′,6-Diamidino-2-phenylindole (DAPI) counter-stain (blue) was used to label the nuclei. (b) Mitochondrial fractions were extracted from HeLa or LNCaP cells in the presence or absence of CT007093 after 48 h of 10 Gy IR. Mitochondrial proteins or whole-cell lysates (WCL) were subjected to immunoblotting (IB) using anti-BAX, prohibitin (a mitochondrial marker), and aldolase (a cytoplasmic marker). (c) LNCaP cells were irradiated with 10 Gy in the presence or absence of CT007093, followed by the preparation of total cell extracts at the indicated times. BAX proteins were immunoprecipitated (IP) using anti-BAX (6A7) antibody, which recognizes only the active form of BAX, and then analyzed by BAX immunoblotting. Anti-IgG antibody was utilized as a negative control
Wip1 interacts with BAX in vitro and in vivo after γ-irradiation
To understand the inhibitory effect of Wip1 on BAX better, the in vitro association of the two proteins was examined. Pull-down analyses were carried out using glutathione S-transferase (GST)-BAX purified from Escherichia coli. As shown in Figure 5a, Wip1 in IR-treated LNCaP cells was associated with recombinant GST-BAX but not with GST. The association was further confirmed by immunoprecipitation assays performed after the transient expression of Wip1-Flag and GFP-BAX in 293T cells. Immunoblot analysis with anti-Wip1 or anti-FLAG antibodies clearly detected Wip1-Flag in the anti-GFP precipitates, indicating the association of BAX with Wip1 following the irradiation of 293T cells (Figure 5b). Next, the ability of endogenous Wip1 to interact with GFP-BAX in LNCaP cells was determined. As shown in Figure 5c, both GFP and BAX specifically co-precipitated with Wip1 after γ-irradiation. Wip1 was originally identified as a nuclear protein, whereas BAX localizes to the cytoplasm and translocates to the mitochondria in response to stress stimuli. To explore the cellular compartmentalization of Wip1/BAX complexes, co-immunoprecipitation experiments using anti-BAX antibodies were performed with nuclear and cytoplasmic fractions of LNCaP cells after 6 h of γ-irradiation. As shown in Figure 5d, BAX-bound Wip1 proteins were detected within the nucleus of non-γ-irradiated cells, whereas the BAX-Wip1 complexes were present mainly in the cytoplasm of γ-irradiated cells, although individual Wip1 and BAX proteins were common in the nucleus and cytoplasm, respectively. These data indicate that Wip1 specifically interacts with BAX and that its localization is tightly regulated. Taken together, these results further confirm the in vivo interaction of Wip1 and BAX.

Wip1 interacts with BAX protein both in vivo and in vitro. (a) Recombinant GST-BAX or GST alone was precipitated with glutathione-Sepharose beads, incubated with IR-treated LNCaP cell lysates, and subjected to immunoblot (IB) analysis using anti-Wip1 or anti-GST antibodies. (b) Wip1-Flag and GFP-BAX constructs were transfected into the 293T cells. Forty-eight hours after transfection, the cells were irradiated with 10 Gy. After 24 h, the cells were lysed and immunoprecipitated using the anti-GFP antibody. The Wip1-BAX interaction was analyzed by immunoblotting, as indicated. (c) LNCaP cells transfected with the GFP-BAX construct for 48 h before stimulation with 10 Gy IR were harvested and lysed. The lysates were analyzed by immunoprecipitation using anti-GFP or anti-BAX and then immunoblotted using Wip1 antibody. (d) After 6 h of 10 Gy IR, LNCaP cells were fractionated into cytoplasm and nucleus, and immunoprecipitated using the anti-BAX antibody. The Wip1-BAX interaction was analyzed via western blotting with indicated antibodies. Histone H3 is a nuclear marker and aldolase is a cytosolic marker
Wip1 dephosphorylates BAX and blocks its mitochondrial translocation
To ascertain whether Wip1 dephosphorylates BAX, recombinant Wip1 was incubated with previously reported BAX-derived phosphopeptides containing Ser87, Ser163, Thr167, and Ser184 in an in vitro phosphatase assay.27, 30 Peptides containing phospho-Thr180 from p38 MAPK and phospho-Thr31 from UNG2 were used as a positive and negative control, respectively.10 As shown in Figure 6a, purified Wip1 did not dephosphorylate the four BAX-derived phosphopeptides. The possible phosphorylation sites of BAX were then analyzed using the NetPhos 2.0 software program (Center for Biological Sequence Analysis, Lyngby, Denmark),31 which identified nine candidates (Figure 6b).

Wip1 dephosphorylates active BAX and suppresses mitochondria-dependent apoptosis. (a) Recombinant Wip1 does not dephosphorylate BAX-derived phosphopeptides encompassing Ser87, Ser163, Ser 184, and Thr167, as shown in an in vitro phosphatase assay. Released free phosphate was measured by absorbance at 630 nm in the presence of molybdate dye. Phosphopeptides p180T from p38 MAPK and p31T from UNG2 were used as positive and negative controls, respectively.10 (b) Phosphorylation sites on Ser and Thr residues in BAX were predicted with the NetPhos 2.0 server.30 (c) Expression vectors carrying GFP wild-type or one of nine mutated GFP-BAX mutant constructs were transfected into HeLa cells. At 48 h post-transfection, the cells were exposed to γ-radiation and then analyzed 24 h later. BAX translocation to the mitochondria results in intense green fluorescence (phosphothreonine and phosphoserine are indicated in bold). (d) BAX p172T, p174T, and p186T phosphopeptides were incubated with recombinant Wip1 protein. Free phosphate released by dephosphorylation was measured. Two polypeptides of p38 MAPK p180T served as the positive control, and UNG2 p31T as the negative control.10 Assays were also performed in the absence of magnesium or Wip1 and in the presence of okadaic acid. Wip1 phosphatase activity is magnesium-dependent and insensitive to okadaic acid. (e) DU145 cells were transfected with BAX or mutated BAX (T172D, T174D, and T186D) expression plasmids. After 48 h, the transfectants were irradiated, and then harvested, and finally assayed for caspase-3 activity. (f) DU145 cells were transfected with wild-type or mutated BAX together with wild-type Wip1 or phosphatase-dead (PD) Wip1 (D314A and D105A) expression vectors for 48 h and then irradiated with 10 Gy. After 24 h, the transfectants were assayed for caspase-3 activity. The mean of three experiments is shown in each column; bars in (d), (e), and (f) correspond to the S.D. Asterisks (*) indicate P<0.05, according to the two-tailed Student’s t-test
To determine the critical residues for BAX translocation, all of the candidate residues were mutated using the GFP-BAX plasmid and the rates of mitochondrial translocation in HeLa cells were then measured. Confocal microscopy showed intense green fluorescence in the wild-type BAX-transfected cells as well as those transfected with the T14D, S55A, T56D, S72A, T85D, and T127D mutants and also indicating the mitochondrial translocation of these mutated BAX proteins after IR. By contrast, in cells transfected with the T172D, T174D, and T186 BAX mutants, diffuse green fluorescence was observed, indicative of inefficient BAX translocation (Figure 6c). These results suggested that phosphorylation at these residues is important for the normal activity of BAX. In a follow-up experiment, the ability of Thr172, Thr174, and Thr186 of BAX to serve as prime candidates for dephosphorylation by Wip1 was tested in an in vitro phosphatase assay. Wip1 strongly dephosphorylated the three phosphopeptides, with the highest efficiency at the site containing Thr172, TPTWQpTVTIFV (phosphothreonine is indicated in bold). The dephosphorylation activity of Wip1 was not affected by okadaic acid, but was greatly sensitive to the absence of magnesium, consistent with the characteristics of Wip1 as a type 2C phosphatase (Figure 6d).
To demonstrate that the dephosphorylation activity of Wip1 directly affects mitochondria-dependent apoptosis, caspase-3 activity was measured in BAX-deficient DU145 cells expressing wild-type BAX or the mutants T172D, T174D, and T186D. Following IR exposure, caspase-3 activity was significantly lower in cells expressing any of these three BAX mutants than in those expressing wild-type BAX. Caspase-3 activity was particularly reduced, by approximately 40%, in the extract prepared from the IR-treated BAX T172D mutant (Figure 6e). Furthermore, overexpression of wild-type Wip1 inhibited caspase-3 activity in BAX-transfected DU145 cells, whereas this was not the case with the phosphatase-dead Wip1 double mutant (D314A and D105A) expressed in the same cells (Figure 6f). These results are consistent with the ability of Wip1 to inhibit caspase-dependent apoptosis specifically via BAX dephosphorylation.
Discussion
Upon DNA damage, p53 is rapidly activated by its phosphorylation and then transactivates several genes, including those of its negative regulators Mdm2 and Wip1. Many studies have reported that the overexpression of Wip1 promotes tumorigenesis, whereas its loss enhances apoptosis in response to DNA damage.21, 29, 32, 33 To date, the role of Wip1 in the DDR has been primarily defined as that of a homeostatic regulator, given that Wip1 dephosphorylates DDR factors, including ATM, ATR, p53, Chk1, Chk2, UNG2, and H2AX, thereby restoring their activities to normal homeostatic levels after DNA damage repair.34 In this study, a novel function of Wip1 was identified, that is, its ability to dephosphorylate directly and thus inactivate apoptotic BAX protein in response to γ-irradiation.
Wip1 was shown to bind BAX directly following γ-irradiation, dephosphorylating the latter protein at threonines 172, 174, and 186, as measured by an in vitro phosphatase assay using purified recombinant Wip1. The absence of magnesium caused a decrease in Wip 1 activity, whereas okadaic acid had no effect, consistent with the characteristics of the PP2C family, to which Wip1 belongs. The three dephosphorylated threonine sites of BAX (WQpTVT at 172, TVpTIF at 174, and SLpTIW at 186) are not concordant with the two well-known Wip1 substrate motifs, pTXpY and pS/pTQ.17, 35 In BAX-deficient cells exposed to IR, the overexpression of BAX bearing T172D, T174D, and T186D mutations resulted in an approximately 40% reduction of caspase-3 activity. Wip1 overexpression reduced caspase-3 activity by as much as half, but phosphatase-dead Wip1 had no effect. These results suggest that the phosphatase activity of Wip1 is important for suppressing BAX activity in apoptosis. Although BAX inactivation, caused by the phosphorylation of protein kinases such as AKT and PKCζ, has been reported previously,24, 25 protein phosphoatase-mediated BAX inactivation is a newly identified mechanism in apoptosis regulation.
Many studies have demonstrated the ability of Wip1 to return the cell to a basal DDR state by lowering upstream ATM/ATR signalings to p53.13, 15 This activity of Wip1 could underlie the protection of normal cells from the deleterious side effects of anticancer agents. Goloudina et al.21, 22 reported that Wip1 overexpression promotes apoptosis by the Wip1-RUNX2-BAX pathway in p53-negative tumors, but represses apoptosis in p53 wild-type tumors. Activating dephosphorylation of RUNX2 by Wip1 increases its transcriptional activity on the Bax promoter. These results suggest that the Wip1-RUNX2-BAX pathway should be more effective in p53 wild-type tumors because p53 transactivates the Bax and Wip1 genes. Accordingly, the dephosphorylated active form of RUNX2 would be more abundant and its high levels attributable to BAX-mediated cell death in p53 wild-type tumors. Based on this assumption, the antiapoptotic activity of Wip1-BAX may outperform the proapoptotic activity of Wip1-RUNX2-BAX in p53 wild-type tumors in response to DNA damage.
BAX is maintained in the cytoplasm of non-apoptotic cells along with other antiapoptotic Bcl-2 family members, such as Bcl-2, Bcl-xL, and Mcl-1, and in a phosphorylated and closed form.36 PP2A complexes dephosphorylate BAX, leading to configurational changes and the dissociation of BAX from its dimerization partner in response to apoptotic stimuli.37 These proapoptotic functions of PP2A are in contrast to Wip1, which acts as an antiapoptotic phosphatase. It will thus be of particular interest to determine whether Wip1 dephosphorylates other Bcl-2 family proteins, as this may reveal other oncogenic roles for the phosphatase.
In this study, we have presented evidence that Wip1-mediated dephosphorylation inhibits BAX translocation to the mitochondria in response to IR. Our findings shed light on the mechanism of the anticancer therapy resistance of Wip1-amplified tumors, particularly those bearing wild-type p53, and suggest the use of Wip1 as a predictive marker of the clinical responses to anticancer therapy.
Materials and Methods
Cell culture
The human prostate cancer cell lines LNCaP, PC3, and DU145 were grown in RPMI 1640 (Invitrogen-GIBCO, Carlsbad, CA, USA). Control HeLa cells were cultured in DMEM supplemented with 10% fetal bovine serum, 50 μg penicillin per ml, and 100 μg streptomycin per ml at 37 °C in a 5% CO2 incubator.
Plasmid constructs
Human BAX (GenBank ID: NM_138761) was amplified using primers 5′-TATGAATTCATGGACGGGTCCGGGGAGCAG-3′ (forward) and 5′-AATCTCGAGTCAGCCCATCTTCTTCCAGATG-3′ (reverse). The PCR products were digested with EcoRI and XhoI and then cloned into the pGEX-4T-1 vector (GE Healthcare, Little Chalfont, UK), yielding the GST-BAX construct. Based on the predicted phosphorylation sites in BAX using the NetPhos 2.0 server (http://www.cbs.dtu.dk/services/NetPhos/),31 BAX mutants were constructed using the following primer sets and the Quickchange mutagenesis kit II (210518; Stratagene, Santa Clara, CA, USA) (mutated sequence is shown in lower-case letters): BAX T14D, 5′-AGAGGCGGGGGGCCCgaCAGCTCTGAGCAGAT-3′ and 5′-ATCTGCTCAGAGCTGtcGGGCCCCCCGCCTCT-3′; BAX S55A, 5′-TGCCTCAGGATGCGgCCACCAAGAAGCTGA-3′ and 5′-TCAGCTTCTTGGTGGcCGCATCCTGAGGCA-3′; BAX T56D, 5′-TGCCTCAGGATGCGTCCGaCAAGAAGCTGAGCGAGT-3′ and 5′-ACTCGCTCAGCTTCTTGtcGGACGCATCCTGAGGCA-3′; BAX S72A, 5′-GGGACGAACTGGACgcTAACATGGAGCTGCAGA-3′ and 5′-TCTGCAGCTCCATGTTAgcGTCCAGTTCGTCCC-3′; BAX T85D, 5′-ATGATTGCCGCCGTGGACgacGACTCCCCCCGAGAGGTCTTT-3′ and 5′-AAAGACCTCTCGGGGGGAGTCgtcGTCCACGGCGGCAATCAT-3′; BAX T127D, 5′-TCAAGGCCCTGTGCgaCAAGGTGCCGGAACTGA-3′ and 5′-TCAGTTCCGGCACCTTGtcGCACAGGGCCTTGA-3′; BAX T172D, 5′-ACGCCCACGTGGCAGgaCGTGACCATCTTTGT-3′ and 5′-ACAAAGATGGTCACGtcCTGCCACGTGGGCGT-3′; BAX T174D, 5′-ACGTGGCAGACCGTGgaCATCTTTGTGGCGGGA-3′ and 5′-TCCCGCCACAAAGATGtcCACGGTCTGCCACGT-3′; and BAX T186D, 5′-AGTGCTCACCGCCTCACTCgaCATCTGGAAGAAGAT-3′ and 5′-ATCTTCTTCCAGATGtcGAGTGAGGCGGTGAGCACT-3′.
qRT-PCR
Total RNA was isolated using the NucleoSpin RNA II kit (Macherey-Nagel, Duren, Germany). First-strand cDNA was synthesized using the SuperScript II first-strand synthesis kit (Invitrogen). qRT-PCR was performed on a Bio-Rad iQ5 (Bio-Rad Laboratories, Hercules, CA, USA) using SYBR Green (Invitrogen) and the following sets of primers: Wip1, 5′-GCCAGAACTTCCCAAGGAAAG-3′ and 5′-GGTTCAGGTGACACCACAAATTC-3′, and GAPDH, 5′-GAAGGTGAAGGTCGGAGTC-3′ and 5′-GAAGATGGTGATGGGATTTC-3′. The expression of Wip1 mRNA was normalized to that of GAPDH.
Western blot analysis
Cell lysates containing 50 μg of protein were analyzed by western blotting using the following primary antibodies: Wip1 (Abgent, San Diego, CA, USA); p53, p38 MAPK, JNK (Upstate, Charlottesville, VA, USA); phospho-ATR (Ser428), phospho-ATM (Ser1981), phospho-MKK4 (Thr261), MKK4, phospho-JNK (Thr183/Tyr185), phospho-c-Jun (Ser73), phospho-p38 (Thr180/Tyr182), and aldolase (Cell Signaling, Beverly, MA, USA); c-Jun, prohibitin, GFP, GST, and BAX (N20) (Santa Cruz, Santa Cruz, CA, USA); and Flag and β-actin (Sigma, St Louis, MO, USA). Blots were developed using the SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL, USA).
Annexin V staining
Cells were transfected with 50 nM Wip1-specific siRNA (D-004554-01) or unrelated scrambled control siRNA (D-001210-01-05) (Thermo Scientific) using the Neon transfection system (Invitrogen). Alternatively, they were treated with 10 μM of Wip1 inhibitor CCT007093 (Merck, Darmstadt, Germany) for 1 h and then exposed to IR. The cells were stained with annexin V-FITC (BD Pharmingen, San Jose, CA, USA) and propidium iodide (Sigma), and then analyzed on a fluorescence-activated cell sorting cytometer.
Immunofluorescence
Cells grown on coverslips were incubated with 100 nM of MitoTracker Orange CMTMRos (M77510; Molecular Probe, Eugene, OR, USA) for 30 min at 37 °C, followed by fixation with 4% paraformaldehyde for 10 min at 37 °C and permeabilization with 0.2% Triton X-100 in TBS. They were then blocked in 2% BSA in TBS–0.1% Triton X-100 for 30 min at room temperature. Anti-BAX (B9) (sc-7480; Santa Cruz) and secondary antibodies conjugated to FITC-mouse (115-095-003; Jackson Immuno Research, West Grove, PA, USA) were used.
Mitochondrial fraction
The cells were harvested and resuspended on ice in MIB buffer (mitochondria isolation buffer; 25 mM Tris (pH 7.4), 250 mM sucrose, 1 mM EDTA), and then homogenized using a 22-G needle attached to a syringe. The homogenates were centrifuged at 1000 × g for 10 min at 4 °C, followed by 10 900 × g for 10 min to collect the crude mitochondrial fraction. The pellets were resuspended in 1 ml of a 50% Optiprep solution (D1556; Sigma-Aldrich, St Louis, MO, USA) and loaded into an ultracentrifuge tube (R5; Hitachi Koki, Tokyo, Japan). Subsequently, 3 ml layers of 50, 30, and 10% Optiprep solution were sequentially added to the suspended solution. Following centrifugation of the samples in a Hitachi Himac swinging bucket rotor at 80 000 × g for 3 h, mitochondrial fractions at the 10/30% Optiprep interface were collected, resuspended in MIB solution, and centrifuged at 3000 × g for 5 min. The supernatants were recovered and re-centrifuged at 10 900 × g for 10 min. The pellets were then resuspended in MIB solution and stored as mitochondrial fractions.
Co-immunoprecipitation
Cells were seeded on six-well plates and transfected with Wip1-Flag, GFP-BAX, or a combination thereof using Lipofectamine 2000 (Invitrogen) in Opti-MEM (Invitrogen). After 6 h of γ-irradiation, the cells were lysed in CHAPS lysis buffer containing 150 mM NaCl, 10 mM HEPES (pH 7.5), 1% CHAPS, and protease inhibitors. GFP-BAX was precipitated overnight with anti-GFP or anti-BAX antibodies, followed by the addition of 20 μl protein A/G beads to the cell lysates overnight at 4 °C. The immunoprecipitates were spun down and subjected to western blot analysis using Flag or Wip1 antibodies. BAX conformational changes were detected by immunoprecipitating the cell lysates with anti-BAX (6A7) (sc-23959; Santa Cruz). The immunoprecipitates were then immunoblotted using BAX (N20) antibody.
GST pull-down assay
GST-BAX expression was induced in Escherichia coli BL21 cells in the presence of 0.4 mM isopropyl-β-D-thio-galactoside. The expressed proteins were lysed in ProFound lysis buffer (Thermo Scientific) for 30 min on ice and then isolated with glutathione-Sepharose beads. The IR-treated LNCaP cell lysates were incubated overnight with GST-BAX fusion protein conjugated with glutathione-Sepharose beads at 4 °C, washed five times with lysis buffer, and eluted with 100 mM glutathione elution buffer. Precipitates from the pull-down assay were detected by western blot analysis.
In vitro Ser/Thr phosphatase assay
The assays were carried out in buffer (250 mM imidazole, 1 mM EGTA, 25 mM MgCl2, 0.1% 2-mercaptoethanol, 0.5 mg BSA per ml) at 25 °C for 20 min upon the addition of purified hWIP1-Δexon6 (30 μg).10 Free phosphate was determined by the malachite-green/molybdate-based assay using the serine/threonine phosphatase assay system (Promega, Madison, WI, USA). Peptides synthesized by Peptron (Daejon, Korea) or Thermo Scientific were resuspended in DMSO and used at 100 μM per reaction. The sequences of the synthesized phosphopeptides were as follows: p38 MAPKp180T (TDDEMpTGYVAT) (positive control), UNG2p31T (AVQGpTGVAGV) (negative control), BAXp87S (AVDTDpSPREVF), BAXp163T (WDGLpSYFGTP), BAXp167T (LSYFGpTPTWQT), BAXp172T (TPTWQpTVTIFV), BAXp174T (TWQTVpTIFVAG), BAXp184T (GVLTApSLTIWK), and BAXp186T (LTASLpTIWKKM) (phosphothreonine and phosphoserine are indicated in bold).
Caspase-3 activity assay
After 24 h of γ-irradiation, DU145 cells were lysed in Triton X-100 lysis buffer (10 mM Tris-HCl, 5 mM EDTA, 320 mM sucrose, 1% Triton X-100, 1 mM PMSF, 2 mM DTT, 1 × proteinase inhibitor cocktail). Aliquots of protein (50 μg) were diluted to 50 μl with Triton X-100 lysis buffer and then incubated for 1 h with an equal volume of 2 × caspase assay buffer (100 mM HEPES, 10% sucrose, 0.1% CHAPS, 1 mM PMSF, 1 mM DTT, 1 × proteinase inhibitor cocktail) containing 50 μM of fluorogenic (Ac-DEVD-AMC) caspase-3 substrate (235425; Calbiochem, Billerica, MA, USA). Cleavage was monitored by excitation at 380 nm and emission at 460 nm using a fluorescence spectrometer (Victor X3, Waltham, MA, USA).
Statistics
Values represent the means±S.D. (n=3 or 4). Comparisons between groups were analyzed using two-tailed Student’s t-tests. Statistical significance is represented in the figures by asterisks (*), indicating P<0.05.
Abbreviations
- Wip1:
-
wild-type p53-induced phosphatase 1
- PPM1D:
-
protein phosphatase, Mg2+/Mn2+ dependent, 1D
- MAPK:
-
mitogen-activated protein kinase
- Chk1:
-
checkpoint kinase 1
- Chk2:
-
checkpoint kinase 2
- UNG:
-
uracil DNA glycosylase
- ATM:
-
ataxia–telangiectasia mutated
- IR:
-
ionizing radiation
- DDR:
-
DNA damage response
- PKCζ:
-
protein kinase Cζ
- JNK:
-
c-Jun NH2-terminal kinase
- GSK-3β:
-
glycogen synthase kinase-3β
- MKK4:
-
mitogen-activated protein kinase kinase 4
- ATR:
-
ataxia–telangiectasia and Rad3-related
- PP2A:
-
protein phosphatase type 2A
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ . Cancer statistics, 2009. CA-Cancer J Clin 2009; 59: 225–249.
Pieters BR, de Back DZ, Koning CC, Zwinderman AH . Comparison of three radiotherapy modalities on biochemical control and overall survival for the treatment of prostate cancer: a systematic review. J Eur Soc Therap Radiol Oncol 2009; 93: 168–173.
Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci USA 1997; 94: 6048–6053.
Choi J, Appella E, Donehower LA . The structure and expression of the murine wildtype p53-induced phosphatase 1 (Wip1) gene. Genomics 2000; 64: 298–306.
Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H et al. P53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J 2000; 19: 6517–6526.
Park JY, Song JY, Kim HM, Han HS, Seol HS, Jang SJ et al. P53-independent expression of wild-type p53-induced phosphatase 1 (Wip1) in methylmethane sulfonate-treated cancer cell lines and human tumors. Int J Biochem Cell Biol 2012; 44: 896–904.
Rossi M, Demidov ON, Anderson CW, Appella E, Mazur SJ . Induction of PPM1D following DNA-damaging treatments through a conserved p53 response element coincides with a shift in the use of transcription initiation sites. Nucleic Acids Res 2008; 36: 7168–7180.
Lu X, Nannenga B, Donehower LA . PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev 2005; 19: 1162–1174.
Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ 2006; 13: 1170–1180.
Lu X, Bocangel D, Nannenga B, Yamaguchi H, Appella E, Donehower LA . The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol Cell 2004; 15: 621–634.
Lu X, Nguyen TA, Donehower LA . Reversal of the ATM/ATR-mediated DNA damage response by the oncogenic phosphatase PPM1D. Cell Cycle (Georgetown, TX) 2005; 4: 1060–1064.
Lu X, Ma O, Nguyen TA, Jones SN, Oren M, Donehower LA . The Wip1 phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell 2007; 12: 342–354.
Macurek L, Lindqvist A, Voets O, Kool J, Vos HR, Medema RH . Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene 2010; 29: 2281–2291.
Moon SH, Lin L, Zhang X, Nguyen TA, Darlington Y, Waldman AS et al. Wild-type p53-induced phosphatase 1 dephosphorylates histone variant gamma-H2AX and suppresses DNA double strand break repair. J Biol Chem 2010; 285: 12935–12947.
Moon SH, Nguyen TA, Darlington Y, Lu X, Donehower LA . Dephosphorylation of gamma-H2AX by WIP1: an important homeostatic regulatory event in DNA repair and cell cycle control. Cell Cycle (Georgetown, TX) 2010; 9: 2092–2096.
Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet 2002; 31: 210–215.
Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA . The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metast Rev 2008; 27: 123–135.
Mendrzyk F, Radlwimmer B, Joos S, Kokocinski F, Benner A, Stange DE et al. Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. J Clin Oncol 2005; 23: 8853–8862.
Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell 2006; 23: 757–764.
Batchelor E, Mock CS, Bhan I, Loewer A, Lahav G . Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol Cell 2008; 30: 277–289.
Goloudina AR, Tanoue K, Hammann A, Fourmaux E, Le Guezennec X, Bulavin DV et al. Wip1 promotes RUNX2-dependent apoptosis in p53-negative tumors and protects normal tissues during treatment with anticancer agents. Proc Natl Acad Sci USA 2012; 109: E68–E75.
Goloudina AR, Mazur SJ, Appella E, Garrido C, Demidov ON . Wip1 sensitizes p53-negative tumors to apoptosis by regulating the Bax/Bcl-xL ratio. Cell Cycle (Georgetown, TX) 2012; 11: 1883–1887.
Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC . Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Academy of Sci USA 1998; 95: 4997–5002.
Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, Frasch SC, Borregaard N et al. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem 2004; 279: 21085–21095.
Xin M, Gao F, May WS, Flagg T, Deng X . Protein kinase Czeta abrogates the proapoptotic function of Bax through phosphorylation. J Biol Chem 2007; 282: 21268–21277.
Kim BJ, Ryu SW, Song BJ . JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem 2006; 281: 21256–21265.
Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA et al. Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. Off J Soc Neurosci 2004; 24: 9993–10002.
Garzotto M, Haimovitz-Friedman A, Liao WC, White-Jones M, Huryk R, Heston WD et al. Reversal of radiation resistance in LNCaP cells by targeting apoptosis through ceramide synthase. Cancer Res 1999; 59: 5194–5201.
Xia Y, Ongusaha P, Lee SW, Liou YC . Loss of Wip1 sensitizes cells to stress- and DNA damage-induced apoptosis. J Biol Chem 2009; 284: 17428–17437.
Wang Q, Sun SY, Khuri F, Curran WJ, Deng X . Mono- or double-site phosphorylation distinctly regulates the proapoptotic function of Bax. PLoS One 2010; 5: e13393.
Blom N, Gammeltoft S, Brunak S . Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 1999; 294: 1351–1362.
Yoda A, Toyoshima K, Watanabe Y, Onishi N, Hazaka Y, Tsukuda Y et al. Arsenic trioxide augments Chk2/p53-mediated apoptosis by inhibiting oncogenic Wip1 phosphatase. J Biol Chem 2008; 283: 18969–18979.
Ali AY, Abedini MR, Tsang BK . The oncogenic phosphatase PPM1D confers cisplatin resistance in ovarian carcinoma cells by attenuating checkpoint kinase 1 and p53 activation. Oncogene 2012; 31: 2175–2186.
Park HK, Panneerselvam J, Dudimah FD, Dong G, Sebastian S, Zhang J et al. Wip1 contributes to cell homeostasis maintained by the steady-state level of Wtp53. Cell Cycle (Georgetown, TX) 2011; 10: 2574–2582.
Yamaguchi H, Durell SR, Chatterjee DK, Anderson CW, Appella E . The Wip1 phosphatase PPM1D dephosphorylates SQ/TQ motifs in checkpoint substrates phosphorylated by PI3K-like kinases. Biochemistry 2007; 46: 12594–12603.
Walensky LD, Gavathiotis E . BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem Sci 2011; 36: 642–652.
Xin M, Deng X . Protein phosphatase 2A enhances the proapoptotic function of Bax through dephosphorylation. J Biol Chem 2006; 281: 18859–18867.
Acknowledgements
This work was supported by the Mid-career Researcher Program through an NRF grant funded by the MEST (no. 2009-0081016), by a grant (A062254) from the Korea Health 21 R&D Project, Ministry of Health, Welfare, and Family Affairs, Republic of Korea, and by a grant from the Asan Institute for Life Science (2008-309).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Raschellá
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Song, JY., Ryu, SH., Cho, Y. et al. Wip1 suppresses apoptotic cell death through direct dephosphorylation of BAX in response to γ-radiation. Cell Death Dis 4, e744 (2013). https://doi.org/10.1038/cddis.2013.252
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.252
Keywords
This article is cited by
-
DNA damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities
Experimental & Molecular Medicine (2022)
-
WIP1 phosphatase as pharmacological target in cancer therapy
Journal of Molecular Medicine (2017)
-
WIP1 regulates the proliferation and invasion of nasopharyngeal carcinoma in vitro
Tumor Biology (2014)
