
Abstract
Sustained endoplasmic reticulum (ER) stress has been linked to cell death and the pathogenesis of many liver diseases, including toxic liver, cholestasis, and infectious liver disease. The cellular pathways that attenuate hepatic ER stress have been the focus of many recent studies, but the role of microRNAs (miRNA) in this process remains unknown. Here, we report that one of the most abundant miRNAs in hepatocytes, miR-199a-5p, was elevated in both bile acid- and thapsigargin (TG)-stimulated cultured hepatocytes, as well as in the liver of bile duct-ligated mice. We identify the misfolded protein chaperone GRP78, as well as the unfolded protein response transducers endoplasmic reticulum to nucleus signaling 1 and activating transcription factor 6 as direct targets of miR-199a-5p, and show that endogenous miR-199a-5p represses the 3′ untranslated regions (UTRs) of their mRNAs. Through gain-of-function and loss of function approaches, we demonstrate that the elevated miR-199-5p disrupts sustained ER stress and prevents hepatocytes from undergoing bile acid- or TG-induced cell death. Furthermore, we reveal that the transcription factor AP-1 is a strong positive regulator of miR-199a-5p. In brief, our study demonstrates that AP-1/miR-199a-5p and ER stress mediators form a feedback loop, which shields hepatocytes from sustained ER stress and protects the liver from injury. On the basis of these findings, we also suggest that the miRNA miR-199a-5p is a potential target for clinical approaches aiming to protect hepatocytes in liver disease.
Similar content being viewed by others


Main
The accumulation of unfolded proteins in the endoplasmic reticulum (ER) constitutes a dangerous form of cellular stress that can culminate in a variety of pathological outcomes, including cell death. At the cellular level, ER stress triggers an evolutionarily conserved series of signal-transduction events that constitutes the unfolded protein response (UPR). At the molecular level, the binding of the chaperone GRP78 (also known as BiP or HSPA5)1 to misfolded proteins activates the proximal UPR transducer proteins eukaryotic translation initiation factor 2-alpha kinase 3, also known as PERK, IRE1α (inositol-requiring enzyme 1α, also called ERN1, ER to nucleus signaling 1), and ATF6 (activating transcription factor 6).2 PERK phosphorylates eIF2α (eukaryotic initiation factor 2), thereby inhibiting global protein translation and promoting the expression of the transcription factor ATF4. IRE1α is a dual-function protein with endoribonuclease and protein kinase activities, and is required for the splicing and activation of XBP1 (X-box-binding protein 1).3 The ER stress-responsive transcription factors ATF4, ATF6, and XBP1 co-operate to regulate the expression of multiple ER stress-responsive genes, including the proapoptotic transcription factor CHOP (C/EBP homologous protein), and chaperone proteins, including GRP78 and GRP94.4
Many recent studies have focused on the role of ER stress in various liver diseases, including toxic, metabolic, and infectious liver disease.5, 6, 7 Hepatocytes with a high burden of protein synthesis depend on efficient protein folding mechanisms to cope with the protein load within the ER. However, ER homeostasis can be profoundly disturbed by defective post-translational protein modifications and the accumulation of misfolded or viral proteins, all of which activate the UPR pathway in the ER. Recently, hepatic ER stress has been proven to have a role in multiple liver diseases, such as insulin resistance,8, 9 hepatic steatosis,10, 11 alcohol-induced injury,12 and cholestatic liver.
Cholestasis is a common feature of many human liver diseases. Increased bile acid concentration in hepatocytes, a hallmark of cholestasis, promotes liver cell death and the development of liver injury,13 and liver cirrhosis.14, 15 In the cholestatic process, toxic bile acid sodium deoxycholate induces the expression of the UPR genes GRP78 and CHOP,16 and CHOP deficiency protects hepatocytes from cell death induced by toxic bile acid in vitro and in vivo.17, 18 These findings suggest that the accumulation of bile acid in the liver can disrupt protein folding in the ER and contribute to liver injury. However, the cellular pathways that regulate the hepatic UPR and ER stress remain undefined.
microRNAs (miRNAs) are small noncoding RNAs that can negatively regulate gene expression post-transcriptionally.19 Hundreds of miRNAs have been identified, and these molecules have important roles in a wide variety of biological processes. The effects of miRNAs on ER stress have only become evident in recent years. miR-181a has been reported to regulate GRP78 and influence cerebral ischemia outcomes.20 Likewise, miR-30d, miR-181a, and miR-199a-5p co-operatively suppress GRP78 in cancer.21 Recently, miR-199a has also been proven to regulate ER stress in cancer cells by targeting GRP78.22, 23 Overall, however, the potential role of miRNAs in the hepatic ER stress responses to bile acid and other agents is just beginning to emerge.
Here, we investigated the roles of miRNAs in the direct repression of ER stress genes in hepatocytes. Through expression analysis and computational predictions, we identified endogenous miR-199a-5p is necessary for the modulation of hepatic ER stress progression by negatively regulating the IRE1α-related pathway. Our study uncovered a direct link between a miRNA and hepatic ER stress, and demonstrated that miR-199a-5p repressed sustained ER stress and prevented hepatocyte apoptosis.
Results
Dicer-knockdown hepatocytes exhibit sustained ER stress and apoptosis during bile acid stimulation
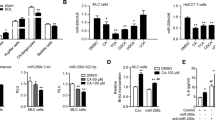
To assess the role of miRNAs in hepatic ER stress, we first investigated the expression levels of ER stress markers after bile acid stimulation in hepatocytes deficient in the miRNA-processing enzyme Dicer. The LV-mediated knockdown of Dicer mRNA and protein levels in transformed HL-7702/L02 human hepatocytes was significant and sustained for 7 days, as confirmed by qualitative reverse transcription (qRT-PCR) and immunoblot in comparison with uninfected and scrambled LV-short hairpin RNA (shRNA) controls (Figure 1a). We then incubated these cells with deoxycholic acid (DCA) or, as a positive control, the chemical thapsigargin (TG), a commonly used inducer of ER stress that inhibits the endoplasmic Ca2+-ATPase SERCA. In uninfected HL-7702/L02 cells, GRP78 (HSPA5) mRNA, and protein levels increased 8 h after DCA and TG incubation, and then rapidly declined after 48 h (Figure 1b). No significant differences were observed in cells infected with a scrambled sequence compared with uninfected controls. LV-Dicer shRNA, however, induced a sustained elevation of GRP78 mRNA and protein levels at 48 h after the treatment. This elevation was significantly higher than uninfected controls (Figure 1b). Furthermore, we found that the apoptosis rate increased 48 h after stimulation in the Dicer-knockdown cells, but not in the control cells (Figure 1c). These results indicated that Dicer knockdown impaired the regression of ER stress and promoted hepatocyte apoptosis during bile acid stimulation, supporting our hypothesis that miRNAs has an essential role in the regulation of hepatic ER stress.

Dicer deficiency promotes sustained ER stress and hepatocyte apoptosis. (a) The knockdown efficiency of lentivirus encoding Dicer-targeting shRNAs (LV-shDicer) was confirmed by comparison to a NC lentivirus (LV-NC). (b) Expression levels of ER stress marker mRNAs in HL-7702/L02 cells treated with TG, DCA or dimethyl sulfoxide (DMSO) after LV-NC or shDicer infections at 10 MOI for 24 h. RNA and protein levels were assayed by real-time RT-PCR and western blot analysis; GAPDH is the normalization control. Data are represented as the mean±S.D. of three independent experiments. **P<0.01, n=3. (c) Apoptosis rates were assayed by apoptosis marker annexin-V flow cytometry in shDicer- and NC-infected HL-7702/L02 cells. Data are representative of three independent experiments
mir-199a-5p levels are elevated during hepatic ER stress
To investigate the role of miRNAs in regulating hepatic ER stress, we sought to functionally characterize specific regulatory miRNAs in bile acid-stimulated hepatocytes, focusing particularly on core factors of the ER stress process, such as GRP78, ATF4, ATF6, IRE1α, and CHOP. To accomplish this, we mined previously published microarray and high-throughput sequencing data for miRNAs elevated in cholestatic livers.24 At the same time, we used the bioinformatics tool TargetScan to search for miRNAs that target the 3′ untranslated regions (UTRs) of these core factors (Supplementary Table S1). Of the miRNAs that fit these criteria, the miR-199a/b-5p cluster emerged as an obvious candidate because of its multiple ER stress regulatory gene targets, including GRP78, ATF6, and IRE1 α (Supplementary Table S1). Although the interaction of GRP78 and miR-199a-5p has been reported recently,21 the other two targets were not yet validated. The miRNA, miR-199a-5p, has been reported as one of the most abundant hepatic miRNAs, and has been implicated in many types of liver diseases,25, 26 although its contribution to ER stress remains undefined. To address whether miR-199a/b-5p contributes to hepatic ER stress, we used real-time PCR to compare miR-199a/b-5p expression in DCA- and TG-stimulated human hepatocytes. Both DCA- and TG-treated HL-7702/L02 cells showed significantly elevated levels of miR-199a-5p, but not miR-199b-5p expression 4 and 24 h after treatment, compared with untreated cells (Figure 2a). We also confirmed the increased expression of miR-199a-5p in DCA-treated BNL-CL2 mouse hepatocytes (Supplementary Figure S1A) and bile duct-ligated mouse liver (Supplementary Figure S1B), which indicated that the functions of hepatic miR-199a-5p was conserved between mouse and human. Furthermore, a qRT-PCR kinetic expression assay indicated that the elevation of miR-199a-5p was maintained at high levels 48 h after DCA and TG treatments (Figure 2b), while GRP78, ATF6 and IRE1 α mRNA expression peaked at 12 h and rapidly decreased at 48 h (Figure 2c). These findings were consistent with our hypothesis that miR-199a-5p suppresses GRP78, ATF6 and IRE1 α mRNA. Thus, miR-199a-5p is a strong candidate to be a repressor of hepatic ER stress.

Identification of mir-199a-5p as a hepatic ER stress-related miRNA. (a) The relative level of miR-199a-5p after TG, DCA, and DMSO treatments in human hepatocyte HL-7702/L02 cells. (b and c) The kinetics of the expression levels of miR-199a-5p (b), GRP78, ATF6, and IRE1α (c) in TG- and DCA-treated HL-7702/L02 cells. The relative expression levels of RNA were quantified by qRT-PCR and normalized to U6 or GAPDH. Data are shown as means±S.D. of three independent experiments. **P<0.01. (d) The miR-199a-5p-binding site predictions for GRP78, ATF6, and IRE1α mRNAs by Targetscan 6.2. The underlined letters indicate the mutant sites. (e) Target validation using luciferase reporters in HEK293 cells. The relative luciferase activities of 3′UTR reporters containing wild-type (WT) or mutant (Mut) transcripts were assayed 48 h after co-transfection with the indicated miRNAs or scrambled NC RNA (NC). (f) QRT-PCR and western blotting analysis of GRP78, ATF6, and IRE1α mRNA and protein expression in HL-7702/L02 cells transfected with miR-199a-5p minics or NC RNA. The relative expression levels of RNA and proteins were normalized to GAPDH. For proteins, data are shown as the rate normalized to GAPDH in each sample. (g) The endogenous miR-199a-5p in HL-7702/L02 cells suppressed the luciferase activities of 3′UTR reporters containing WT, Mut or control transcripts 48 h after transfection. Data are shown as means±S.D. of three independent experiments. **P<0.01
Ectopic mir-199a-5p directly targets GRP78, ATF6, and IRE1 α 3′UTRs
To confirm the possibility that GRP78, ATF6, and IRE1α were directly targeted by miR-199a-5p, we engineered luciferase reporters that had either the wild-type 3′ UTRs of these genes, or mutant UTRs with 6-base pair deletions in the target sites (Figure 2a). These luciferase reporters were co-transfected with miRNA mimics into HEK293 cells with high-expression efficiency (Supplementary Figure S2). Scrambled RNAs were used as negative controls (NC). The miR-199a-5p mimic significantly reduced the luciferase activities of the wild-type GRP78, ATF6, and IRE1 α reporters compared with the NC (Figure 2e). In contrast, the mutant reporters were not repressed by miR-199a-5p, which indicates that the repression was directly mediated by the target sites (Figure 2e). We also analyzed the luciferase mRNA expression levels after miR-199a-5p transfection and found a similar result to luciferase activities, which indicates that miR-199a-5p may influence its target genes’ mRNA levels (Supplementary Figure S3). We also investigated the influence of ectopic miR-199a-5p on GRP78, ATF6, and IRE1 α expression in the hepatocyte cell line, HL-7702/L02. We found that the mimic of miR-199a-5p significantly reduced the mRNA and protein expression of GRP78, ATF6, and IRE1 α compared with the NC RNA (Figure 2f). Taken together, these results show that miR-199a-5p suppresses GRP78, ATF6, and IRE1 α expression by directly targeting their 3′ UTRs.
Endogenous mir-199a-5p directly targets GRP78, ATF6, and IRE1 α 3′UTRs in hepatocytes
We next studied the role of endogenous miR-199a-5p in repressing the GRP78, ATF6, and IRE1 α 3′ UTR reporters in HL-7702/L02 cells. A luciferase reporter construct with no UTR was used as a NC, as this modification makes the construct resistant to any hepatic miRNA repression. In normal hepatocytes, at 24 h after transfection, there was significant repression of the wild-type GRP78, ATF6, and IRE1 α 3′ UTR luciferase reporter activities in comparison with the no-UTR control, and these effects persisted after 48 h (Figure 2g). In contrast, the mutant reporters were significantly less repressed than the wild-type reporters (Figure 2g). We conclude that endogenous miR-199a-5p in hepatocytes is sufficient to directly repress GRP78, ATF6, and IRE1 α 3′ UTR reporters.
Endogenous mir-199a-5p directly targets GRP78, ATF6, and IRE1 α 3′UTRs during hepatic ER stress
To gain insight into the biological role of miR-199a-5p in hepatic ER stress, we studied the repression of GRP78, ATF6, and IRE1 α 3′ UTR reporters by miR-199a-5p in hepatocytes under ER stress conditions. Six hours after the transfection of the reporters, we stimulated the cells with either DCA or TG to induce ER stress. We found that compared with the no-UTR control, there was significant repression of the GRP78, ATF6, and IRE1 α wild-type 3′ UTR luciferase reporter activities 24 h after transfection, and that these effects persisted at the 48-h timepoint (Figure 3a). In contrast, the mutant reporters were significantly less repressed than the wild-type reporters. Furthermore, we used an miRNA antisense inhibitor to abolish the elevation of miR-199a-5p induced by DCA and TG. We confirmed by qRT-PCR that miR-199a-5p levels were significantly downregulated 24 h after inhibitor transfection in normal and DCA- and TG-treated hepatocytes (Figure 3b). We then treated miR-199a-5p inhibitor-transfected hepatocytes with DCA and TG 24 h after transfection with scrambled RNA as a control. The depletion of miR-199a-5p in hepatocytes significantly reversed the decrease in wild-type 3′UTR reporter luciferase activity 24 h after DCA (Figure 3c) and TG treatment (Figure 3d), indicating that the targeted UTR reporter repression during hepatic ER stress was mainly mediated by miR-199a-5p. All these results demonstrated that endogenous miR-199a-5p exerts post-transcriptional repression on GRP78, ATF6, and IRE1 α during hepatic ER stress.

Endogenous mir-199a-5p directly regulates GRP78, ATF6, and IRE1α 3′UTRs during hepatic ER stress. (a) The endogenous miR-199a-5p in HL-7702/L02 (short to L02) cells suppressed the luciferase activities of WT 3′UTR reporters compared with Mut or control transcripts 24 h after TG, DCA, or DMSO treatments. (b) The miR-199a-5p inhibitor (inh) suppressed the elevation of endogenous miR-199a-5p compared with NC RNA (NC) 24 h after DCA or TG treatments. (c and d) The endogenous miR-199a-5p in HL-7702/L02 (short to L02) cells suppressed the luciferase activities of the WT 3′UTR but not the mutant or control reporters 24 h after DCA (c) and TG (d) treatments. The relative luciferase activities of 3′UTR reporters were assayed with dual-luciferase assays. Data are shown as means±S.D. of three independent experiments with similar results. *P<0.05, **P<0.01
mir-199a-5p suppression facilitated sustained ER stress and cell death
Next, we examined the effect of miR-199a-5p loss on the regulation of hepatic ER stress. We employed the miRNA inhibitor to abolish the elevation of miR-199a-5p, and then treated miR-199a-5p inhibitor-transfected hepatocytes with DCA or TG 24 h after transfection, using scrambled RNA as a control. At 8 h after treatment, we found that GRP78 and CHOP mRNA levels were all significantly increased in both miR-199a-5p inhibitor- and scramble RNA-transfected cells, which indicated the induction of ER stress (Figures 4a and b). At 48 h after treatment, these mRNAs were significantly decreased in control cells. However, in TG- and DCA-treated miR-199a-5p-deficient cells, the mRNA levels of GRP78 and CHOP remained elevated, indicating sustained ER stress (Figures 4a and b). We also confirmed the extent of ER stress by GRP78 and CHOP protein levels, and also found a sustained ER stress in miR-199a-5p-deficient cells after 48 h, which is consistent with the observed mRNA levels (Figure 4c). Furthermore, the miR-199a-5p-deficient cells had a higher rate of apoptosis, as assessed by Annexin A5 straining (Figure 4d) and a higher rate of cell death, as accessed by LDH release (Figure 4f) than the control cells after DCA or TG treatment. Our results indicate that the disruption of miR-199a-5p upregulation during bile acid or TG stimulation leads to the direct deregulation of target expression and the indirect disruption of ER stress, and apoptosis progression. Thus, miR-199a-5p is necessary to inhibit sustained ER stress and apoptosis.

Suppression of miR-199a-5p promoted sustained ER stress and apoptosis in DCA and TG treated hepatocytes. (a and b) Quantitative PCR analysis of GRP78 and CHOP mRNA levels in miR-199a-5p inh- or scrambled RNA (NC)-transfected hepatocytes after TG, DCA, or DMSO treatment for the indicated times. The mRNA levels were normalized to GAPDH in each sample. Data are shown as means±S.D. of three independent experiments. **P<0.01. (c) Western blotting analysis of GRP78 and CHOP proteins in miR-199a-5p inh- or scrambled RNA (NC)-transfected hepatocytes HL-7702/L02 (short to L02) after DCA or TG treatment for the indicated times. Data are shown as the level normalized to GAPDH in each sample. (d) Apoptotic rates were assayed by flow cytometry in miR-199a-5p inh- or scrambled RNA (NC)-transfected hepatocytes after DCA or TG treatment for 48 h. Data are representative of three independent experiments. (e) Cell death levels of siRNAs-(si-) or NC-RNA-transfected HL-7702/L02 cells were assayed by LDH release after DCA or TG treatment for 48 h. Data are shown as means±S.D. of three independent experiments. **P<0.01, ref to NC-RNA-transfected cells. (f) Cell death levels of miR-199a-5p inh- or NC-RNA-transfected HL-7702/L02 cells were assayed by LDH release after DCA or TG treatment for 48 h. The efforts of miR-199a-5p inh were also rescued by siRNAs targeting to GRP78, ATF6 or IRE1α with scramble RNA as a control. Data are shown as means±S.D. of three independent experiments. **P<0.01
mir-199a-5p protects hepatocytes from cell death by suppressing IRE1α
We next tested whether the protective effects of miR-199a-5p were mediated directly by the suppression of GRP78, ATF6, and IRE1α expression. As such, we employed siRNA-mediated knockdown approaches to investigate the role of GRP78, ATF6, or IRE1α in the DCA or TG-induced cell death. We found that only IRE1α knockdown (about 80% depletion) (Supplementary Figure S4) resulted in decreased cell death rates in DCA- or TG-treated cells (Figure 4e). In contrast, GRP78 and ATF6 knockdown (about 76 and 81% depletion, respectively) (Supplementary Figure S4) increased cell death rate after both DCA and TG treatment (Figure 4e). This finding indicated that although GRP78, ATF6, and IRE1α are all targeted by miR-199a-5p, these three genes may have different roles during ER stress and the subsequent reactions. IRE1α may function to promote cell death during ER stress, which is consistent with the previous reports.
At the same time, we also tried to use GRP78, ATF6, or IRE1α-specific siRNAs to abolish the sustained elevation of these genes during miR-199a-5p inhibition, and then observe whether the loss of these genes could rescue the effects of miR-199a-5p inhibitors. Compared with control cells, we found that loss of IRE1α partially rescued the DCA- or TG-induced cell death in miR-199a-5p-deficient cells, whereas loss of GRP78 and ATF6 significantly enhanced the rates of cell death in miR-199a-5p-deficient cells (Figure 4f). Our results indicate that the disruption of miR-199a-5p upregulation during bile acid or TG stimulation leads to the sustained ER stress, and that cell death progression is mainly through the direct regulation to IRE1α.
Transcriptional regulation of miR-199a-5p by AP-1
To determine the exact mechanisms of DCA- and TG-induced miR-199a upregulation, we further analyzed primary (pri-) and premature (pre-) miR-199a-5p transcripts in treated hepatocytes. Mature miR-199a-5p has two genetic loci. Six hours post-treatment, both pri- and pre-miR-199A2 were significantly upregulated (Figure 5a), suggesting de novo miR-199a transcription. At the same time, pri- and pre-miR-199A1 were not significantly increased, indicating that MIR-199A2 was the major contributing genetic locus for miR-199a5p production in these cell lines. We next investigated, which pathways participate in the transcription of pri-miR-199a2 in DCA- and TG-treated hepatocytes. Computational transcription factor-binding site prediction in the promoter region suggested that AP-1 may regulate MIR-199A2 (Supplementary Figure S5). Additionally, AP-1 has been reported to be activated during bile acid stimulation,27 and moreover, c-Jun protects against sustained hepatic ER stress to promote hepatocyte survival.28 We then validated this prediction by direct promoter analysis (Figure 5b) using luciferase reporter assays.. We found that the wild-type MIR-199A2 promoter showed elevated transcription during DCA stimulation, while mutant promoters with one or two mutant AP-1 binding sites had significantly weaker activity (Figure 5b, Supplementary Figure S6). We also performed chromatin immunoprecipitation (ChIP) assays to confirm that AP-1 was present at the promoter region of MIR-199A2. We found that AP-1 bound to the MIR-199A2 promoter under basal conditions, but the amount of bound AP-1 was greatly increased 12 h after both DCA and TG stimulation (Figure 5c). We therefore confirmed that AP-1 is a strong positive regulator of the MIR-199A2 gene during ER stress. As AP-1 is directly activated by JNK (c-Jun N-terminal kinase), we also inhibited AP-1 activation with the JNK chemical inhibitor SP600125. We observed that the elevation of pri-, pre-, and mature miR-199a2 expression (Figure 5d) and the increased promoter reporter activity (Figure 5e) during ER stress was abolished by SP600125 treatment. These data indicate that DCA and TG-induced de novo miR-199a expression in hepatocytes primarily through the AP-1 pathway.

AP-1 directly regulates miR-199a-2 transcription. (a) Quantitative PCR analysis of primary (pri-) and pre-mature (pre-) miR-199a-1 and miR-199a-2 levels in DCA- and TG-treated hepatocytes after 24 h. The mRNA levels were normalized to GAPDH in each sample. (b) Predicted AP-1-binding sites in the miR-199A2 promoter are shown. Reporter vectors containing wild-type or deleted or mutant promoters were constructed, and luciferase activities were detected in hepatocytes after TG, DCA, or DMSO treatment. (c) ChIP analysis showed high AP-1 (c-Jun) enrichment at the MIR199A2 promoter in HL-7702/L02 cells. Enriched DNA was assayed with qRT-PCR and relative enrichment is normalized to control IgG. The positions of the PCR amplicon are labeled according to the predicted AP-1 binding sites. **P<0.01, n=3. (d) Quantitative PCR analysis of pri- or pre-miR-199a2, and mature miR-199a-5p levels in AP-1 inh SP600125-treated hepatocytes after DCA or TG treatment for 24 h. The mRNA levels were normalized to GAPDH in each sample. (e) The relative luciferase activities of promoter reporters in AP-1 inh SP600125-treated hepatocytes after DCA or TG treatment for 24 h. Data are shown as means±S.D. of three independent experiments. **P<0.01
Ectopic miR-199-5p partly rescued AP-1-deficient hepatocytes from ER stress and cell death
Previous studies have highlighted that AP-1 activation protects hepatocytes from the excessive activation of the ER stress response and subsequent cell death. We therefore examined whether miR-199a-5p elevation has a role in the AP-1 pathway during hepatic ER stress. We confirmed that miR-199a-5p suppression sensitized hepatocytes to bile acid-induced apoptosis, which suggests that miR-199a-5p elevation may be essential for AP-1-mediated prevention of cell death. To further investigate the role of miR-199a-5p in the protective effects of AP-1, we used SP600125 to inhibit AP-1 activation and investigated the ability of miR-199a-5p mimics to ameliorate cell death in response to ER stress, using scrambled RNA as a control. After culturing hepatocytes with DCA or TG for 3 days, SP600125 treatment in LO2 cells triggered a strong increase in GRP78 and CHOP mRNA levels, both of which are markers of ER stress (Figure 6a). At the same time, cell death also increased in SP600125-treated LO2 cells, as shown by LDH (lactate dehydrogenase) release assays (Figure 6b). However, miR-199a-5p significantly suppressed the elevation of ER stress markers compared with controls (Figure 6a). Accordingly, cell death was inhibited in LO2 cells transfected with miR-199a-5p mimics (Figure 6b). These data provide further evidence that ectopic miR-199a-5p is sufficient to rescue the phenotype of AP-1-inhibited cells during ER stress, and suggests that miR-199a-5p may be a novel endogenous protective agent for hepatocytes during hepatic ER stress.

Ectopic miR-199-5p protects hepatocytes from bile acid-induced ER stress and cell death. HL-7702/L02 (short to L02) cells were transfected with miR-199a-5p mimics or NC with or without SP600125 treatment after culture with TG, DCA, or DMSO for 48 h. (a) Detection of the mRNA ER stress markers GRP78 and CHOP with quantitative RT-PCR. Data are shown as means±S.D. of three independent experiments. **P<0.01. (b) HL-7702/L02 (short to L02) cell death levels were assayed by LDH release. Data are shown as means±S.D. of three independent experiments. **P<0.01
Discussion
In this study, we observed that miR-199a-5p levels are strongly elevated in human and mouse hepatocytes following bile acid and TG stimulation. MiR-199a-5p is one of the most broadly conserved miRNAs and is generally believed to be a multifunctional miRNA involved in the regulation of angiogenesis,29 cell proliferation,30, 31 autophagy32, and many other cell functions.33 Interestingly, miR-199a-3p, another miRNA generated from the same stem-loop RNA as miR-199a-5p, is also involved in the regulation of many biological processes and diseases.30, 34, 35 In particular, miR-199a-3p has been identified as one of the most abundant miRNAs in normal hepatocytes,36 where it regulates apoptosis, cell growth, and invasion.34, 35, 36 These findings also suggest a role for hepatic miR-199a-5p in the same biological and pathological processes. Here, we reported that under bile acid or TG stimulation, miR-199a-5p may also act to reduce hepatic ER stress.
Recently, Su et al.21 demonstrated that miR-30d, miR-181a, and miR-199a-5p co-operatively suppress GRP78 in cancer. We also validated their finding that miR-199a-5p targets GRP78. However, we found that miR-199a-5p also targets the UPR transducers IRE1α and ATF6. The multiple substrates of miR-199a-5p further indicate its key role in managing ER stress. Interestingly, we also found that these three target genes have different roles during hepatic ER stress, and that IRE1α contributes to cell death induced by sustained ER stress. We therefore suggest that the protective effects of miR-199a-5p are mainly mediated through suppression of IRE1α expression.
We also predicted other targets for miR-199a-5p by Targetscan. Interesting, 33 genes among the 496 well-conserved, predicted targets of miR-199a-5p were related to cell death as predicted by the DAVID database (http://david.abcc.ncifcrf.gov/). Among these genes, DNA-damage regulated autophagy modulator 1 (DRAM1)37 and cyclin-dependent kinase inhibitor 1B (p27, also known as Kip1)38 are known to function as positive regulators of apoptosis and cell death. We therefore hypothesize that miR-199a-5p also suppresses DRAM1 and p27 expression to protect hepatocytes from ER stress-induced apoptosis; this may be the focus of future studies.
Previous research had indicated that AP-1 activation protects hepatocytes from the excessive activation of the ER stress response and subsequent cell death.28 In our current study, we also found that the induction of miR-199a-5p requires activation of AP-1. We therefore suggest that the elevation of miR-199a-5p contributes to the AP-1-mediated attenuation of the ER stress response and subsequent cell death. We further suggest that there may be feedback regulation during the hepatic ER stress response (Figure 7). First, ER stress could induce JNK and AP-1 activation, and, in-turn, AP-1 could induce miR-199a-5p expression. Elevated miR-199a-5p could then inhibit ER stress by targeting ER chaperones and signal transducers. This feedback loop may shield hepatocytes from sustained ER stress and protect the liver from injury.

Model for the AP-1/miR-199a-5p/ER-stress-mediators regulatory loop in the modulation of hepatic ER stress. Unfolded protein-induced ER stress response could result in both the elevation of the ER mediators’ mRNA and the activation of AP-1, which could in turn induce miR-199a-5p expression. (a) In normal hepatocytes, elevated miR-199a-5p could negatively regulate ER stress by targeting the mRNAs of IRE1α, and then promotes hepatocytes survival. (b) In hepatocytes treated with miR-199a-5p inh, this feedback loop may be broken and unfolded protein may induce uncontrolled sustained ER stress, which results in hepatocyte apoptosis and cell death
In summary, our results demonstrate that bile acid can induce the de novo expression of miR-199a-5p, most likely through the JNK/AP-1 pathway. AP-1-induced miR-199a-5p then directly targets the 3′UTRs of GRP78, IRE1 α, and ATF6 mRNAs, and prevents sustained ER stress and cell death, mainly through the negative regulation of IRE1α. Inhibitors of miR-199a-5p abolished this protective effect and triggered a more potent ER stress response, which promoted cell death in vitro. Our data suggest that miR-199a-5p and AP-1 work together to mediate ER stress during bile acid and TG stimulation. The miRNA miR-199a-5p may constitute a potential target for clinical approaches aiming to protect hepatocytes in liver disease.
Materials and Methods
Mice, cell lines and reagents
Female C57BL/6 mice (at 6–8 weeks of age) were purchased from the SMMU Laboratory Animal Center. The mice were maintained and used in accordance with the Institutional guidelines for animal care. The human embryonic kidney epithelial cell line HEK293 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in DMEM medium containing 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA). The transformed human hepatocyte line HL-7702/L02 and mouse hepatocyte line BNL-CL2 (ATCC) were maintained in minimum essential medium (MEM) supplemented with 10% FBS, 50 μg/ml streptomycin, 50 U/ml penicillin, 100 μM MEM nonessential amino acids, 2 mM l-glutamine, and 1 mM sodium pyruvate (all from Invitrogen. Chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, USA), except where specifically indicated. MicroRNA mimics and inhibitors were purchased from Genepharma (Shanghai, China). Anti-GRP78 (Bip), anti-ATF6, anti-IRE1A, and anti-CHOP antibodies were purchased from Cell Signaling Technology (CST, Beverly, MA, USA).
ER stress models
ER stress was induced by incubating cells with either thapsigargin (TG, 1 μM, Sigma) or deoxycholic acid (DCA, 200 μM, Sigma) for at least 4 h. For the in vivo cholestatic liver model, bile duct-ligated mouse liver and sham operation mouse liver were obtained 24 h after treatment.
Cytotoxicity assays
Hepatocyte damage was determined by the analysis of LDH in cell culture supernatant using semiautomated and routine clinical methods.
RNA preparation, reverse transcription and quantitative real time-PCR
Total RNA was extracted using Trizol (Invitrogen). The miRNA levels were assayed with Taqman probes and primer sets (AB: Foster City, CA, USA) according to the manufacturer’s instructions. For mRNA analysis, first-strand cDNA was generated using the Reverse Transcription System Kit Promega (Madison, WI, USA) with random primers for RT-PCR or real-time PCR using Power SYBR Green PCR Master Mix (Applied Biosystems) in a StepOne Plus system (Applied Biosystems). U6 snoRNA or GAPDH mRNA levels were used as internal normalization controls. The primers used for PCR were: GRP78: 5′-GGAGGAGGACAAGAAGGA-3′ and 5′-AGTGAAGGCGA CATAGG A-3′; CHOP: 5′-CCTCACTCTCCAGATTCC A-3′ and 5′-AGCCGTTCATTCTCTTCAG-3′; IRE1A: 5′-GGTCTGAGGAAGGTGATG-3′ and 5′-CATAGAGGCTGGTAGAGTATT-3′; ATF6: 5′-AACAAGACCACAAGACCAA-3′ and 5′-AGGAGGAACTGACGAACT-3′; XBP1(S): 5′-ATGGATTCTGGCGGTATTG-3′ and 5′-GGAGGCTGGTAAGGAACT-3′; GAPDH: 5′-CGGATTTGGTCGTATTGGG-3′ and 5′-CTG GAAGATGGTGATGGGATT-3′; Firefly luciferase: 5′- GCATACGACGATTCTGTGATTTG-3′ and 5′-GATACGCCCTGGTTCCTGG-3′; pri-miR-199A1: 5′-CCGCTCTGTCCCTTCTGACG-3′ and 5′-AAACCCTGCCTCCTGCTCC-3′; pre-miR-199A1: 5′-GCCAACCCAGTGTTCAGACTAC-3′ and 5′-GCCTAACCAATGTGCAGACTACT-3′; pri-miR-199A2: 5′-TGCCCAGTCTAACCAATGTGC-3′ and 5′-AGCTGAATGCAACCCCTGG-3′; and pre-miR-199A2: 5′-AGCTTCTGGAGATCCTGCTCC-3′ and 5′-TGCTCTCCCTTGCCCAGTCT-3′.
Luciferase reporter assay
For luciferase reporter assays, wild-type GRP78, ATF6, or IRE1A mRNA 3′-UTRs were amplified from HL-7702/L02 total cDNA and cloned into the pGL3-promoter vector (Promega) using the Xba I site. Wild-type and AP-1-binding-site-1-deleted promoters of MIR199A2 were amplified from HL-7702/L02 genomic DNA and cloned into the pGL3 basic vector (Promega) using the XhoI and HindIII site. The following primers were used: GRP78 3′UTR: 5′-TCTAGACTTTTCATTAGCAGTTGCTCACA-3′ and 5′-TCTAGACCCAACATACCAAATACTCCCTC-3′; ATF6 3′UTR: 5′-TCTAGAGATCAATGGGCAGGACTACGA-3′ and 5′-TCTAGACCAAATAGATGGGTAGATGATGAAA-3′; and IRE1A 3′UTR: 5′-TCTAGAGACCTACAGGGAACTGGGAAGA-3′ and 5′-TCTAGAATGGCTCCTGCTGCCTCTG-3′. Pro-MIR199A2 WT: 5′-CTCGAGGCACAGCATTCAGAGACCCAG-3′ and 5′-AAGCTTCAGTCTTGACGTGGCACATTTG-3′; Δ-1: 5′-CTCGAGGCTTTTCCATACTGGGGCCCAC-3′ and 5′-AAGCTTCAGTCTTGACGTGGCACATTT G-3′. The pGL3-Report constructs containing these 3′UTRs with 6-base mutations in the seed sequence and the constructs containing Pro-MIR199A2 Δ-2 (construct Δ-1 with 7-base mutations in the AP-1 binding site 2) were synthesized with QuikChange site-directed mutagenesis kits (Agilent Technologies, Palo Alto, CA, USA). These reporters were transfected into HEK293 and HL-7702/L02 cells using Lipofectamine 2000 (Invitrogen). During transfection, cells (5 × 103) were seeded into 96-well plates and incubated overnight. Cells were then co-transfected with 80 ng wild-type (WT) or mutant (Mut) reporter vector, 8 ng internal control pRL-TK-Renilla-luciferase plasmid and the indicated RNAs (final concentration, 50 nM). After 24 or 48 h, luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Data were normalized by dividing firefly luciferase activity by Renilla luciferase activity, as reported previously.
Chromatin immunoprecipitation assay
ChIP assays were performed according to the manufacturer’s instructions (EZ-Magna ChIP A/G Kit—Merck Millipore Headquarters (Billerica, MA, USA)). Chromatin was immunoprecipitated using anti-c-Jun (Abcam, Cambridge, MA, USA) with control total human IgG Santa Cruz Technologies (Santa Cruz, CA, USA). ChIP-derived DNA was quantified using real time PCR with SYBR Green incorporation (Applied Biosystems). The promoter region of MIR199A2 was analyzed according to the previous reports and information from the UCSC website. Two pairs of primers were designed to detect the enriched genomic DNA fragments. Fold enrichments were calculated from the apparent IP efficiency (ratio of ChIP enriched DNA over control IgG input DNA) and normalized to the level at a control region. The primer sequences are as follows: primer pair 1 sense: 5′-CAAATGAGC TCTGCC TTCCAGTG-3′; anti-sense: 5′-GGGCCCCAGTATGGAAAAGC-3′; primer pair 2 sense: 5′-AGAGCAGGCGATTCTAGCGGT-3′; anti-sense: 5′-TCCAGTCTTGACGTGGCACATT-3′; control prime sense: 5′-GAGGTCTCGTATTTGCTGCATCGTA-3′; anti-sense: 5′-GCTAATTTCCTTCTCCACCCCAACCA-3′.
Lentivirus preparation and transduction
Lentiviral vectors were purchased from Genepharma (Shanghai, China). All lentivirus batches used for experiments had comparable titers ranging from 1 × 108 to 1 × 109 transducing functional U/ml. Viral suspensions were stored at −80 °C until use and briefly centrifuged and kept on ice immediately before use. For infection, 2 × 105 cells were transduced 1 day after initial seeding of the cells at a multiplicity of infection of 10. Cells were incubated in pluripotent maintenance media containing lentiviral particles and 4 μg/ml polybrene (Sigma Aldrich St. Louis, MO, USA) for 18 h at 37 °C in a humidified atmosphere containing 5% CO2. The sequences of the shRNAs are as follows: dicer-shRNA sense: 5′-CCGGTAAGGGCACCCATCTCTAATTACTCGAGTAATTAGAGATGGGTGCCCTTTTTTTG-3′; antisense: 5′-AATTCAAAAAAAGGGCACCCATCTCTAATTACTCGAGTAATTAGAGATGGGTGCCCTTA-3′; NC-shRNA sense: 5′-CCGGTTTCTCCGAACGTGTCACGTCTCGAGACGTGACACGTTCGGAGAATTTTTG-3′; antisense: 5′-AATTCAAAAATTCTCCGAACGTGTCACGTCTCGAGACGTGACACGTTCGGAGAAA-3′.
Flow cytometry analysis of apoptosis
For apoptosis analysis, 1–5 × 105 cells from each sample were processed with Annexin V-PE (Annexin V-PE apoptosis detection kit, BioVision, Inc. (Palo Alto, CA, USA)) according to the manufacturer’s instructions. The cell population of interest was determined and dead cells were excluded using forward and side scatter parameters. A total of 30 000 events were acquired per sample. The data were analyzed with FACSDiva software (version 4.1.2; BD Biosciences). Triplicate samples were analyzed in each experiment. All the samples were analyzed using BD FACSCalibur equipment BD Bioscience (San Jose, CA, USA) according to instructions from facility instrument technicians.
Western blotting analysis
Total cell lysates were prepared in a 1x sodium dodecyl sulfate buffer. Identical quantities of proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes.
Statistical analysis
Data are presented as the mean±S.D. of three independent experiments with similar results. For qRT-PCR, WB and luciferase activity data, statistical comparisons between experimental groups were performed with ANOVA analysis and Fisher’s exact test or two-tailed Student’s t-test. P<0.01 was considered statistically significant.
Abbreviations
- ER:
-
endoplasmic reticulum
- UPR:
-
unfolded protein response
- EIF2AK3:
-
eukaryotic translation initiation factor 2-alpha kinase 3
- IRE1A:
-
endoplasmic reticulum to nucleus signaling 1
- ATF6:
-
activating transcription factor 6
- XBP1:
-
X-box-binding protein 1
- CHOP:
-
C/EBP homologous protein
- miRNA:
-
microRNAs
- UTR:
-
untranslated region
- ShRNA:
-
hairpin rna
- DCA:
-
deoxycholic acid
- TG:
-
thapsigargin
- qRT:
-
qualitative reverse transcription
- JNK:
-
c-Jun N-terminal kinase
- LDH:
-
lactate dehydrogenase
References
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D . Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2000; 2: 326–332.
Oikawa D, Kimata Y, Kohno K, Iwawaki T . Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res 2009; 315: 2496–2504.
Sidrauski C, Cox JS, Walter P . tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 1996; 87: 405–413.
Cox JS, Shamu CE, Walter P . Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993; 73: 1197–1206.
Messner DJ, Kowdley KV . Biting the iron bullet: endoplasmic reticulum stress adds the pain of hepcidin to chronic liver disease. Hepatology 2010; 51: 705–707.
Ji C, Kaplowitz N . ER stress: can the liver cope? J Hepatol 2006; 45: 321–333.
Malhi H, Kaufman RJ . Endoplasmic reticulum stress in liver disease. J Hepatol 2011; 54: 795–809.
Wang Y, Vera L, Fischer WH, Montminy M . The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 2009; 460: 534–537.
Birkenfeld AL, Lee HY, Majumdar S, Jurczak MJ, Camporez JP, Jornayvaz FR et al. Influence of the hepatic eukaryotic initiation factor 2alpha (eIF2alpha) endoplasmic reticulum (ER) stress response pathway on insulin-mediated ER stress and hepatic and peripheral glucose metabolism. J Biol Chem 2011; 286: 36163–36170.
Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J 2011; 30: 1357–1375.
Ai D, Baez JM, Jiang H, Conlon DM, Hernandez-Ono A, Frank-Kamenetsky M et al. Activation of ER stress and mTORC1 suppresses hepatic sortilin-1 levels in obese mice. J Clin Invest 2012; 122: 1677–1687.
Ramirez T, Tong M, Chen WC, Nguyen QG, Wands JR, de la Monte SM . Chronic alcohol-induced hepatic insulin resistance and ER stress ameliorated by PPAR-delta agonist treatment. J Gastroenterol Hepatol 2012; 28: 179–187.
Hofmann AF . Cholestatic liver disease: pathophysiology and therapeutic options. Liver 2002; 22 (Suppl 2): 14–19.
Reinehr R, Graf D, Haussinger D . Bile salt-induced hepatocyte apoptosis involves epidermal growth factor receptor-dependent CD95 tyrosine phosphorylation. Gastroenterology 2003; 125: 839–853.
Schmucker DL, Ohta M, Kanai S, Sato Y, Kitani K . Hepatic injury induced by bile salts: correlation between biochemical and morphological events. Hepatology 1990; 12: 1216–1221.
Bernstein H, Payne CM, Bernstein C, Schneider J, Beard SE, Crowley CL . Activation of the promoters of genes associated with DNA damage, oxidative stress, ER stress and protein malfolding by the bile salt, deoxycholate. Toxicol Lett 1999; 108: 37–46.
Bochkis IM, Rubins NE, White P, Furth EE, Friedman JR, Kaestner KH . Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nat Med 2008; 14: 828–836.
Tamaki N, Hatano E, Taura K, Tada M, Kodama Y, Nitta T et al. CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am J Physiol Gastrointest Liver Physiol 2008; 294: G498–G505.
Bartel DP . MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–297.
Ouyang YB, Lu Y, Yue S, Xu LJ, Xiong XX, White RE et al. miR-181 regulates GRP78 and influences outcome from cerebral ischemia in vitro and in vivo. Neurobiol Dis 2012; 45: 555–563.
Su SF, Chang YW, Andreu-Vieyra C, Fang JY, Yang Z, Han B et al. miR-30d, miR-181a and miR-199a-5p cooperatively suppress the endoplasmic reticulum chaperone and signaling regulator GRP78 in cancer. Oncogene 2012.
O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D . Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol 2010; 10: 111–122.
Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O'Leary JJ, Ruan Q et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol 2010; 11: 141–147.
Kanda T, Ishibashi O, Kawahigashi Y, Mishima T, Kosuge T, Mizuguchi Y et al. Identification of obstructive jaundice-related microRNAs in mouse liver. Hepatogastroenterology 2010; 57: 1013–1023.
Murakami Y, Toyoda H, Tanaka M, Kuroda M, Harada Y, Matsuda F et al. The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS One 2011; 6: e16081.
Shen Q, Cicinnati VR, Zhang X, Iacob S, Weber F, Sotiropoulos GC et al. Role of microRNA-199a-5p and discoidin domain receptor 1 in human hepatocellular carcinoma invasion. Mol Cancer 2010; 9: 227.
Bernt C, Vennegeerts T, Beuers U, Rust C . The human transcription factor AP-1 is a mediator of bile acid-induced liver cell apoptosis. Biochem Biophys Res Commun 2006; 340: 800–806.
Fuest M, Willim K, MacNelly S, Fellner N, Resch GP, Blum HE et al. The transcription factor c-Jun protects against sustained hepatic endoplasmic reticulum stress thereby promoting hepatocyte survival. Hepatology 2012; 55: 408–418.
Chan YC, Roy S, Huang Y, Khanna S, Sen CK . The microRNA miR-199a-5p down-regulation switches on wound angiogenesis by de-repressing the v-ets erythroblastosis virus E26 oncogene homolog 1 -matrix metalloproteinase-1 pathway. J Biol Chem 2012; 287: 41032–41043.
Shatseva T, Lee DY, Deng Z, Yang BB . MicroRNA miR-199a-3p regulates cell proliferation and survival by targeting caveolin-2. J Cell Sci 2011; 124 (Pt 16): 2826–2836.
Tsukigi M, Bilim V, Yuuki K, Ugolkov A, Naito S, Nagaoka A et al. Re-expression of miR-199a suppresses renal cancer cell proliferation and survival by targeting GSK-3beta. Cancer Lett 2012; 315: 189–197.
Xu N, Zhang J, Shen C, Luo Y, Xia L, Xue F et al. Cisplatin-induced downregulation of miR-199a-5p increases drug resistance by activating autophagy in HCC cell. Biochem Biophys Res Commun 2012; 423: 826–831.
Haghikia A, Missol-Kolka E, Tsikas D, Venturini L, Brundiers S, Castoldi M et al. Signal transducer and activator of transcription 3-mediated regulation of miR-199a-5p links cardiomyocyte and endothelial cell function in the heart: a key role for ubiquitin-conjugating enzymes. Eur Heart J 2011; 32: 1287–1297.
Henry JC, Park JK, Jiang J, Kim JH, Nagorney DM, Roberts LR et al. miR-199a-3p targets CD44 and reduces proliferation of CD44 positive hepatocellular carcinoma cell lines. Biochem Biophys Res Commun 2010; 403: 120–125.
Fornari F, Milazzo M, Chieco P, Negrini M, Calin GA, Grazi GL et al. MiR-199a-3p regulates mTOR and c-Met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res 2010; 70: 5184–5193.
Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell 2011; 19: 232–243.
Crighton D, Wilkinson S, Ryan KM . DRAM links autophagy to p53 and programmed cell death. Autophagy 2007; 3: 72–74.
Zhang J, Cheng C, He CL, Zhou YJ, Cao Y . The expression of Bcl-XL, Bcl-XS and p27Kip1 in topotecan-induced apoptosis in hepatoblastoma HepG2 cell line. Cancer Invest 2008; 26: 456–463.
Acknowledgements
This work was supported by Grant 81172304 and Grant 31201110 from the National Natural Science Foundation of China; Grant 2010127 from Science Foundation of Shanghai Municipal Public Health Bureau, China.
Author contributions
Bing-Hua Dai, Li Geng, Yue Wang: Conception and design, collection of data, data analysis and interpretation, manuscript writing, financial support; Cheng-Jun Sui, Feng Xu: Provision of study material, collection of data; Tong Kan: Collection of data, interpretation; Wei-Feng Shen: Provision of study material; Jia-Mei Yang: Conception and design, data analysis and interpretation, administrative support, final approval of manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by Y Shi
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Dai, BH., Geng, L., Wang, Y. et al. microRNA-199a-5p protects hepatocytes from bile acid-induced sustained endoplasmic reticulum stress. Cell Death Dis 4, e604 (2013). https://doi.org/10.1038/cddis.2013.134
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.134
Keywords
This article is cited by
-
The protective microRNA-199a-5p-mediated unfolded protein response in hypoxic cardiomyocytes is regulated by STAT3 pathway
Journal of Physiology and Biochemistry (2019)
-
Involvement of methylation of MicroRNA-122, −125b and -106b in regulation of Cyclin G1, CAT-1 and STAT3 target genes in isoniazid-induced liver injury
BMC Pharmacology and Toxicology (2018)
-
MiR-199a Inhibits Secondary Envelopment of Herpes Simplex Virus-1 Through the Downregulation of Cdc42-specific GTPase Activating Protein Localized in Golgi Apparatus
Scientific Reports (2017)
-
microRNA let-7c is essential for the anisomycin-elicited apoptosis in Jurkat T cells by linking JNK1/2 to AP-1/STAT1/STAT3 signaling
Scientific Reports (2016)
-
Identification of a microRNA (miR-663a) induced by ER stress and its target gene PLOD3 by a combined microRNome and proteome approach
Cell Biology and Toxicology (2016)
