
Abstract
MLN4924, a newly discovered small molecule inhibitor of NEDD8-activating enzyme (NAE), inactivates Cullin-RING E3 ubiquitin Ligases (CRLs) by blocking cullin neddylation. As a result, MLN4924 causes accumulation of several key substrates of CRLs and effectively suppresses tumor cell growth by inducing apoptosis and senescence. However, the role of MLN4924 in induction of autophagy and its biological significance are totally unknown. Here we showed that MLN4924 effectively induces autophagy in both time- and dose-dependent manners in multiple human cancer lines, indicating a general phenomenon. Mechanistically, by inactivating CRLs, MLN4924 causes accumulation of DEPTOR and HIF1α. The siRNA knockdown and gene KO studies showed that DEPTOR and the HIF1-REDD1-TSC1 axis are responsible for MLN4924-induced autophagy via inhibiting mTORC1. Biologically, autophagy is a survival signal to tumor cells, and blockage of autophagy via siRNA knockdown, gene KO and small molecule inhibitor remarkably enhanced MLN4924-induced apoptosis. Our study reveals an uncharacterized mechanism of MLN4924 action and provides the proof-of-concept evidence for strategic drug combination of MLN4924 with an autophagy inhibitor for maximal killing of tumor cells via enhancing apoptosis.
Similar content being viewed by others


Main
Cullin-RING ligases (CRLs) are the largest family of E3 ubiquitin ligases.1 By promoting ubiquitination and degradation of various key substrates, CRLs control several important biological processes, including cell-cycle progression, signal transduction, gene transcription, embryonic development, genomic integrity and tumor suppression.1 Among eight members of the cullin family,2 cullin-1 binds to adaptor protein SKP1 and an F-box protein at the N-terminus, and a RING protein, RBX1 or RBX2 (also known as ROC2 or SAG) at the C-terminus, thus forming the Skp1, Cullin and F-box protein (SCF) E3 complex,3 which is the largest member within the CRL family.1 It is well-established that the substrate specificity of the SCF complex is determined by the F-box proteins,3, 4 whereas the core SCF E3 ubiquitin ligase is a complex of cullin-1 and RBX1/2. Activity of SCF E3 ubiquitin ligases requires (1) RING protein, RBX1 or RBX2, which binds to E2 and facilitates ubiquitin transfer from E2 to substrates,5 and (2) cullin neddylation, which disrupts inhibitory binding by CAND1 and retains the SCF in an active conformation.6 Like ubiquitination, neddylation requires E1 NEDD8-activating enzyme (NAE), E2 NEDD8-conjugating enzyme (UBC12), and E3 NEDD8 ligase, which catalyzes the transfer of NEDD8 to a target molecule.7, 8 Thus, inhibition of any neddylation enzyme would cause deneddylation of cullins, leading to inactivation of CRLs/SCF E3 ligases.
MLN4924 is a newly discovered investigational small molecule inhibitor of NAE.9 MLN4924 binds to NAE to create a covalent NEDD8–MLN4924 adduct, which cannot be further utilized in subsequent intraenzyme reactions, thus blocking NAE enzymatic activity.10 By blocking cullin neddylation, MLN4924 inactivates CRLs/SCF E3 ligase and causes accumulation of CRLs/SCF E3 substrates to suppress tumor cell growth both in vitro and in vivo.9, 10 A potential advantage of MLN4924 over bortezomib, the first and only FDA-approved general proteasome inhibitor for the treatment of multiple myeloma,11 is that MLN4924 selectively inhibits degradation of a specific set of cellular proteins regulated by CRLs/SCF E3 ligases, whereas bortezomib blocks degradation of all proteins through the 26S proteasome, leading to greater general cytotoxicity.11 In fact, MLN4924 was well-tolerated when administrated to mice.9, 12 With promising preclinical efficacy, MLN4924 has been advanced to several Phase I clinical trials against a number of human malignancies.8, 13
Mechanistic studies of MLN4924 action in growth suppression of tumor cells revealed that MLN4924 effectively induced apoptosis9, 14, 15, 16, 17 and senescence18, 19, 20 in several human cancer cell lines. A potential role for MLN4924 in regulation of autophagy, a type II-programmed cell death, is, however, unknown. We have recently shown that DEPTOR, a naturally occurring inhibitor of mTORC1 and mTORC2,21 is a physiological substrate of SCFβTrCP E3 ubiquitin ligase and that DEPTOR accumulation in response to glucose deprivation inhibited mTORC1 to induce autophagy.22 We therefore hypothesized that by inactivating SCF E3 ubiquitin ligase to cause DEPTOR accumulation, MLN4924 may induce autophagy. Here we report that MLN4924 effectively induced autophagy in both dose- and time-dependent manners in multiple human cancer cell lines. Mechanistic studies revealed that via inactivating CRLs/SCF E3s, MLN4924 caused accumulation of DEPTOR and HIF1α to inhibit mTORC1 activity directly (DEPTOR) and via the HIF1–REDD1–TSC1 axis (HIF1α). We further demonstrated that mTORC1 inactivation and subsequent autophagy induction act as an overall survival signal, as abrogation of autophagy led to an increased suppression of tumor growth by enhancing apoptosis induction. Our study provides proof-of-concept evidence for a novel drug combination of MLN4924 with an autophagy inhibitor for more effective cancer therapy.
Results
MLN4924 induces autophagy in multiple human cancer cells
It was documented that MLN4924 effectively killed cancer cells via inducing apoptosis9, 14, 15, 16, 17 and senescence.18, 19, 20 To precisely determine the effect of MLN4924 on cell growth, we used the time-resolved video to dynamically observe how cancer cells were killed by MLN4924, using HeLa cells expressing EGFP-LC3. We found that MLN4924 at 0.1 or 1 μM within a period of 24 h effectively induced autophagy in a dose-dependent manner (Supplementary Videos A and B). We followed up this initial observation and found that like rapamycin (a known autophagy inducer), MLN4924 effectively induced autophagy punctate structures23 in EGFP-LC3-expressing HeLa (Figure 1a), SK-BR3 (Figure 1b) and MDA-MB-231 cells (Supplementary Figure S1A). MLN4924-induced autophagy was further demonstrated by acridine orange staining (AOS), which measures autophagic acidic vesicular organelles in SK-BR3 cells (Figure 1c) and U87 cells (Supplementary Figure S1B). In addition, MLN4924 induction of autophagy was biochemically demonstrated by a time-dependent conversion of LC3-I to LC3-II and degradation of p62, two well-established measurements of autophagy23 in multiple human cancer cell lines, including HeLa, SK-BR3, MCF7, HCT116 cells (Figure 1d) and in MDA-MB-231 and U87 cells (Supplementary Figure S1C). Furthermore, MLN4924-induced autophagy was blocked by BAF A1 (bafilomycin A1), an inhibitor of vacuolar (H+)-ATPase that inhibited acidification and protein degradation in lysosomes,24 as evidenced by accumulation of both LC3-I and LC3-II and abrogation of p62 degradation in MCF7 and HCT116 cells (Figure 1e). Finally, autophagosomes were readily detectable in HeLa and SK-BR3 cells upon MLN4924 exposure by electron microscopy (Figure 1f). Taken together, these results clearly indicated that MLN4924 is a potent and universal inducer of autophagy in multiple cancer cell lines derived from different tumor tissues.

MLN4924 induces autophagy: (a and b) Autophagy measured by appearance of punctate vesicle structure: HeLa (a) or SK-BR3 cells (b) stably expressing EGFP-LC3 were treated with indicated concentrations of MLN4924, along with rapamycin (100 nM) as a positive control, for 24 h and 48 h before photography under a fluorescent microscope (left panels). Cells with punctate vesicle structures of EGFP-LC3 were counted and expressed as percentage of autophagy (right panel). *P<0.05, **P<0.01. (c) Autophagy measured by acridine orange staining (AOS): SK-BR3 cells were treated with MLN4924 at indicated concentrations for 24 h, followed by flow cytometry (top) to quantify autophagy by AOS, using the ratio between geo-mean fluorescence intensity of red versus green fluorescence (FL3/FL1). Shown are mean±S.E.M. from two independent experiments with statistical values listed (bottom). (d and e) Autophagy measured by LC3-II conversion and p62 degradation: Cells were treated with MLN4924 (1 μM for SK-BR3 and MCF7, and 0.5 μM for HCT116) in the absence (d) or presence of BAF (50 nM for 48 h) (e), followed by IB with indicated Abs. The band density was quantified and expressed as fold change, compared with the control, by arbitrarily setting the control value as 1. (f) Detection of autophagosomes by EM: HeLa and SK-BR3 cells were treated for 24 h with MLN4924 (1 μM), along with DMSO vehicle control, followed by the EM analysis. Autohpagosomes (arrows) were shown in MLN4924-treated cells. Direct magnification: × 46 000. Size bar=100 nm
MLN4924 selectively inhibits mTORC1 activity
We next determined whether MLN4924-induced autophagy is mediated by mTORC1, a well-established negative regulator of autophagy.25, 26 Indeed, in multiple cancer lines, MLN4924 treatment for 24 h caused a dose-dependent inhibition of mTOR auto-phosphorylation at the S2481, and of mTORC1 activity, as reflected by a reduced phosphorylation of S6K1 and 4E-BP1 (Figure 2a). Effect of MLN4924 on mTORC2 activity, as reflected by AKT phosphorylation at S473, was minimal, if any, among all tested cancer lines (Figure 2a). We next determined precisely the time course of mTORC1 inactivation upon MLN4924 exposure for up to 12–24 h in MCF7, SK-BR3, HCT116 (Figure 2b), HeLa (Supplementary Figure S2A) and MDA-MB-231 cells (Supplementary Figure S2B). In all the lines tested, MLN4924 remarkably inactivated mTORC1 (reduced phosphorylation of mTOR, S6K1 and 4E-BP1), starting as early as 4–8 h and lasting up to 12–24 h in the testing periods, although the extent of inactivation varied among different lines. Furthermore, MLN4924 had little, if any, effect on AKT phosphorylation and no effect on MAPK activity (Figures 2a and b, and Supplementary Figures S2A and B). Thus, MLN4924 is highly selective against mTORC1 activity. Finally, we determined directly whether MLN4924 inactivated mTORC1 kinase activity using an in vitro kinase assay and found that mTORC1-mediated S6K1 phosphorylation (25-fold higher than the background noise) was completely abrogated if mTORC1 complex was isolated from cells pretreated with MLN4924 or rapamycin (as positive control; Figure 2c). We concluded from these cell-based and cell-free assays that MLN4924 effectively and selectively inhibited mTORC1 activity. We therefore focused on mTORC1 for the remaining experiments.

MLN4924 inhibits mTORC1 activity: (a) Dose and (b) time dependent: Cells were treated with various concentrations of MLN4924 for 24 h (a) or with 1.0 μM MLN4924 for MCF7 and SK-BR3 cells, or 0.5 μM MLN4924 for HCT116 cells (b) for indicated time periods, followed by IB with indicated Abs. (c) The in vitro mTORC1 kinase activity assay. HA-tagged S6K1 was transfected into 293 cells, followed by immune-affinity purification by IP using bead-conjugated HA antibody. After elution with HA peptide, HA-S6K1 was added into a kinase reaction mixture containing mTORC1 complex, which was immunoprecipitated by an mTOR Ab from HCT116 cells after a 24-h treatment with MLN4924 (0.5 μM), along with rapamycin control (100 nM). The reaction mixture was incubated for 90 min at 30°C with constant shaking, followed by IB with indicated Abs. The whole-cell extract (WCE) of HCT116 cells were also subjected to IB with indicated Abs to show expected effects of MLN4924 and rapamycin. LEX, longer exposure. The band density was quantified and expressed as fold change, compared with the control, by arbitrarily setting the control value as 1
MLN4924 caused accumulation of DEPTOR: DEPTOR is necessary but not sufficient for MLN4924-induced autophagy
We next determined the underlying mechanism by which MLN4924 inactivated mTORC1. We first focused on DEPTOR, a naturally occurring inhibitor of both mTORC1 and mTORC2,21 given very recent observations by us and the others that DEPTOR is a substrate of SCFβTrCP E3 ubiquitin ligase.22, 27, 28 We treated SK-BR3 and MDA-MB-231 cells, which expressed very low levels of DEPTOR (Figures 3a and b) with MLN4924 (1 μM) or DMSO for various time points up to 36 h. In both lines, cullin-1 neddylation was inhibited at 6 h post-treatment when DEPTOR started to increase, reaching the peak levels at 12 or 36 h in a cell line-dependent manner (Figures 3a and b). No accumulation of DEPTOR was found in DMSO-treated cells within the testing periods (Figures 3a and b). We also determined whether MLN4924 altered the levels of mTORC components, RAPTOR, RICTOR and GβL, and mTORC1 inhibitors, TSC1 and TSC2. Although no effect was found on GβL, TSC1 and TSC2, MLN4924 did reduce the levels of RAPTOR, but to a less extent, of RICTOR, which may also contribute to its preferred inhibition of mTORC1 activity (Figures 3a and b). In addition, we found that in cells expressing the higher basal levels of DEPTOR, such as in MCF7 and HeLa cells, MLN4924 had little, if any, effect on DEPTOR levels, but in both the lines, MLN4924 reduced RAPTOR levels (data not shown).

MLN4924 induces accumulation of DEPTOR, which is necessary but not sufficient for MLN4924-induced autophagy: (a and b) Accumulation of DEPTOR by MLN4924. Cells were treated with 1.0 μM MLN4924 or DMSO vehicle control for indicated time periods, followed by IB with indicated Abs. (c and d) Attenuation of MLN4924-induced autophagy by siRNA knockdown of DEPTOR. Cells were transfected with siRNA targeting DEPTOR or scrambled control siRNA, for 24 h and subjected to MLN4924 (1.0 μM) treatment for 1 or 2 days, followed by IB with indicated Abs. The band density was quantified and expressed as fold change, compared with the control, by arbitrarily setting the control value as 1
To determine whether DEPTOR is required for MLN4924-induced autophagy, we silenced DEPTOR via siRNA in SK-BR3 and MDA-MB-231 cells, and found that in both the lines, DEPTOR knockdown partially restored mTORC1 activity, as reflected by a partial recovery of 4E-BP1 phosphorylation, and incompletely inhibited MLN4924-induced autophagy, as demonstrated by partial recovery of LC3-II conversion and p62 degradation (Figures 3c and d, lanes 7 and 8 versus 3 and 4). A similar observation, but to a lesser extent, was made in HeLa cells, in which MLN4924 treatment failed to cause DEPTOR accumulation (Supplementary Figure S3, lanes 3 and 4 versus 1 and 2; and 7 and 8 versus 3 and 4). Interestingly, although much less effective than MLN4924, DMSO treatment caused a moderate increase of DEPTOR to inactivate mTORC1 (reduced 4E-BP1 phosphorylation) after an extended culture period for 48 h, when cells became confluent (Figures 3c and d, lanes 1 versus 2 and 3 versus 4), suggesting that high cell density could also trigger DEPTOR expression to inactivate mTOR proliferation signals.21 Taken together, these results indicate that DEPTOR is necessary but not sufficient to mediate MLN4924-induced autophagy, suggesting the involvement of additional regulators of mTORC1.
MLN4924 causes the accumulation of HIF1 α but not REDD1 and TSC2
We next focused on other known substrates of CRL/SCF E3 ligases in the mTOR signaling pathway for additional regulators that may mediate MLN4924-induced autophagy. Although mTOR itself and mTOR inhibitor TSC2 were reported to be degraded by SCF-FBXW729 and Cul4A-DDB1-FBXW5,30 respectively, we did not observe any accumulation of mTOR and TSC2 upon MLN4924 treatment in multiple cancer cell lines (Figures 2 and 3 and Supplementary Figure S2), thus, excluding their involvement. We then measured HIF1α, a well-known substrate of Cul2-VHL-RBX1 E331 which negatively regulates mTORC1 signal via REDD1–TSC1/2 axis.25 Indeed, MLN4924 inhibited cullin-2 neddylation and caused a remarkable accumulation of HIF1α in a dose-dependent manner (Figure 4a). As MLN4924 at 0.1 μM is sufficient to deneddylate Cul2 and inactivate Cul2-VHL-RBX1 E3, we used this low drug concentration and determined the time course (up to 24 h) of HIF1α accumulation. As shown in Figures 4b–d and Supplementary Figures S4A and B, in all the five cancer lines tested, MLN4924 caused a time-dependent accumulation of HIF1α, although the peak induction varied among the lines. Thus, HIF1α is likely involved in the process of MLN4924-induced autophagy. Surprisingly, although REDD1 was reported to be a hypoxia/HIF1 downstream target32, 33 and a known substrate of Cul4A-DDB1 E3 ligase,34 we did not observe any MLN4924-selective REDD1 accumulation in all the five cancer lines tested, even MLN4924-induced Cul4A deneddylation is evident (Figures 4b–d and Supplementary Figures S4A and B). However, consistent with a previous report that REDD1 increased under high cell-density condition,32 we did observe increased REDD1 levels in DMSO-treated cells at later time points when cell density became high (Figures 4b–d and Supplementary Figures S4A and B). Thus, REDD1 may not be a direct target of CRL ligases. Rather, the expression of REDD1 is very sensitive to the culture conditions.

MLN4924 induces accumulation of HIF1α, but not REDD1 in both dose- and time-dependent manners: Cells were treated with various concentrations of MLN4924 for (a) 24 h, or with 0.1 μM MLN4924 for (b–d) indicated time periods, followed by IB with indicated Abs. The band density was quantified and expressed as fold change, compared with the control, by arbitrarily setting the control value as 1
HIF1 α –REDD1–TSC1 axis contributes to MLN4924-induced autophagy
We next determined whether the HIF1α–REDD1–TSC1 axis causally affected MLN4924-induced authophagy using siRNA knockdown and gene-knockout approaches. HIF1α knockdown in SK-BR3 and MCF7 cells partially restored mTORC1 activity (as reflected by partial recovery of S6K1 and 4E-BP1 phosphorylation), and partially abrogated MLN4924-induced autophagy (as demonstrated by partial inhibition of LC3-II conversion and p62 degradation) (Figure 5a, lanes 3 and 4 versus 7 and 8; and 11 and 12 versus 15 and 16). Likewise, we used paired Hif1α+/+ versus Hif1α−/− MEFs35 and found a reduction of MLN4924-induced autophagy in Hif1α−/− MEF cells, as compared with the Hif1α+/+ control cells, particularly after 3-day MLN4924 treatment, as MEFs are more resistant than cancer cells to MLN492418 (Figure 5b, lanes 3 and 4 versus 7 and 8; and 11 and 12 versus 15 and 16). We confirmed the identity of Hif1α+/+ versus Hif1α−/− MEFs by PCR genotyping35 and RT-PCR (Supplementary Figures S5A and B), and showed Hif1α accumulation by MLN4924 at the earlier time points in Hif1α+/+ but not in Hif1α−/− MEFs (Supplementary Figure S5C). Furthermore, siRNA knockdown of REDD1 partially restored mTORC1 activity and inhibited MLN4924-induced autophagy in SK-BR3 and MCF7 cells (Figure 5c, lanes 3 and 4 versus 7 and 8; and 11 and 12 versus 15 and 16), and to a lesser extent in HCT116 cells, likely due to less effective REDD1 knockdown (Supplementary Figure S5D, lanes 3 and 4 versus 7 and 8). Lastly, using paired Tsc1+/+ versus Tsc1−/− MEF cells, we found that both the MLN4924-induced mTOR inactivation and autophagy induction were largely abrogated in Tsc1-null cells (Figure 5d, lanes 1–4 versus 5–8). Taken together, these results indicated that the HIF1α–REDD1–TSC1 axis also contributes to MLN4924-induced mTORC1 inactivation and autophagy induction.

The HIF1–REDD1–TSC1 axis is required for MLN4924-induced autophagy. Cancer cells were transfected with siRNA targeting (a) HIF1α, (c) REDD1 or scrambled control siRNA, for 24 h and subjected to 1.0 μM MLN4924 treatment for 1 (M1) or 2 (M2) days, using DMSO vehicle as the control for 1 (D1) or 2 (D2) days. Paired MEF cells of (b) Hif1α+/+ versus Hif1α−/− or (d) Tsc1+/+ versus Tsc1−/− were treated with 2.5 μM MLN4924, or DMSO control for up to 3 days (M1-3/D1-3). Cell lysates were then prepared and subjected to IB with indicated Abs. M, MLN4924; D, DMSO; LEX, longer exposure. The band density was quantified and expressed as fold change, compared with the control, by arbitrarily setting the control value as 1
Abrogation of autophagy enhanced MLN4924-induced apoptosis
Finally, we determined whether manipulation of autophagy by approaches including siRNA knockdown, gene knockout and small molecule inhibitors would affect apoptosis, given the fact that apoptosis is a well-known mechanism for MLN4924-induced growth suppression.9, 14, 15, 16, 17 Using a paired Atg5+/+ versus Atg5−/− MEF cells in the ATP-lite cell growth assay, we found that autophagy-deficient Atg5−/− cells36 were much more sensitive than autophagy-competent Atg5+/+ cells to MLN4924-induced growth suppression with ∼threefold lower IC50 value (3 μM in Atg5−/− versus 8 μM in Atg5+/+ cells) (Figure 6a). Interestingly, MLN4924 at 2–2.5 μM (which induced autophagy in Atg5+/+ MEF cells, see Figure 6b) stimulated the growth up to 30% in autophagy-competent Atg5+/+ MEFs, but not at all in autophagy-deficient Atg5−/− MEFs (Figure 6a), suggesting that induction of autophagy have a survival role. To determine the nature of enhanced growth suppression in Atg5−/− MEFs, we first confirmed that MLN4924 at 2.5 μM indeed induced autophagy in autophagy-competent Atg5+/+ MEFs, but not in autophagy-deficient Atg5−/− MEFs, as demonstrated by LC3-I to LC3-II conversion and p62 degradation (Figure 6b, top three panels, lanes 1–4 versus 5–8). We then used western blotting to determine the cleavage of PARP and caspase-3 as the readouts for apoptosis, and found that MLN4924 induced apoptosis in Atg5−/− but not in Atg5+/+ MEFs (Figure 6b, panels 4–6, lanes l–4 versus 5–8). Furthermore, we used FACS analysis and showed that MLN4924 induced remarkably more apoptosis in Atg5−/− MEFs than that in Atg5+/+ cells, as reflected by a much higher fold induction of sub-G1 population in Atg5−/− MEFs (Figure 6c, top panel, a representative FACS profile; and bottom panel, fold change, n=3). Similar results, but to a lesser extent, were seen in MCF7 cells when ATG5 was partially silenced to ∼50% levels (Figures 6d and e). Lastly, we used BAF A1 to further demonstrate that blockage of autophagy would enhance cell killing via switching to apoptosis. Although BAF A1 at nanomolar concentration remarkably increased the levels of LC3 and p62 (Figure 6f and Supplementary Figure S6, top two panels) as expected, it had minor, if any, effect on inducing apoptosis (Figure 6f and Supplementary Figure S6, lanes 8 and 9). Combination of BAF A1 with MLN4924 significantly increased MLN4924-induced apoptosis in a dose-dependent manner, as evidenced by increased cleavage of PARP and caspase-3/7 (Figure 6f and Supplementary Figure S6, lanes 4–7 versus 2 and 3 and 8 and 9). Note that MCF7 cells lack the expression of caspase-3 and the cleavage/activation of caspase-7 was used instead. Taken together, these results indicated that induction of autophagy in this setting is an overall survival signal for tumor cells, and autophagy abrogation increases growth suppression by MLN4924 via enhancing apoptosis.

Inhibition of autophagy enhances MLN4924-induced apoptosis: (a–c) Autophagy deficient cells are more sensitive to MLN4924-induced apoptosis. Paired MEFs (Atg5−/− versus Atg5+/+) were treated with MLN4924 at various concentrations, along with DMSO control, for up to 3 days, followed by ATPlite cell proliferation assay (a, mean±S.E.M., n=3, *P<0.05; **P<0.01), IB (b, 2.5 μM) or FACS analysis for sub-G1 apoptotic population (c, Top, a representative FACS profile and bottom, fold of change, mean±S.E.M., n=3, *P<0.05). LEX, longer exposure. (d and e) siRNA knockdown of ATG5 sensitizes MCF7 breast cancer cells to MLN4924-induced apoptosis. MCF7 cells were transfected with siRNA oligo targeting ATG5, along with scrambled control siRNA for 24 h, then subjected to MLN4924 treatment for 1 or 2 days, followed by ATPlite cell proliferation assay (d, mean±S.E.M., n=3) *P<0.05 or IB (e). (f) Blockage of autophagy sensitizes MCF7 cells to MLN4924-induced apoptosis. MCF7 cells were treated with MLN4924 alone or in combination with BAF A1 (after 24-h pre-treatment with MLN4924) for another 24 h, followed by IB. The band density was quantified and expressed as fold change, compared with the control, by arbitrarily setting the control value as 1
Discussion
MLN4924 is an investigational small molecule currently in several Phase I clinical trials as a novel agent against 4–5 types of human cancers.8, 13 MLN4924 was identified in a chemical screening for inhibitors of NAE, an enzyme which catalyzes the first-step reaction of protein neddylation.9 By inhibiting NAE activity via forming a covalent binding,10 MLN4924 blocks cullin neddylation, a process required for CRL E3 ligase activity,7, 8 and inactivates CRLs. Subsequent accumulation of several key regulatory substrates of CRLs, such as IκBα and CDT1, causes the suppression of tumor growth mainly by inducing apoptosis.9, 14, 15, 16, 17 Few recent studies also showed that MLN4924 induced senescence18, 19, 20 via inducing p21.18 Involvement of MLN4924 in other types of cell death, such as autophagy, was totally unknown. In this study, we reported our novel finding that MLN4924 is a general inducer of autophagy in all tested human cancer lines derived from carcinomas of breast, colon, cervix and brain (Figure 1 and Supplementary Figure S1).
Autophagy is a stress-responsive cell survival program by which cytoplasmic materials, including dysfunctional organelles and misfolded proteins, reach lysosomes for degradation.37 Autophagy is involved in many physiological and pathological processes.23, 37 Although multiple signaling pathways and associated proteins are known to regulate autophagy, either positively or negatively,38 the mTOR pathway is a well-established negative regulator of autophagy, given that mTORC1 inhibits autophagosome formation, whereas mTORC2 represses the expression of some autophagy-related genes and other autophagy regulators.25, 26 Likewise, rapamycin, a potent inhibitor of mTORC1, is commonly used as an autophagy inducer.25 Our mechanistic study revealed that MLN4924-induced autophagy is largely mediated by inactivation of mTORC1 (reflected by reduced mTOR/S6K1/4E-BP1 phosphorylation), but unlikely by inactivation of mTORC2 (reflected by lack of major change in AKT phosphorylation; Figure 2 and Supplementary Figure S2). We validated a causal effect of mTORC1 inactivation on autophagy induction by both genetic and pharmaceutical approaches, including siRNA knockdown, gene KO and small molecule inhibitors (Figures 3, 5 and 6, and Supplementary Figures S3, S5 and S6). We further demonstrated mechanistically that mTORC1 inactivation by MLN4924 mainly involves (1) DEPTOR, a direct inhibitor of mTORC1, and (2) the HIF1–REDD1–TSC1 axis, a known negative regulatory pathway of mTORC1. Furthermore, downregulation of RAPTOR (Figure 3), an mTORC1 component essential for its activity,39 may also contribute to mTORC1 inhibition, although the underlying mechanism is unclear.
DEPTOR is a recently discovered naturally occurring inhibitor of mTOR, which directly binds to mTOR and inhibits activities of both mTORC1 and mTORC2.21 By doing so, DEPTOR acts in general as a tumor suppressor via inhibiting protein synthesis, cell proliferation and survival effects of AKT. However, under certain circumstances, DEPTOR could act as an oncogene by relieving the feedback inhibition from S6K1 to PI3K, thus activating AKT.21 This could also explain why DEPTOR appears to be associated with suppression of mTORC1, but not mTORC2, in our study. As a physiological substrate of SCFβTrCP E3 ligase,22, 27, 28 DEPTOR was accumulated upon MLN4924 exposure in tumor cell lines expressing a low basal level (Figures 3a and b), but not in other lines with a high basal expression (data not shown). The underlying mechanism likely involves either deficiency of SCF E3 components or inactivation of DEPTOR kinases (RSK1 and S6K1)22 in high expressing lines. Functionally, we found that DEPTOR is necessary but not sufficient for MLN4924-mediated mTORC1 inhibition and autophagy induction in both low and high expressing lines (Figures 3c and d and Supplementary Figure S3), as only a partial rescue of mTORC1 inactivation and autophagy induction were observed upon DEPTOR knockdown.
In addition to DEPTOR, we found that the HIF1–REDD1–TSC1 axis is actively involved in mediating mTORC1 inhibition and autophagy induction by MLN4924. As a well-known substrate of Cul2-VHL E3,31 HIF1α was remarkably accumulated by MLN4924. Unexpectedly, however, MLN4924 failed to cause REDD1 accumulation in any of the five lines tested (Figures 4b–d and Supplementary Figures S4A and B), although REDD1 is a known hypoxia and HIF-1 target,32, 33 and a known substrate of Cul4A-DDB1 E3 ligase.34 By contrast, we observed a lower REDD1 level in MLN4924-treated cells than that in DMSO-control cells. Given the observation that REDD1 was induced at high cell density,32 it is likely that MLN4924-induced growth suppression prevented density-dependent REDD1 induction in MLN4924-treated cells. Although REDD1 levels were not accumulated upon MLN4924 treatment, REDD1, as well as HIF1 and TSC1, was found to be necessary for mTORC1 inactivation and autophagy induction by MLN4924, as demonstrated by the rescuing experiments using siRNA knockdown of REDD1 or HIF1α, and gene knockout of Hif1α or Tsc1 (Figure 5 and Supplementary Figure S5). Thus, both DEPTOR and the HIF1–REDD1–TSC1 axis are responsible for MLN4924-mediated mTORC1 inactivation and autophagy induction.
Unlike apoptosis, which directly kills tumor cells, autophagy could, in addition to cell killing, allow survival of tumor cells under a nutrient-poor environment by providing free amino acids and metabolites from degraded proteins and subcellular organelles.37, 38 It becomes clear that apoptosis and autophagy are intertwined processes, and cross-talk between them determines overall fate of the cell.40 Indeed, in some cases, autophagy potentiates and enhances apoptosis, whereas in other cases, autophagy is cytoprotective and offers drug resistance. Under the later situation, autophagy inhibition potentiates apoptosis and sensitizes cancer cells to anticancer drugs (for review, see Chen and White37 and Notte et al.41). In this study, we showed that MLN4924-induced autophagy has a survival role, as its abrogation via various approaches increases growth suppression via enhancing apoptosis.
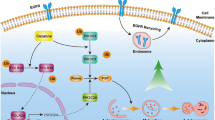
In summary, our study supports the following model. By inactivation of CRLs/SCF E3 ligases, MLN4924 causes accumulation of DEPTOR and HIF1α, two well-known substrates of Cul1-βTrCP and Cul2-VHL E3s. DEPTOR directly binds to and inhibits mTORC1 (occurring relatively late—12–24 h post treatment, Figures 3a and b), whereas HIF1α activates REDD1–TSC1 axis to block mTORC1 (occurring relatively early—4 h post treatment, Figures 4b–d), leading to induction of survival autophagy. Blockage of autophagy sensitizes tumor cells to apoptosis induced by MLN4924 (Figure 7). Our most recent results showed that inactivation of CRL E3 ligase by MLN492442 or by RBX1 knockdown43 could also induce protective autophagy in two human liver cancer cell lines. Taken together, our study reveals that MLN4924 can effectively target mTOR pathway in cancer cells44 and suggests a rational drug combination of MLN4924 with an autophagy inhibitor for effective tumor cell killing, likely leading to an increased efficacy of cancer therapy.

A model for MLN4924 induces protective autophagy (see text for details)
Materials and Methods
Cell lines
The following human cancer cell lines were obtained from ATCC and used in this study: HeLa, human cervical cancer cells; SK-BR3, MCF7, MDA-MB-231 human breast cancer cells; HCT116, human colon cancer cells; U87 human glioblastoma cells; and 293 human embryonic kidney cells. Cells were cultured in DMEM, supplied with 10% fetal calf serum. All MEFs cells with various gene knockout were also cultured in the same 10% DMEM.
Establishment of EGFP-LC3 lines and MLN4924 treatment
Several stable lines expressing EGFP-LC3 were established after transfection with an expression vector encoding EGFP-LC3 and puromycin or hygromycin selection. Cells were treated with MLN4924 (a gift from Millennium Pharmaceuticals, Inc., Cambridge, MA, USA) at various concentrations for various periods of time, followed by photography under fluorescence microscopy.
Acridine orange (AO) immunofluorescent staining
Quantification of autophagy by AO staining using flow cytometry was performed. Briefly, cells after being treated with MLN4924 at different concentrations for 24 h were stained with 1 μM AO for 15 min at 37°C, trypsinized, washed and collected in phenol red-free growth medium. Green (510–530 nm) and red (650 nm) fluorescence emission from cells illuminated with blue (488 nm) excitation light was measured by flow cytometry. Depending on their acidity, autophagic lysosomes appeared as the orange/red fluorescent cytoplasmic vesicles, while nuclei were stained green. Autophagy was quantified as a ratio between geomean fluorescence intensity of red versus green fluorescence (FL3/FL1), and the data are presented as the fold changes with an arbitrary setting of autophagy in cells treated with DMSO control as 1.
Transmission electron microscopy
HeLa and SK-BR3 cells were treated with MLN4924 at 1 μM for 24 h, respectively, and rinsed with 0.1 M Sorensen’s phosphate buffer (pH 7.4) before fixing with 2.5% glutaraldehyde in Sorensen’s phosphate buffer at room temperature for 30 min. The cell dishes were then processed at the Microscopy and Image-analysis Core at the University of Michigan for TEM analysis with photos taken at various magnifications.
Immunoblotting analysis
After the indicated treatments, cells were lysed in a Triton X-100 or RIPA buffer with phosphatase inhibitors.45 The antibodies were purchased as follows: CUL1, human HIF1α, S6K1, mTOR (Santa Cruz Biotechnology, Santa Cruz, CA, USA), β-actin, HA, (Sigma, St. Louis, MO, USA), DEPTOR, ATG5 (Millipore, Billerica, MA, CA, USA), Phospho-4E-BP1, 4E-BP1, Phospho-ERK1/2 (Thr202/Tyr204), ERK1/2, Phospho-S6K1 (Thr389), Phospho-AKT (Ser473), AKT, LC3-I/II, p62, mTOR, phosphor-mTOR-S2481, RICTOR, RAPTOR, GβL, TSC1, TSC2, Cul4A, PARP, caspase-3 and caspase-7 (Cell Signaling Technology, Danvers, MA, USA), Cul-2 and human HIF1α (BD Biosciences, San Jose, CA, USA), REDD1 (Proteintech, Chicago, IL, USA), and mouse HIF1α (Novus Biologicals, Littleton, CO, USA). The band density in western blots was quantified by Image J software.46
The in vitro kinase assay
The in vitro kinase assay was performed as described.47 Briefly, HA-tagged S6K1 was transfected into HEK293 cells; after 48 h, the cells were treated with 20 μM LY294002 for 1 h (without medium change) before cell harvesting and lysis. HA-tagged S6K1 was pulled down by bead-conjugated anti-HA Ab and eluted with HA peptide. The mTORC1 complex was isolated and purified from HCT116 cells pretreated with MLN4924 (1 μM), rapamycin (100 nM, as positive control) or DMSO vehicle control for 24 h by immunoprecipitation using anti-mTOR antibody. A kinase reaction was initiated by incubating bead-conjugated mTORC1 complex with HA-S6K1 in a kinase reaction buffer (25 mM HEPES (pH 7.4), 50 mM KCl, 10 mM MgCl2, 250 μM ATP) at 30°C with constant vortexing for 90 min. Phosphorylation of HA-S6K1 by mTORC1 was detected by IB with anti-S6K1 phospho-specific Ab.
Genotype and RT-PCR analysis of Hif1α+/+ and Hif1α−/− MEF cells by PCR
Identification of Hif1α−/− and Hif1α+/+ MEF was confirmed by genotyping as described35 and RT-PCR analysis.48 For genotype, the sequences for wild-type Hif1α were 5′-TGTAGTCTCCTGCTAAAAG-3′ (forward) and 5′-TTATTCGAGTTAAGACAAAC-3′ (reverse). The PCR condition was as follows: annealing at 94°C for 4 min, followed by 32 cycles of amplification at 94°C for 30 s, 52°C for 25 s, and 72°C for 25 s, and extension at 72°C for 7 min. The sequences for mutant allele were 5′- ACTGGCTGCTATTGGGCGAAGTG-3’ (forward) and 5′-GTAAAGCACGAGGAAGCGGTCAG-3′ (reverse) with the PCR condition of annealing at 94°C for 4 min; amplification for 32 cycles at 94°C for 30 s, 67°C for 25 s, and 72°C for 25 s; and extension at 72°C 7 for min. For RT-PCR, the sequences of the primer sets are as follows: Hif1α, 5′-AGATGAGTTCTGAACGTCGAAA-3′ (forward) and 5′-TCACTGTCTAG ACCACCGGC-3′ (reverse); GAPDH, 5′-GTATGACTCCACTCACGGCAAA-3′ (forward) and 5′-GGTCTCGCTCCTGGAAGATG-3′ (reverse).
SiRNA silencing
Cells were transfected with the following siRNA oligonucleotides by Lipofectamine 2000. ATG5: 5′-GGATGAGATAACTGAAAGG-3′; REDD1: 5′-GTGGAGACTAGAGGCAGGAGC-3′; HIF1α: 5′-CTAACTGGACACAGTGTGT-3′; DEPTOR: 5′-GCCATGACAATCGGAAATCTA-3′; and siCont: 5′-ATTGTATGCGATCGCAGACTT-3′.
ATPlite cell proliferation assay
Cells were seeded in 96-well plates and treated with various concentrations of MLN4924 for 72 h, followed by ATP-lite assay for cell proliferation.18 The results from three independent experiments, each run in triplicate were plotted.
FACS (fluorescence-activated cell sorting) analysis
Cells were treated with various concentration of MLN4924 for 24 or 48 h, followed by flow cytometry, as described.12
Statistical analysis
Paired or homoscedastic two-tailed Student’s t-test was performed.
Abbreviations
- 4E-BP1:
-
eukaryotic translation initiation factor 4E-binding protein-1
- ATG-5:
-
autophagy-related gene 5
- AO:
-
acridine orange
- BAF A1:
-
bafilomycin A1
- CRLs:
-
Cullin-RING ligases
- DEPTOR:
-
DEP domain containing mTOR-interacting protein
- EGFP:
-
enhanced green fluorescent protein
- ERK:
-
extracellular signal-regulated kinases
- FACS:
-
fluoresence-activated cell sorting
- HIF1:
-
hypoxia-inducible factor 1
- LC-3:
-
microtubule-associated protein light chain 3
- MAPK:
-
mitogen-activated protein kinases
- mTORC:
-
mammalian target of rapamycin complex
- NAE:
-
NEDD8-activating enzyme
- NEDD8:
-
neural precursor cell expressed, developmentally down-regulated 8
- RAPTOR:
-
regulatory-associated protein of mTOR
- RBX1/2:
-
RING box protein-1/2
- REDD1:
-
regulated in development and DNA damage responses-1
- ROC1:
-
regulator of cullins-1
- SAG:
-
Sensitive to Apoptosis Gene
- SCF:
-
Skp1, Cullin and F-box protein
- S6K1:
-
ribosomal S6 kinase 1
- TSC1/2:
-
tuberous sclerosis protein 1/2
References
Deshaies RJ, Joazeiro CA . RING domain E3 ubiquitin ligases. Annu Rev Biochem 2009; 78: 399–434.
Sarikas A, Hartmann T, Pan ZQ . The cullin protein family. Genome Biol 2011; 12: 220.
Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 2002; 416: 703–709.
Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW . Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev 2004; 18: 2573–2580.
Wu K, Fuchs SY, Chen A, Tan P, Gomez C, Ronai Z et al. The SCF(HOS/beta-TRCP)-ROC1 E3 ubiquitin ligase utilizes two distinct domains within CUL1 for substrate targeting and ubiquitin ligation. Mol Cell Biol 2000; 20: 1382–1393.
Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA . Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell 2008; 134: 995–1006.
Deshaies RJ, Emberley ED, Saha A . Control of cullin-ring ubiquitin ligase activity by nedd8. Subcell Biochem 2010; 54: 41–56.
Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE . The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer 2010; 1: 708–716.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009; 458: 732–736.
Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell 2010; 37: 102–111.
Orlowski RZ, Kuhn DJ . Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res 2008; 14: 1649–1657.
Wei D, Li H, Yu J, Sebolt JT, Zhao L, Lawrence TS et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res 2012; 72: 282–293.
Soucy TA, Smith PG, Rolfe M . Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res 2009; 15: 3912–3916.
Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood 2010; 116: 1515–1523.
Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood 2010; 115: 3796–3800.
Tan M, Li Y, Yang R, Xi N, Sun Y . Inactivation of SAG E3 ubiquitin ligase blocks embryonic stem cell differentiation and sensitizes leukemia cells to retinoid acid. PLoS One 2011; 6: e27726.
Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B . Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res 2011; 71: 3042–3051.
Jia L, Li H, Sun Y . Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia 2011; 13: 561–569.
Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 2010; 464: 374–379.
Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A . NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res 2010; 70: 10310–10320.
Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009; 137: 873–886.
Zhao Y, Xiong X, Sun Y . DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell 2011; 44: 304–316.
Mizushima N, Yoshimori T, Levine B . Methods in mammalian autophagy research. Cell 2010; 140: 313–326.
Bowman EJ, Siebers A, Altendorf K . Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci USA 1988; 85: 7972–7976.
Jung CH, Ro SH, Cao J, Otto NM, Kim DH . mTOR regulation of autophagy. FEBS Lett 2010; 584: 1287–1295.
Sengupta S, Peterson TR, Sabatini DM . Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell 2010; 40: 310–322.
Duan S, Skaar JR, Kuchay S, Toschi A, Kanarek N, Ben-Neriah Y et al. mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Mol Cell 2011; 44: 317–324.
Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell 2011; 44: 290–303.
Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008; 321: 1499–1502.
Hu J, Zacharek S, He YJ, Lee H, Shumway S, Duronio RJ et al. WD40 protein FBW5 promotes ubiquitination of tumor suppressor TSC2 by DDB1-CUL4-ROC1 ligase. Genes Dev 2008; 22: 866–871.
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001; 292: 464–468.
Jin HO, An S, Lee HC, Woo SH, Seo SK, Choe TB et al. Hypoxic condition- and high cell density-induced expression of Redd1 is regulated by activation of hypoxia-inducible factor-1alpha and Sp1 through the phosphatidylinositol 3-kinase/Akt signaling pathway. Cell Signal 2007; 19: 1393–1403.
Regazzetti C, Bost F, Le Marchand-Brustel Y, Tanti JF, Giorgetti-Peraldi S . Insulin induces REDD1 expression through hypoxia-inducible factor 1 activation in adipocytes. J Biol Chem 2010; 285: 5157–5164.
Katiyar S, Liu E, Knutzen CA, Lang ES, Lombardo CR, Sankar S et al. REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A-DDB1 ubiquitin ligase. EMBO Rep 2009; 10: 866–872.
Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 1998; 12: 149–162.
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432: 1032–1036.
Chen HY, White E . Role of autophagy in cancer prevention. Cancer Prev Res (Phila) 2011; 4: 973–983.
Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P . Autophagy: for better or for worse. Cell Res 2012; 22: 43–61.
Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002; 110: 177–189.
Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A . Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ 2009; 16: 966–975.
Notte A, Leclere L, Michiels C . Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem Pharmacol 2011; 82: 427–434.
Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res 2012; 72: 3360–3371.
Yang D, Li L, Liu H, Wu L, Luo Z, Li H et al. Induction of autophagy and senescence by knockdown of ROC1 E3 ubiquitin ligase to suppress the growth of liver cancer cells. Cell Death Differ 2012 doi:10.1038/cdd.2012.113.
Zhao Y, Sun Y . Targeting the mTOR-DEPTOR pathway by CRL E3 ubiquitin ligases: therapeutic application. Neoplasia 2012; 14: 360–367.
Bockbrader KM, Tan M, Sun Y . A small molecule Smac-mimic compound induces apoptosis and sensitizes TRAIL-and etoposide-induced apoptosis in breast cancer cells. Oncogene 2005; 24: 7381–7388.
Abramoff MD, Maglhaes PJ, Ram SJ . Image processing with ImageJ. Biophotonics Int 2004; 11: 36–42.
Sancak Y, Thoreen CC, Peterson TR, RA Lindquist, Kang SA, Spooner E et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 2007; 25: 903–915.
Wang Z, Sun Y . Identification and characterization of two splicing variants of human Noxa. Anticancer Res 2008; 28: 1667–1674.
Acknowledgements
We thank Drs Noboru Mizushima and Xiao-Ming Yin for providing us Atg5+/+ and Atg5−/− MEF cells; Dr Jun-Lin Guan for Tsc1+/+ and Tsc1−/− MEF cells; and Dr Greg L Semenza for HIF1α+/+ and HIF1α−/− MEF cells. We also thank Mr Chris A Edwards at the Microscopy & Image-analysis Laboratory, University of Michigan for his help in movie capture of autophagy. We also thank Millennium Pharmaceuticals, Inc. for providing MLN4924. This work was supported by the NCI grants (CA118762 and CA156744) to YS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by RA Knight
Supplementary Information accompanies the paper on Cell Death and Disease website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Zhao, Y., Xiong, X., Jia, L. et al. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis 3, e386 (2012). https://doi.org/10.1038/cddis.2012.125
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.125
Keywords
This article is cited by
-
Deciphering the role of neddylation in tumor microenvironment modulation: common outcome of multiple signaling pathways
Biomarker Research (2024)
-
KLHL3-dependent WNK4 degradation affected by potassium through the neddylation and autophagy pathway
BMC Nephrology (2023)
-
Targeting NEDD8-activating enzyme for cancer therapy: developments, clinical trials, challenges and future research directions
Journal of Hematology & Oncology (2023)
-
Nuclear ErbB2 represses DEPTOR transcription to inhibit autophagy in breast cancer cells
Cell Death & Disease (2021)
-
Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment
Signal Transduction and Targeted Therapy (2021)
