
Abstract
Traumatic brain injury is a major public health concern and is characterised by both apoptotic and necrotic cell death in the lesion. Anatomical imaging is usually used to assess traumatic brain injuries and there is a need for imaging modalities that provide complementary cellular information. We sought to non-invasively image cell death in a mouse model of traumatic brain injury using a near-infrared fluorescent conjugate of a synthetic heat shock protein-90 alkylator, 4-(N-(S-glutathionylacetyl) amino) phenylarsonous acid (GSAO). GSAO labels both apoptotic and necrotic cells coincident with loss of plasma membrane integrity. The optical GSAO specifically labelled apoptotic and necrotic cells in culture and did not accumulate in healthy organs or tissues in the living mouse body. The conjugate is a very effective imager of cell death in brain lesions. The optical GSAO was detected by fluorescence intensity and GSAO bound to dying/dead cells was detected from prolongation of the fluorescence lifetime. An optimal signal-to-background ratio was achieved as early as 3 h after injection of the probe and the signal intensity positively correlated with both lesion size and probe concentration. This optical GSAO offers a convenient and robust means to non-invasively image apoptotic and necrotic cell death in brain and other lesions.
Similar content being viewed by others


Main
Traumatic brain injury is estimated to affect 2% of the US population and cost about $60 billion annually.1, 2 The nature of the injuries depends on the type, severity and location of the insult but are usually characterised by primary mechanical damage and haemorrhage followed by secondary damage because of disruption of the blood–brain barrier and associated inflammation.3 The secondary insult occurs hours to days after the primary insult and is amenable to therapeutic intervention. Computer tomography and magnetic resonance imaging are used to assess morphological changes in traumatic brain injury that occur in the later stages of the injury.4 Treatment of the secondary damage would be facilitated by imaging modalities that report on changes in the lesion at the cellular level. Traumatic brain injury is characterised by both apoptotic and necrotic cell death in the lesion3 and this is what we sought to image in this study.
An established model of traumatic brain injury in mice is the cryolesion. A sharp delineated area of neuronal cell death is induced in the mouse brain by applying a liquid nitrogen cooled rod to the skull.5 Cell death within brain cryolesions occurs via two distinct mechanisms. First, intracellular ice crystals rupture the cell membrane causing direct cell damage.6 Second, injury to endothelial cell junctions results in increased vascular permeability, oedema, thrombosis and ultimately failure of the microcirculation, leading to a secondary ischaemic assault.7 Although the centre of the lesion is characterised by tissue necrosis, apoptosis is also observed in the periphery as early as 12-h post injury.5 The latter is thought to occur via mitochondrial permeabilisation following increased expression of Bax.8 Upregulation of genes involved in neuronal survival or regeneration is also observed.9, 10 This lesion bears many features of neurotrauma.6, 7 Here we have non-invasively imaged the cell death in these lesions using a heat shock protein-90 (Hsp-90) alkylator.11
4-(N-(S-glutathionylacetyl) amino) phenylarsonous acid (GSAO) is a tripeptide trivalent arsenical that rapidly accumulates in the cytoplasm of dying cells in culture and in murine tumors.11 It is retained in the cell predominantly through covalent reaction with the Cys597 and Cys598 residues of Hsp-90, which is the most abundant protein chaperone in mammalian cells.12 The As(III) atom of GSAO cross-links the Hsp-90 Cys597 and Cys598 sulphur atoms forming a stable cyclic dithioarsinite.13, 14 This complex is effectively irreversible in biological milieu. There are very few appropriately spaced cysteine thiols in the extracellular environment so GSAO is largely unreactive until it crosses the plasma membrane. GSAO does not cross the intact plasma membrane of viable or early-stage apoptotic cells but enters mid- to late-stage apoptotic cells when the plasma membrane is compromised.
In this study, we have investigated the utility of a near-infrared fluorescent conjugate of GSAO to non-invasively detect cell death in mouse brain cryolesions.15, 16, 17, 18, 19 GSAO was conjugated to the near-infrared fluorophore, Alex Flour 750 (GSAO-AF750), to maximise tissue penetration of the fluorescent signal and to minimise the complications of tissue auto-fluorescence.20
Results
GSAO-AF750 specifically labels apoptotic and necrotic cells in culture
Conjugation of AF750 to the primary amine of the λ-glutamyl residue of GSAO did not affect its capacity to specifically label dying cells. The specificity and sensitivity of labelling of dying/dead cells by AF750-tagged GSAO was examined in in vitro models of apoptotic (human Jurkat T cells treated with staurosporine) and necrotic (4T1-luc2 mouse breast cancer cells treated with dry ice) cell death. Staurosporine is a microbial alkaloid and broad spectrum protein kinase inhibitor that triggers mitochondrial-mediated apoptotic cell death, whereas snap freezing of cells ruptures the plasma membrane resulting in necrotic cell death. The probe was detected from the fluorescence intensity of the AF750 tag.
Apoptotic Jurkat T cells were labelled with the late-stage apoptosis/necrosis marker, propidium iodide (PI), and either GSAO-AF750 or the phosphatidylserine ligand, PSVue794.18 GSAO-AF750 and PSVue794 labelled dying and dead PI-positive cells in staurosporine-treated samples (Figure 1a). There were sub-populations of cells that labelled with PSVue794 but not PI (Figure 1a, top panels) and labelled with PI but not GSAO-AF750 (Figure 1a, middle panels). PSVue794, like annexin V, binds exteriorised phosphatidylserine on cell membranes of early- and late-stage apoptotic cells,21 so the PSVue794-positive, PI-negative population likely represents early-stage apoptotic cells.

GSAO-AF750 specifically labels apoptotic and necrotic cells in culture. (a) Apoptotic death in Jurkat A3 T cells was triggered with staurosporine for 24 h and the cells subsequently incubated with PI (all panels) and either with PSVue794 (upper panels), GSAO-AF750 (middle panels) or control GSCA-AF750 (lower panels) and labelling analysed by flow cytometry. In some samples (right panels), cells were pre-incubated with the trivalent arsenic ligand, DMP, before GSAO-AF750 or PSVue794 staining. The numbers depicted in the scatter plots represent the percentage of cells in the quadrants. (b) Necrotic death in confluent monolayers of 4T1-luc2 mouse breast cancer cells was induced by applying dry ice to the underside of the culture well and the cells subsequently incubated either with GSAO-AF750, GSCA-AF750 or PSVue794. In some wells, cells were pre-treated with DMP before incubation with GSAO-AF750. Cells were imaged for fluorescence (right panels) and then for bioluminescence following by incubation with D-luciferin (left panels)
The selectivity of GSAO-AF750 for dying and dead cells is mediated by the trivalent arsenic moiety. This was confirmed using two different controls. The synthetic dithiol, dimercaptopropanol (DMP), sequesters the trivalent arsenic moiety of GSAO, whereas 4-(N-((S-glutathionyl)acetyl)amino)benzoic acid (GSCA)-AF750 contains a chemically inert carboxylic acid group in place of the reactive trivalent arsenic moiety. Pre-treatment with DMP effectively ablated labelling of dying/dead cells by GSAO-AF750, but not by PSVue794 (Figure 1a, right panels). The mean GSAO-AF750 fluorescence intensity of PI-positive cells was ∼60-fold less when cells were pre-treated with DMP. In addition, GSCA-AF750 did not label dying/dead PI-positive cells in staurosporine-treated samples. The mean GSCA-AF750 fluorescence intensity of PI-positive cells was >250-fold less than cells stained with GSAO-AF750 (Figure 1a, bottom panels). Viable untreated cells did not stain with any of the compounds. These findings are consistent with our initial characterisation of the compound11 and are in accordance with its reaction with Hsp-90.
Necrotic 4T1 mouse breast cancer cells expressing the luc2 luciferase reporter gene were incubated with either GSAO-AF750, GSCA-AF750 or PSVue794. Necrotic cell death was induced by applying dry ice to the underside of the culture well, resulting in a discrete central region of dead cells surrounded by an external ring of healthy cells. The external ring of viable cells was imaged by bioluminescence following incubation with D-luciferin (Figure 1b, indicated by blue/green). Necrotic cells are deficient in ATP, which is a cosubstrate for the luciferin/luciferase reaction, so do not emit light. Fluorescence imaging revealed that both GSAO-AF750 and PSVue794, but not control GSCA-AF750, selectively labelled the area of dead cells in the centre of the well (Figure 1b). The ring of healthy cells was not stained by any probe. Pre-treatment of the cultures with DMP totally abolished staining by GSAO-AF750.
In vivo and ex vivo imaging of brain lesion cell death in mice with GSAO-AF750
The suitability of GSAO-AF750 for non-invasive imaging of apoptotic and necrotic cell death in the living animal was examined in a murine model of traumatic brain injury. A focal area of cell death is induced in the mouse brain by applying a liquid nitrogen insult to the skull.5 The cryolesion is characterised by both apoptotic and necrotic cell death and shares many features with neurotrauma.6, 7
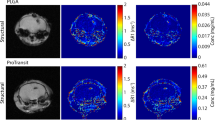
A 60-s brain cryolesion was induced in the front part of the right parietal lobe, followed by tail vein injection of GSAO-AF750 or GSCA-AF750 (1 mg/kg). Whole-body fluorescence imaging was performed at 1, 3, 6 and 24 h after probe injection. GSAO-AF750 and GSCA-AF750 were, with the exception of the kidneys, eliminated from the body within 3 h of administration (Figure 2a). Both compounds were observed to be excreted in the urine, in accordance with the kidney localisation at earlier time points. A strong GSAO-AF750 signal, but not for control GSCA-AF750, was observed at all times points at the brain lesion site (Figure 2a). In one mouse, a 3D sectional view was formed to depict the relative concentration of GSAO-AF750 in the brain lesion (Figure 3). The GSAO-AF750 signal in brain lesions remained steady between 1 and 6 h after probe injection and dropped by 24 h (Figure 2b). The control GSCA-AF750 signals were 50- to 100-fold lower than those obtained with GSAO-AF750 at all-time points (P<0.001). The signal to background ratio for GSAO-AF750 (lesion versus adjacent healthy hemisphere) was approximately 3 : 1 at all-time points.

In vivo and ex vivo imaging of brain lesion cell death in mice with GSAO-AF750. (a) A 60-s brain cryolesion was induced in the front part of the right parietal lobe, followed by tail vein injection of 1 mg/kg GSAO-AF750 (left panels) or GSCA-AF750 (right panels) and whole-body fluorescence imaging was performed at 1, 3, 6 and 24 h after probe injection. (b) Quantification of the integrated mean intensity of GSAO-AF750 and GSCA-AF750 signals in the brain lesion over time. GSAO-AF750 versus GSCA-AF750 intensities were calculated at 1, 3, 6 and 24 h (***P<0.001). (c) The brains of the mice were excised for ex vivo imaging 24 h after probe injection. Bright field images of a representative GSAO-AF750 and control GSCA-AF750 brain, showing a red discolouration of the lesion site, are shown in the top panel, whereas the fluorescence images of the same brains are shown in the bottom panel. (d) Quantification of the ex vivo integrated mean intensity of the AF750 signal. GSAO-AF750 versus GSCA-AF750 fluorescence intensity was calculated (***P<0.001)

3D sectional view depicting the relative concentration of GSAO-AF750 in a brain cryolesion. A concentration gradient of GSAO-AF750 radiates from the centre of the lesion
The brains of the mice were excised for ex vivo imaging 24 h after probe injection. Accumulation of GSAO-AF750 in the lesion site was clearly evident (Figure 2c). Some GSCA-AF750 was also detected in the lesion site, however, the signal intensity was ∼7-fold lower than that for GSAO-AF750 (P<0.001; Figure 2d).
The specificity of GSAO-AF750 for dying and dead brain cells was confirmed ex vivo by analysis of brain sections. Fluorescence imaging and immunohistochemistry revealed colocalisation of GSAO-AF750 with TdT-mediated dUTP nick-end labelling (TUNEL)-positive cells in the cryolesion (Figure 4).

GSAO-AF750 labels dying/dead brain lesion cells in mice. The brains of mice subjected to a cryolesion were excised and sectioned 24 h after GSAO-AF750 or control GSCA-AF750 injection. The sections were examined for AF750 fluorescence (left panels) and cell death using TUNEL staining (right panels). GSAO-AF750, but not control GSCA-AF750, was observed in the lesion and colocalised with TUNEL-positive areas
In vivo and ex vivo imaging of brain lesion cell death with GSAO-AF750 is a function of lesion size and probe concentration
The influence of brain lesion size and the dose of GSAO-AF750 on the signal intensity was evaluated. Mice bearing a 60-s cryolesion were administered 0.1 or 1 mg/kg GSAO-AF750, and mice bearing a 5-s cryolesion were administered 1 mg/kg GSAO-AF750 (Figure 5a). All mice were imaged at 1, 3 and 6 h following probe injection. At all-time points, total fluorescence intensity in the lesions of mice injected with 0.1 mg/kg GSAO-AF750 was 30- to 50-fold less than in those injected with 1 mg/kg GSAO-AF750 (P<0.01 at 1 and 6 h and P<0.05 at 3 h). In addition, the total fluorescence intensity of lesions in mice injected with 1 mg/kg GSAO-AF750 was ∼5-fold lower in mice with a 5-s cryolesion compared with those with a 60-s cryolesion (P<0.05 at all-time points). These findings were confirmed ex vivo by measuring the fluorescence intensity of the excised brains 6 h after GSAO-AF750 injection (Figure 5b).

In vivo and ex vivo imaging of brain lesion cell death with GSAO-AF750 is a function of lesion size and probe concentration. (a) In vivo quantification of GSAO-AF750 signal intensity in the cryolesion over time. GSAO-AF750 fluorescence intensity was compared between mice bearing 5 and 60-s cryolesions (**P<0.01; *P<0.05), or injected with a lower concentration of GSAO-AF750 (*P<0.05 at 1, 3 and 6 h). (b) The brains of the mice were excised for ex vivo imaging 6 h after probe injection and the integrated mean intensity of the AF750 signal was measured. GSAO-AF750 fluorescence intensity was significantly lower in brains bearing a 5-s cryolesion or injected with 0.1 mg/kg GSAO-AF750 (**P<0.01)
Fluorescence lifetime imaging of GSAO-AF750 bound to dying/dead cells in culture and in brain lesions
The fluorescence lifetime of unbound GSAO-AF750 versus bound to dying/dead cells was measured in necrotic breast cancer cells and in brain cryolesions. The lifetime of free GSAO-AF750 and control GSCA-AF750 in phosphate-buffered saline was 0.58–0.61 ns, whereas the weighted average single fluorescence lifetime of GSAO-AF750 lengthened to 0.89 ns when bound to necrotic breast cancer cells (Figure 6a). Negative controls were no binding of GSAO-AF650 in the presence of DMP and no binding of GSCA-AF750 (Figure 6a). A single exponential fit of the time point spread function curve produced a weighted average fluorescence lifetime of 0.97–1.05 ns for GSAO-AF750 in brain cryolesions (Figure 6b), which is comparable to the lifetime measured in cultured dying/dead cells.

Fluorescence lifetime imaging of GSAO-AF750 bound to dying/dead cells. (a) Single exponential fluorescence lifetime analysis of GSAO-AF750 labelling of dying/dead cells in culture. Focal cell death in confluent monolayers of 4T1-luc2 mouse breast cancer cells was induced by applying dry ice to the underside of the culture well and the cells subsequently incubated with GSAO-AF750 (top left well), GSAO-AF750 with DMP (top right well), GSCA-AF750 (bottom left well) or GSCA-AF750 with DMP (bottom right well). (b) Single exponential fluorescence lifetime analysis of GSAO-AF750 labelling of dying/dead cells in brain cryolesions of two different mice
Discussion
The effectiveness of a non-invasive imaging agent is mostly a product of the selectivity for its biological target and the volume of its distribution. Imaging sensitivity and resolution is enhanced when the reporter agent accumulates in high concentration in a small volume. We used herein a small molecule that targets Hsp-90 of apoptotic and/or necrotic cells.
Hsp-90 is, in principle, an attractive imaging target as it is one of the most abundant proteins in the mammalian cell cytoplasm. Alkylation of Hsp-90 by GSAO in dying/dead cells results in high concentration of the compound in the largest compartment of the cell.11 This is in contrast to the ligands that target phosphatidylserine exposed on the exterior of the plasma membrane during apoptosis,22, 23, 24 for instance, which are restricted to the cell surface compartment. Conjugation of the near-infrared fluorophore, AF750, to the λ-glutamyl amine of GSAO did not affect its specific labelling of apoptotic and necrotic cells in culture. In accordance with previous studies,11 the labelling was a function of the dithiol alkylating chemistry of the As(III) moiety of GSAO-AF750.
Importantly, as shown herein, GSAO-AF750 has very favourable pharmacokinetics and biodistribution in mice. It is secreted via the renal circulation and does not accumulate in healthy organs or tissues. Three hours after intravenous administration it is found only in the kidneys. Less than optimal pharmacokinetics and biodistribution has limited the development of a number of cell death imaging agents to date. This has been the case for the protein ligands that recognise phosphatidylserine,25, 26 although a new generation of small molecule phosphatidylserine ligands may overcome these issues.18, 27
GSAO-AF750 specifically labels dying and dead cells in brain cryolesions. The AF750 signal was apparent as early as 1 h in the lesions and remained unchanged for approximately 6 h. The signal to background ratio was ∼3 : 1 at all-time points and signal intensity was a function of both lesion size and probe concentration. The specificity of GSAO-AF750 for dying and dead brain cells was confirmed ex vivo. Notably, cells in the periphery of the brain lesion stained strongly by TUNEL, whereas cells in the centre stained weakly. This is indicative of the apoptotic and necrotic cells, respectively, in the lesion.5 Both cell types in the lesion stained with GSAO-AF750, which is in accordance with the cell culture results showing that GSAO-AF750 labels both apoptotic and necrotic cells.
An advantage that optical agents can have over other reporter groups is a change in the fluorescence properties of the fluorophore when it is bound to its target.28, 29, 30, 31, 32, 33 Alteration of the microenvironment of the fluorescent moiety on target binding can result in a shortening or lengthening of the fluorescence lifetime. This difference can be used to distinguish between unbound and bound agent in the target tissue as it is independent of intensity variations or local concentrations of the fluorophore. The fluorescence lifetime of GSAO-AF750 increased from 0.58–0.61 to 0.89 ns when bound to necrotic breast cancer cells and to 0.97–1.05 ns when bound in brain cryolesions. The significantly longer fluorescence lifetime when bound to its target will be a useful parameter when optimising the signal-to-background contrast in future studies that explore other experimental and clinical applications of GSAO-AF750.
Optical imaging technologies, including fluorescence-guided surgery and endoscopy, intravascular fluorescence imaging and photoacoustic applications in cardiovascular disease, dermatology and cancer, have advanced rapidly in recent years.34, 35 In the last decade, optical imaging has moved from gross assessment of structure and morphology to imaging of biological processes that underlie disease. Although, the development of the hardware to measure light in the human body has outpaced the development of specific probes that report on cellular events. GSAO-AF750 shows promise for the pre-clinical and clinical imaging of cell death in brain and other lesions.
Materials and methods
Ethical statement
All animal experiments were approved for animal health, ethics, and research by the Animal Welfare Committee of Leiden University Medical Centre, the Netherlands (Approval DEC number 11198). All the mice were purchased from Charles River Laboratories, Lille, France and received humane care and maintenance in compliance with the ‘Code of Practice Use of Laboratory Animals in Cancer Research’ (Inspectie W&V, July 1999).
Synthesis of near-infrared GSAO and GSCA conjugates
A solution of AF750 (λex:752 nm, λem:776 nm) succinimidyl ester (10 mg/ml in DMSO, Life Technologies, Mulgrave, VIC, Australia) was added to GSAO (4.2 mg/ml) or control GSCA (5.9 mg/ml), in 0.1 M bicarbonate buffer, pH 8.3 and incubated for 60 min in the dark in the presence of nitrogen. The molar ratio of fluorophore to pendant was ∼1 : 1. Unreacted fluorophore was quenched by adding 10 mM glycine. The purity of the conjugates was evaluated as described11 and determined to be >90%.
Cell culture
Jurkat A3 cells (ATCC, Manassas, VT, USA) and mouse breast cancer cells 4T1-luc2 expressing the codon-optimised luciferase gene luc2 (Caliper Life Sciences, Hopkinton, MA, USA) were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum and 1 U/ml penicillin/streptomycin. All other cell culture reagents were from Gibco, Invitrogen, Merelbeke, Netherlands.
In vitro validation of GSAO-AF750 and GSCA-AF750 via flow cytometry
Jurkat A3 cells were seeded at a density of 5 × 105 cells per ml and incubated with or without 4 μM staurosporine (Sigma) for 24 h. Cells were washed twice with ice-cold phosphate-buffered saline and incubated at room temperature with 1 μM GSAO-AF750 or control GSCA-AF750 for 15 min with shaking. Alternatively, cells were washed in phenol red free RPMI-1640 medium and stained with PSVue794 (Polysciences, Inc., West Chester, PA, USA), as per the manufacturer’s instructions. On some occasions, the cells were pre-treated with DMP (50 μM, Sigma), for 1 min. Cells were washed again and incubated with PI (1 μg/ml, Invitrogen) for 15 min in the dark. Flow cytometry was performed using a BD LSR II Flow Cytometer with a high-power 200 mW, 628 nm red excitation laser, a 40 mW, 786 nm near-infrared excitation laser and a 100 mW, 488 nm excitation laser (BD Biosciences, Sydney, Australia). Data were analysed using FlowJo software version 8.7, Ashland, OR, USA.
In vitro cryo-induced cell death assay
4T1-luc2 cells were seeded onto 12-well plates and grown to confluency in complete RPMI-1640 medium. The medium was discarded and a bar of dry ice 3–5 mm in diameter applied to the underside of the culture well for 15 s. GSAO-AF750, GSCA-AF750 or PSVue794 (0.4 μM in 500 μl medium) was added to the cells and incubated for 15 min at 37 °C. The cells were gently washed twice with phosphate-buffered saline and imaged using the IVIS Spectrum (Caliper Life Sciences; excitation 710 nm, emission 780 nm). Subsequently, D-luciferin (150 mg/kg in 5 μl PBS; SynChem, Inc., Elk Grove Village, IL, USA) was added to each well for 10 min and bioluminescence images acquired using the same machine. To complement the intensity measurements, fluorescence lifetime images of 4T1-luc2 cells on 12-well plates in the presence of GSAO-AF750 or control GSCA-AF750 were determined using a time domain optical imager (Optix MX3, Montreal, QC, Canada). Tubes of GSAO-AF750 and GSCA-AF750 in their free state were also scanned in order to establish a fluorescence lifetime reference for the subsequent in vivo studies.
In vivo murine model of cryo-induced traumatic brain injury
A brain cryolesion was applied as described previously with minor modifications.18 In short, 6-week-old male BALB/c nu/nu mice (Charles River Laboratories) were sedated with 2–3% isoflurane and a cryolesion induced in the parietal lobe of the right cerebral hemisphere. A metal rod of 3 mm in diameter was pre-cooled with liquid nitrogen and applied to the skull for 5 or 60 s. Mice were allowed to recover from the anaesthetic and observed for any adverse symptoms resulting from the cryolesion. Mice were then injected intravenously with either GSAO-AF750 or control GSCA-AF750 (0.1 mg/kg or 1 mg/kg in 100 μl PBS) and imaged with the Pearl Impulse Small Animal Imaging System (LI-COR Biosciences, Lincoln, NE, USA) at 1, 3, 6 and 24 h. Before imaging, mice were wiped with 70% v/v ethanol to remove any of the compounds excreted in the urine and contaminating the skin. At the experiment end point, the mice were killed by cervical dislocation and the brains excised for ex vivo fluorescence imaging. Images were acquired at 800 nm at a resolution of 85 μm.
The fluorescent signal was digitised and electronically displayed as a pseudocolor overlay on a grey scale white light image of the animal. The data were analysed using Pearl Impulse Software, version 2.0 (LI-COR Biosciences, Lincoln, NE, USA). Total AF750 fluorescence intensity was determined by drawing a region of interest (ROI) over the area in which the cryolesion was induced. The size and shape of the ROI was the same for each time point. An ROI of equivalent size was then drawn over the adjacent hemisphere to determine the background signal and this was subtracted from the lesion signal. Where integrated mean fluorescence intensity of AF750 is shown, the value has been corrected for the area of the ROI. Results are depicted as mean±S.E.M.
Whole mouse fluorescence imaging was performed using the Optix MX3. Animals were placed on a heated five-mouse bed imaging platform so that mice administered GSAO-AF750 and control GSCA-AF750 could be compared side-by-side. A 735 nm, 80 MHz pulse diode laser was used for excitation and raster scanning, performed in 1 mm steps. Photon emission was captured by time-correlated single photon counting with a 755 nm long pass filter and data were recorded as temporal point spread functions. Image analysis and fluorescence lifetime calculations were performed using Optiview 3.0 software (ART, Montreal, QC, Canada). Rectangular ROIs were drawn around the head region to initially determine the single exponential fluorescence lifetime. This is the weighted average of all the fluorescence lifetime values.
Histological validation
Brains were fixed in 4% formaldehyde, embedded in paraffin and 8 μm sections were prepared and imaged using the LI-COR Odyssey Infrared Imager 9120 (LI-COR Biosciences) at 800 nm. Afterwards the sections were subjected to TUNEL staining (Promega, Leiden, Netherlands) to confirm accumulation of GSAO-AF750 in dying and dead cells.
Statistical analysis
We used two-tailed, unpaired Student’s t-test.
Abbreviations
- DMP:
-
dimercaptopropanol
- GSAO:
-
4-(N-(S-glutathionylacetyl) amino) phenylarsonous acid
- GSCA:
-
4-(N-((S-glutathionyl)acetyl)amino)benzoic acid
- AF750:
-
Alex Flour 750
- Hsp-90:
-
heat shock protein-90
- PI:
-
propidium iodide
- ROI:
-
region of interest
- TUNEL:
-
TdT-mediated dUTP nick-end labelling
References
Faul M, Xu L, Wald MM, Coronado VG . Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths, 2002–2006. National Center for Injury Prevention and Control, Centers for Disease Control and Prevention 2010: 1–71.
Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE . Traumatic brain injury in the United States: a public health perspective. J Head Trauma Rehabil 1999; 14: 602–615.
Werner C, Engelhard K . Pathophysiology of traumatic brain injury. Br J Anaesth 2007; 99: 4–9.
Kim JJ, Gean AD . Imaging for the diagnosis and management of traumatic brain injury. Neurotherapeutics 2011; 8: 39–53.
Steinbach JP, Weissenberger J, Aguzzi A . Distinct phases of cryogenic tissue damage in the cerebral cortex of wild-type and c-fos deficient mice. Neuropathol Appl Neurobiol 1999; 25: 468–480.
Meryman HT . Mechanics of freezing in living cells and tissues. Science 1956; 124: 515–521.
Gage AA, Baust JM, Baust JG . Experimental cryosurgery investigations in vivo. Cryobiology 2009; 59: 229–243.
Wen J, Duan Y, Zou Y, Nie Z, Feng H, Lugnani F et al. Cryoablation induces necrosis and apoptosis in lung adenocarcinoma in mice. Technol Cancer Res Treat 2007; 6: 635–640.
Margulies S, Hicks R . Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma 2009; 26: 925–939.
Quintana A, Giralt M, Rojas S, Penkowa M, Campbell IL, Hidalgo J et al. Differential role of tumor necrosis factor receptors in mouse brain inflammatory responses in cryolesion brain injury. J Neurosci Res 2005; 82: 701–716.
Park D, Don AS, Massamiri T, Karwa A, Warner B, Macdonald J et al. Noninvasive imaging of cell death using an hsp90 ligand. J Am Chem Soc 2011; 133: 2832–2835.
Nardai G, Sass B, Eber J, Orosz G, Csermely P . Reactive cysteines of the 90-kDa heat shock protein, Hsp90. Arch Biochem Biophys 2000; 384: 59–67.
Adams E, Jeter D, Cordes AW, Kolis JW . Chemistry of organometalloid complexes with potential antidotes - structure of an organoarsenic(Iii) dithiolate r. Inorg Chem 1990; 29: 1500–1503.
Dilda PJ, Hogg PJ . Arsenical-based cancer drugs. Cancer Treat Rev 2007; 33: 542–564.
Grzybicki D, Moore SA, Schelper R, Glabinski AR, Ransohoff RM, Murphy S . Expression of monocyte chemoattractant protein (MCP-1) and nitric oxide synthase-2 following cerebral trauma. Acta Neuropathol 1998; 95: 98–103.
Hotta N, Aoyama M, Inagaki M, Ishihara M, Miura Y, Tada T et al. Expression of glia maturation factor beta after cryogenic brain injury. Brain Res Mol Brain Res 2005; 133: 71–77.
Siren AL, Radyushkin K, Boretius S, Kammer D, Riechers CC, Natt O et al. Global brain atrophy after unilateral parietal lesion and its prevention by erythropoietin. Brain 2006; 129: 480–489.
Smith BA, Xie BW, van Beek ER, Que I, Blankevoort V, Xiao S et al. Multicolor fluorescence imaging of traumatic brain injury in a cryolesion mouse model. ACS Chem Neurosci 2012; 3: 530–537.
Stoffel M, Blau C, Reinl H, Breidt J, Gersonde K, Baethmann A et al. Identification of brain tissue necrosis by MRI: validation by histomorphometry. J Neurotrauma 2004; 21: 733–740.
Xie BW, Mol IM, Keereweer S, van Beek ER, Que I, Snoeks TJ et al. Dual-wavelength imaging of tumor progression by activatable and targeting near-infrared fluorescent probes in a bioluminescent breast cancer model. PloS One 2012; 7: e31875.
Hanshaw RG, Smith BD . New reagents for phosphatidylserine recognition and detection of apoptosis. Bioorg Med Chem 2005; 13: 5035–5042.
Blankenberg FG . In vivo imaging of apoptosis. Cancer Biol Ther 2008; 7: 1525–1532.
Zhao M . In vivo apoptosis imaging agents and strategies. Anticancer Agents Med Chem 2009; 9: 1018–1023.
Niu G, Chen X . Apoptosis imaging: beyond annexin V. J Nucl Med 2010; 51: 1659–1662.
Beekman CA, Buckle T, van Leeuwen AC, Valdes Olmos RA, Verheij M, Rottenberg S et al. Questioning the value of (99 m)Tc-HYNIC-annexin V based response monitoring after docetaxel treatment in a mouse model for hereditary breast cancer. Appl Radiat Isot 2011; 69: 656–662.
Lederle W, Arns S, Rix A, Gremse F, Doleschel D, Schmaljohann J et al. Failure of annexin-based apoptosis imaging in the assessment of antiangiogenic therapy effects. EJNMMI Res 2011; 1: 26.
Xiong C, Brewer K, Song S, Zhang R, Lu W, Wen X et al. Peptide-based imaging agents targeting phosphatidylserine for the detection of apoptosis. J Med Chem 2011; 54: 1825–1835.
Akers WJ, Berezin MY, Lee H, Achilefu S . Predicting in vivo fluorescence lifetime behavior of near-infrared fluorescent contrast agents using in vitro measurements. J Biomed Opt 2008; 13: 054042.
Ardeshirpour Y, Chernomordik V, Zielinski R, Capala J, Griffiths G, Vasalatiy O et al. In vivo fluorescence lifetime imaging monitors binding of specific probes to cancer biomarkers. PloS one 2012; 7: e31881.
Erten A, Hall D, Hoh C, Tran Cao HS, Kaushal S, Esener S et al. Enhancing magnetic resonance imaging tumor detection with fluorescence intensity and lifetime imaging. J Biomed Opt 2010; 15: 066012.
Goiffon RJ, Akers WJ, Berezin MY, Lee H, Achilefu S . Dynamic noninvasive monitoring of renal function in vivo by fluorescence lifetime imaging. J Biomed Opt 2009; 14: 020501.
Hall DJ, Han SH, Chepetan A, Inui EG, Rogers M, Dugan LL . Dynamic optical imaging of metabolic and NADPH oxidase-derived superoxide in live mouse brain using fluorescence lifetime unmixing. J Cereb Blood Flow Metab 2012; 32: 23–32.
Wang J, Daphu I, Pedersen PH, Miletic H, Hovland R, Mork S et al. A novel brain metastases model developed in immunodeficient rats closely mimics the growth of metastatic brain tumours in patients. Neuropathol Appl Neurobiol 2011; 37: 189–205.
Taruttis A, Ntziachristos V . Translational optical imaging. AJR Am J Roentgenol 2012; 199: 263–271.
Herron TJ, Lee P, Jalife J . Optical imaging of voltage and calcium in cardiac cells & tissues. Circ Res 2012; 110: 609–623.
Acknowledgements
This study is supported by the Dutch Centre for Translational Molecular Medicine, project MUSIS (grant 03O-202), the ENCITE project 201842, the National Health and Medical Research Council of Australia and the Cancer Council New South Wales.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Xie, BW., Park, D., Van Beek, E. et al. Optical imaging of cell death in traumatic brain injury using a heat shock protein-90 alkylator. Cell Death Dis 4, e473 (2013). https://doi.org/10.1038/cddis.2012.207
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.207
Keywords
This article is cited by
-
Biodistribution and imaging of an hsp90 ligand labelled with 111In and 67Ga for imaging of cell death
EJNMMI Research (2020)
-
Cardioprotection by minocycline in a rabbit model of ischemia/reperfusion injury: Detection of cell death by in vivo 111In-GSAO SPECT
Journal of Nuclear Cardiology (2018)
-
Targeted nanoparticles for the non-invasive detection of traumatic brain injury by optical imaging and fluorine magnetic resonance imaging
Nano Research (2016)
-
Pre-clinical Evaluation of a Cyanine-Based SPECT Probe for Multimodal Tumor Necrosis Imaging
Molecular Imaging and Biology (2016)
-
Noninvasive Molecular Imaging of Cell Death in Myocardial Infarction using 111In-GSAO
Scientific Reports (2014)
