
Abstract
Post-lactational involution of the mammary gland is initiated within days of weaning. Clearing of cells occurs by apoptosis of the milk-secreting luminal cells in the alveoli and through stromal tissue remodeling to return the gland almost completely to its pre-pregnant state. The pathways that specifically target involution of the luminal cells in the alveoli but not the basal and ductal cells are poorly understood. In this study we show in cultured human mammary alveolar structures that the involution process is initiated by fresh media withdrawal, and is characterized by cellular oxidative stress, expression of activated macrophage marker CD68 and finally complete clearing of the luminal but not basal epithelial layer. This process can be simulated by ectopic addition of reactive oxygen species (ROS) in cultures without media withdrawal. Cells isolated from post-involution alveoli were enriched for the CD49f+ mammary stem cell (MaSC) phenotype and were able to reproduce a complete alveolar structure in subcultures without any significant loss in viability. We propose that the ROS produced by accumulated milk breakdown post-weaning may be the mechanism underlying the selective involution of secretory alveolar luminal cells, and that our culture model represents an useful means to investigate this and other mechanisms further.
Similar content being viewed by others
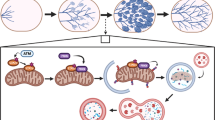

Main
The non-lactating but mature mammary epithelium in humans consists of a simple ductal network that is supported within the mammary stroma. The stroma is comprised of fibroblasts that either form dense connective tissue next to the ducts or fatty adipocytes between the lobules of the gland.1, 2 In response to hormonal changes during pregnancy, the epithelium undergoes massive expansion, differentiation and branching morphogenesis to generate the complex lobulo-alveolar structures that produce and secrete milk.3, 4 Although differentiation of the gland takes almost the full period of pregnancy to occur, subsequent involution and clearing of the secretory epithelium is initiated within several days of weaning and is almost complete within several weeks.5
The process of involution in mouse models involves a highly orchestrated series of molecular and physical events that can be divided into two distinct phases.6 Accumulation of milk in the alveolar lumen is required to initiate the first (reversible) phase during which the secretory cells begin to enter apoptosis 2 days post-weaning. At around 4 days post-weaning, the second (irreversible) phase of involution begins when matrix remodeling enzymes are upregulated.7 Expression of matrix remodeling factors is largely restricted to the stromal fibroblasts surrounding the lobulo-alveolar network, and is likely to be regulated by epithelial-mesenchymal cross-talk originating in the glandular epithelium.2, 8 By 10 days post-weaning the gland is regressed almost completely to its pre-pregnant state, with the exception of a small population of lactation-associated cells that remains at the basal surface and are believed to serve as a ‘memory’ for subsequent lactations.9, 10 Although there is a growing body of knowledge of the specific signals that drive luminal but not basal cell clearing during involution of animal models, such pathways are poorly understood in human systems.11, 12
Synthesis of milk by the secretory epithelial cells is regulated by negative feedback, as the lumen becomes filled with milk and prevents overfilling.13 Several mechanisms to initiate involution have been proposed, and experiments in animal models indicate that involution is not solely a mechano-sensory mechanism but also a biochemical mechanism that is driven by a component of the milk.14 In the mammary gland and other cell types, oxidative stress resulting from excess reactive oxygen species (ROS) has been shown to be an initiator of apoptosis.15, 16
We questioned whether ROS produced by the breakdown of the accumulated milk might be involved in the induction of apoptosis in the first phase of mammary involution. In this study, we show in vitro that involution of mammary epithelial alveoli occurs by the accumulation of ROS after media withdrawal in a pattern similar to post-weaning in vivo, and targets cell death in the luminal but not basal population. Furthermore, we show that involution can be induced by addition of ectopic ROS to fresh culture media. At the onset of irreversible luminal cell death, a population of cells expressing the macrophage marker CD68 emerges in the luminal population. We propose that ROS is a signal to initiate cell death in the ROS-sensitive luminal population, whereas the ROS-insensitive population is preserved, and that completion of clearing is maintained by a signal originating in the CD68+ luminal population.
Results
Primary mammary epithelial cells (PMEC) from lactating human tissue form differentiated, functional alveoli in culture
We generated alveolar structures in reconstituted biomatrix using a multipotent human primary mammary epithelial cell line (PMEC) isolated from lactating tissue via breastmilk (Figure 1a).17, 18 This multipotent population expresses the mammary stem cell marker CD49f, and in culture around 40% of CD49f+ cells were able to produce differentiated alveolar structures; however, cell viability and alveolar size decreased with passage number (Figure 1b). Differentiated structures consisted of a basal layer of CD49f+ cells and a luminal layer of CK18+ cells (Figure 1c). Overall growth of the alveolar units reached a plateau after around 12 days in culture, and with repetitive media changes could be maintained for over 1 month in homeostasis. At this stage luminal cells represented approximately two thirds of the total cell number per alveolus (Figure 1d). In response to prolactin treatment, subsets of the CK18+ luminal cells produced hallmark milk proteins β-casein, α-lactalbumin and human milk fat globule (HMFG) protein (Figure 1e). Both β-casein and α-lactalbumin appeared localized in the luminal cells; however, HMFG appears to be actively transported into the luminal space from the polarized luminal cells (Figure 1f).

Epithelial cells isolated from human breastmilk form differentiated alveolus-like structures that secrete milk proteins in vitro. (a) Bright-field image of alveolus-like structures in three-dimensional Matrigel culture. (b) Number of alveoli formed per 100 single cells plated in Matrigel and mean alveolus size from early to late passage primary cells. (c) Differentiation of CD49f+ (yellow) cells in Matrigel from day 4 into a bilayer with a luminal layer of CK18+ (magenta) cells by day 12. Scale=20 μM. (d) Proportion of basal (CD49f+) and luminal (CK18+) cells per total alveolar cell number. (e) Percentage of luminal cells expressing secretory markers β-casein, α-lactalbumin and HMFG. (f) Localization of secretory marker proteins in alveolar structures. Scale=100 μM (β-casein and α-lactalbumin) and 20 μM (HMFG). ***P=0.001. Graphs represent mean±S.D. of at least 20 alveoli from each of five independent experiments
Oxidative stress is associated with luminal-specific cell death in involuting alveoli
When media changes were withdrawn after complete differentiation at day 12, the cultures began to show signs of partial degradation by day 18, coinciding with a drop in extracellular pH. By day 26 the alveolar structures consisted of a single CD49f+ epithelial layer (Figure 2a). Absolute cell counts of CK18+ luminal cells indicate that the rate of cell death was not constant, and a massive loss in luminal cell integrity occurred at day 20 of culture (Figure 2b). The rate of luminal cell death could be delayed to day 22 by at least a 25-μM increase in the concentration of hydrocortisone, a known inhibitor of involution in vivo.6, 19 Accumulation of milk is a known pre-requisite for the initiation of involution, and during the breakdown of this milk ROS are produced. As ROS can induce apoptosis in sensitive cell types, we questioned whether this mechanism could also be involved in the involuting epithelial cells. Indeed, when fresh media was withdrawn alveolar cells began to undergo oxidative stress within 2 days as shown by the marker DCFDA, which is oxidized in the presence of ROS, generating a fluorescent product (Figure 2c). Notably, oxidative stress was not restricted to the luminal population (far right panel Figure 2c) and in fact occurred first in the basal cell population. Nevertheless, we only observed cell death in the luminal cells and not in the basal population. To test whether cell death was mediated by increased ROS and not other by products of cell metabolism or expiring media, we treated cultures with the reactive oxygen scavenger N-acetyl cysteine (NAC) after media withdrawal on day 12. At a minimum concentration of 5 μM NAC, initiation of luminal cell death was delayed to day 22, and this was extended with increasing concentrations of NAC (Figure 2d).

Luminal but not basal cells regress in response to fresh media withdrawal. (a) Regression of luminal (magenta) but not CD49f+ basal (yellow) compartment from day 12 (withdrawal of media changes or application of ectopic ROS) to day 26. Scale=100 μM. (b) Quantitative count of luminal cell regression per alveolus from day 12 to day 26 when media changes are withdrawn on day 12 in the presence of 15 μM, 20 μM, 25 μM or 30 μM hydrocortisone (HC). (c) Bright-field (top panel) images of alveoli on days 12, 14 and 16 after media withdrawal on day 12. Corresoponding heat maps (bottom panel) show relative oxidative stress (red=high ROS; blue=low ROS). Scale=80 μM (days 12, 14 and 16) or 60 μM (day 14 zoom). (d) Quantitative count of luminal cell regression per alveolus from day 12 to day 26 when media changes are withdrawn on day 12 in the presence of 5 μM, 10 μM, 15 μM or 20 μM N-acetyl cysteine (NAC). Graphs represent mean±S.D. of at least 20 alveoli from each of 12 independent cultures distributed evenly across three separate experiments
Ectopic addition of ROS simulates luminal cell death by fresh media withdrawal
To further investigate the response to increasing ROS concentration, we supplemented differentiated alveoli with hydrogen peroxide at day 12 of growth but continued fresh media changes. Minimal cell death occurred below 12 μM H2O2, but increased across the range to 18 μM where cell death occurred specifically in the luminal population at a rate similar to media withdrawal (Figure 3a). Increasing the concentration further to 24 μM induced cell death in both the luminal and basal compartments within 4 days. To test if this simulated cell death induced by media withdrawal we supplemented either 24 μM HC or 20 μM NAC to cultures treated with 18 μM H2O2 on day 12. Compared with control cultures treated with H2O2 but not HC or NAC, HC-treated cultures showed a delay in initiation but not subsequent rate of luminal cell death to day 22, as observed in cultures with media withdrawn (Figure 3b). Conversely, NAC-treated cultures did not show significant luminal cell death through to day 30 of culture as observed for healthy cultures with fresh media changes. When cells isolated from media-withdrawal (MW) or H2O2-treated (ROS) alveolar cultures were subcultured into fresh ECM, a subtle but significant loss in ability to form new alveoli and a decrease in alveolar size was observed, and this could be rescued by NAC-treatment but not HC (Figure 3c).

Luminal cell loss can be simulated by ectopic addition of ROS. (a) Quantitative count of luminal cell regression per alveolus from day 12 to day 30 when media was supplemented with 12 μM, 18 μM or 24 μM H2O2 on day 12. (b) Quantitative count of luminal cell regression per alveolus from day 12 to day 30 when media was supplemented with 18 μM H2O2 without inhibitor (control) or with 20 μM HC or 15 μM NAC on day 12. (c) Cells isolated from alveolar cultures with media withdrawal (MW; top panels) or treated with H2O2 (ROS; bottom panels) and supplemented with either HC or NAC were subcultured into fresh culture. Charts show quantitative changes in size of alveoli generated and percentage of cells capable of forming new alveoli compared with controls (healthy cultures with fresh media changes). Graphs represent mean±S.D. of at least 20 alveoli from each of 12 independent cultures distributed evenly across three separate experiments. *P=0.01, ***P=0.001
Subpopulations of luminal cells express macrophage marker CD68 coinciding with the onset of irreversible involution
Recent evidence has indicated that in vivo a proportion of the luminal cell compartment adopt phagocytic-like properties during involution and participate in clearing of the gland.8, 20 Some of the phagocytic population also begins to express the activated macrophage marker CD68.8, 21 To investigate this process in the clearing of luminal cells in cultures after media changes are withdrawn (natural ROS) or media changes are supplemented with H2O2 (ectopic ROS), we co-stained for CD68+ cells along with the basal and luminal markers. We observed that on day 14 (2 days after withdrawal of media or application of ectopic ROS) a proportion of the luminal layer adopted a CK18+CD68+ phenotype (Figure 4a). In the proceeding days while CK18+ expression decreased, the number of CD68+ cells remained relatively constant until days 22–26, when luminal cells were cleared to leave only a CD49f+ layer (Figure 4b). Ki67 staining of the same cultures shows that no proliferation in basal or luminal cells occurred during this process (not shown), but that there was an increase in apoptosis as indicated by an increase in the number of apoptotic cells shown by JC-1 assay (Figure 4c).

Initiation of luminal cell regression coincides with emergence of a CD68+ subpopulation and an increase in apoptotic cell markers. (a) CD68+ (white; arrows), CK18+ (magenta) and CD49f+ (yellow) cells in alveolar structures from day 14 to 26 when media changes are withdrawn on day 12. (b) Mean CD68+ cell number per alveolus from day 14 to 26 when media changes are withdrawn on day 12. (c) Percentage of total alveolar cells with depolarized mitochondria marking apoptosis on days 12, 14, 16 and 18 after media was withdrawn on day 12. Graphs represent mean±S.D. of at least 20 alveoli from each of 12 independent cultures distributed evenly across three separate experiments
Discussion
Collectively the data presented in this study provide evidence of a response of specific alveolar cell types to increasing concentrations of ROS, as would be present at the onset of involution at weaning. We propose that the luminal-specific cell death we observed in response to increased ROS supports our hypothesis that ROS-induced cell death is a potential initiator of involution in the post-weaning gland.
Although increased ROS represents a mechanism to initiate luminal cell death, it is clear that completion of clearing is mediated by other pathways, as it is irreversible after a specific timepoint post-initiation, even when ectopic ROS are withdrawn (data not shown). This temporal switch has been implicated previously in vivo and in vitro in other models.6, 22 We speculate that this timepoint may be determined by a signal originating in the CD68+ population, as this population outlives the luminal population and its emergence coincides with the timepoint at which clearing becomes irreversible.
The basal population of cells that remained after involution of the cultures was significantly less sensitive to ROS-associated cell death than the luminal population. However upon sub-cultivation it is clear that this population suffers some side effects including reduced viability and differentiation capacity. In vivo it seems that the basal cells that remain after involution survive unaffected, as they are capable of giving rise to sequential healthy lactations. It is possible that the effects we observed in culture may be alleviated if the cells were recovered for a longer time in fresh media before subcultivation. The ability of these cells to survive and recover from the stress of involution is a priority of our future research, as cells that survive but may sustain damage or mutations could be targets for transformation and tumor formation in some circumstances.
It is known that the tumor microenvironment has a high concentration of ROS and that many tumor cells are resistant to this exposure.23, 24 It has been proposed that some cancers may arise from mutations in the stem cell population (cancer stem cell hypothesis).25 In the model, we propose in Figure 5, a ROS-resistant basal stem cell that has escaped normal cell-cycle regulation can selectively survive and proliferate to form a tumor.

Schematic representation of alveolar differentiation during pregnancy and involution. In this model a CD68+ luminal population emerges at the stage of mammary involution in response to increased ROS produced by the breakdown of accumulated milk post-weaning. The CD68+ population then feeds back to promote cell death in the ROS-sensitive luminal compartment but not the ROS-insensitive basal population. In vivo this would temporally coincide with phase two of involution and irreversible regression of the lactating epithelium. We speculate that luminal progenitors that remain in the post-involution gland because they failed to acquire ROS-sensitivity during differentiation may be a target for transformation, as they are innately proliferative in response to ovarian hormones that fluctuate with the estrus cycle
The luminal progenitor population has also been shown to give rise to breast tumors.26, 27, 28 It has been shown that small populations of progenitor cells that proliferate to fuel glandular expansion during pregnancy remain in the post-involution gland. It is possible that these cells remain because they have retained the property of ROS-resistance during differentiation. These cells are subject to monthly stimulation of growth with each menstrual cycle and also underlie the massive increase in cells during pregnancy.29, 30 It is therefore possible that some tumors, particularly those associated with pregnancy, are derived from these cells and further study of them is warranted.
In conclusion it is clear that there are distinct differences between the basal and luminal populations in response to alveolar clearing during involution. Our method represents a valuable tool to begin to translate the mechanistic explanations for this from animal models to human systems.
Materials and Methods
Cell culture
Cells were isolated from expressed human breastmilk by centrifugation and propagated in adherent monolayer culture as previously described. The purified epithelial monolayer was qualified by expression of the epithelial specific antigen EpCAM and absence of fibroblast marker vimentin. Cells were maintained in monolayer culture in T75 adherent cell flasks (Sarsdedt, Germany) in HuMEC Ready Media (Invitrogen, Mulgrave, Victoria, Australia) and incubated at 37°C and 5% CO2. Three-dimensional alveolar cultures were generated by subcultivation of monolayer cells into Matrigel by the ‘on-top’ method. Lee 200718 In brief, a thin coat of Matrigel is applied to Co-Star ultra-low binding culture plates (Sigma, Sydney, New south wales, Australia) and allowed to gel at 37°C for 20 min before a single-cell suspension in HuMEC media is applied. Cells are allowed to settle for 1 h at 37°C before addition of an equal volume of 10% Matrigel in HuMEC media. Fresh media changes were carried out every 4 days with 10% Matrigel in HuMEC. All experiments were carried out on at least five biological replicates derived from the same primary cell line.
Three-dimensional cell isolation
Cell structures were isolated from extracellular matrix (ECM) culture by addition of 50 μg/ml dispase dissolved in phosphate-buffered saline (PBS) in a volume equivalent to the culture media in the well. Cultures were incubated at 37°C for 30 min–1 h (depending on volume of culture) with gentle agitation every 5 min. When intact cell structures were clearly dissociated from the surrounding matrix by gentle pipetting, the cell suspension was added to a 15-ml polypropylene centrifuge tube with 2 × volumes of 10 mM EDTA in PBS. Suspensions were mixed by inversion for 1 min then centrifuged at 300 × g for 10 min. The supernatant was carefully aspirated with a serological pippete and the cell pellet (consisting of intact alveolar structures and single cells) was resuspended in 1 ml of 10 mM EDTA, transferred to a microfuge tube and centrifuged at 500 × g for 5 min. The washed cell pellet was fixed by resuspending cells in 1 ml of 10% formaldehyde (Sigma) in PBS for 10 min at room temperature. Fixed cells were isolated by centrifugation at 500 × g for 5 min and washed once in PBS before transfer to a fresh tube for antibody staining.
Immunofluorescent antibody staining
Fixed cells were stained in solution in 1.5-ml microfuge tubes, and incubations were carried out on a rotating rocker at 4°C in the dark. Antibody dilutions and washes were carried out in staining solution consisting of 2% normal goat serum (NGS; Invitrogen) in 0.1% Triton-X (Sigma) in PBS. Cells were initially blocked and permeabilized in 10% NGS in 0.1% Triton-X in PBS for 30 min, then incubated in 500 μl of primary antibody diluted 1 : 250 overnight. Cells were washed twice for 20 min per wash, then incubated in 500 μl of secondary antibody diluted 1 : 500 plus 2 mM Hoescht 33342 (Molecular Probes, Mulgrave, Victoria, Australia) for 6 h. Finally cells were washed three times in 0.1% Triton-X in PBS for 20 min per wash then mounted in ProLong Gold Antifade Reagent (Molecular Probes) on glass microscope slides and sealed with number 1.5 glass coverslips (ProSciTech, Kirwan, Queensland, Australia). Mounted cells were allowed to cure overnight at room temperature before storage at 4°C in a sealed container. Primary antibodies used were all monoclononal anti-human antibodies from AbCam (Cambridge, MA, USA) including rat CD49f, rabbit CK18, mouse β-casein, mouse α-lactalbumin, mouse human milk fat globule (HMFG) and rabbit CD68. Secondary antibodies used were all goat antibodies purchased from Molecular Probes including anti-rat AF680, anti-rabbit AF488 or AF547, and anti-mouse AF488 or AF547.
Oxidative stress analysis
H2DCFDA general oxidative stress indicator was used as previously described.31 Briefly, growth medium was removed from cultures and replaced with CM-H2DCFDA (Invitrogen) diluted to 100 μM in MEM. Cells were incubated for 30 min at 37°C and 5% CO2 in the dark, then washed three times in PBS before transfer to a 30-mm imaging dish with coverslip bottom (Mattek, Ashland, MA, USA). Cells were allowed to settle for 30 min in the microscope live-imaging chamber (Tokai Hit, Japan) at 37°C and 5% CO2 before confocal sections were collected with excitation at 488 nm and emission at 530 nm. Cellular oxidative stress was quantified by the mean fluorescence intensity in the non-nuclear cytoplasmic space and expressed relative to extracellular space by heat mapping in Image J (National Institutes of Health, Bethesda, MD, USA) software.
Cellular apoptosis assay
JC-1 membrane potential indicator dye was used as previously described.31 Briefly, media was removed from cultures and replaced with 10 μg/ml JC-1 reagent (Invitrogen) diluted in MEM. Cells were incubated for 10 min at 37°C and 5% CO2 in the dark, then washed once in PBS before transfer to a 30-mm imaging dish. Cells were allowed to settle for 30 min in the microscope live-imaging chamber at 37°C and 5% CO2 before confocal sections were collected with excitation at 488 nm and simultaneous emission at 530 nm (monomer) and 590 nm (J-aggregate). Image J software was used to calculate the ratio of monomer/J-aggregate by dividing each 8-bit 530 nm image by its corresponding 8-bit 590 nm image. Mitochondria area was defined using the 530 nm image and all three images were integrated to compute mean pixel intensity.
Fluorescent microscopy and cell analysis
Stained cells were visualized using a Nikon (Melville, NY, USA) confocal microscope and data were collected with the Nikon NIS Elements (Melville, NY, USA) software package. Raw images were exported to 8-bit TIFF files and analyzed using Image J software. Positively expressing cells were validated using light intensity cutoffs relative to positive and negative control cell samples. Autofluorescence and background was removed by normalizing the light offset to no-primary antibody controls. Automated positive-cell counts per optical confocal section were collected after adjusting the light intensity threshold for each wavelength empirically on a minimum of three positive and three negative control samples. Total cell counts per section were collected on the Hoescht channel and marker-positive cell counts were expressed as a proportion of this.
Statistical analysis
Cell count data were expressed as the mean±S.D. Statistical comparisons were carried out by GraphPad Prism5 (La Jolla, CA, USA) software. The students’ t-test was used for two-group comparisons. The statistical difference between samples for multiple groups was determined using analysis of variance followed by Tukey's multiple-comparison tests.
Author Contributions
ET conducted the experimental work and wrote the initial manuscript. NZ provided extensive advice and intellectual input to data interpretation and presentation, and contributed to and reviewed the manuscript. PR supervised and assisted microscopy data collection and analysis and reviewed the manuscript. PEH provided laboratory space and materials and funding for experimental work, ongoing intellectual advice and contributed detailed criticism of the manuscript.
Abbreviations
- MaSC:
-
mammary stem cell
- ROS:
-
reactive oxygen species
- PMEC:
-
primary mammary epithelial cells
- HMFG:
-
human milk fat globule
- MW:
-
media withdrawal
- α-Lac:
-
α-lactalbumin
- β-cas:
-
β-casein
- Prl:
-
prolactin
- CK18:
-
cytokeratin 18
- CK14:
-
cytokeratin 14
- DCFDA:
-
dichlorofluoroscein diacetate
- NAC:
-
N-acetyl cysteine
- ECM:
-
extracellular matrix
References
Watson CJ . Post-lactational mammary gland regression: molecular basis and implications for breast cancer. Expert Rev Mol Med 2006; 8: 1–15.
Wiseman BS, Werb Z . Stromal effects on mammary gland development and breast cancer. Science 2002; 296: 1046–1049.
Visvader JE, Lindeman GJ . Mammary stem cells and mammopoiesis. Cancer Res 2006; 66: 9798–9801.
Watson CJ, Khaled WT . Mammary development in the embryo and adult: a journey of morphogenesis and commitment. Development 2008; 135: 995–1003.
Li M, Liu X, Robinson G, Bar-Peled U, Wagner KU, Young WS et al. Mammary-derived signals activate programmed cell death during the first stage of mammary gland involution. Proc Natl Acad Sci U S A 1997; 94: 3425–3430.
Lund LR, Romer J, Thomasset N, Solberg H, Pyke C, Bissell MJ et al. Two distinct phases of apoptosis in mammary gland involution: proteinase-independent and -dependent pathways. Development 1996; 122: 181–193.
Talhouk RS, Bissell MJ, Werb Z . Coordinated expression of extracellular matrix-degrading proteinases and their inhibitors regulates mammary epithelial function during involution. J Cell Biol 1992; 118: 1271–1282.
Monks J, Henson PM . Differentiation of the mammary epithelial cell during involution: implications for breast cancer. J Mammary Gland Biol Neoplasia 2009; 14: 159–170.
Monks J, Geske FJ, Lehman L, Fadok VA . Do inflammatory cells participate in mammary gland involution? J Mammary Gland Biol Neoplasia 2002; 7: 163–176.
Wagner KU, Boulanger CA, Henry MD, Sgagias M, Hennighausen L, Smith GH . An adjunct mammary epithelial cell population in parous females: its role in functional adaptation and tissue renewal. Development 2002; 129: 1377–1386.
Watson CJ . Immune cell regulators in mouse mammary development and involution. J Anim Sci 2009; 87 (13 Suppl): 35–42.
Baxter FO, Neoh K, Tevendale MC . The beginning of the end: death signaling in early involution. J Mammary Gland Biol Neoplasia 2007; 12: 3–13.
Pang WW, Hartmann PE . Initiation of human lactation: secretory differentiation and secretory activation. J Mammary Gland Biol Neoplasia 2007; 12: 211–221.
Sutherland KD, Lindeman GJ, Visvader JE . The molecular culprits underlying precocious mammary gland involution. J Mammary Gland Biol Neoplasia 2007; 12: 15–23.
Miranda SG, Wang YJ, Purdie NG, Osborne VR, Coomber BL, Cant JP . Selenomethionine stimulates expression of glutathione peroxidase 1 and 3 and growth of bovine mammary epithelial cells in primary culture. J Dairy Sci 2009; 92: 2670–2683.
Monastyrskaya K, Tschumi F, Babiychuk EB, Stroka D, Draeger A . Annexins sense changes in intracellular pH during hypoxia. Biochem J 2008; 409: 65–75.
Thomas E, Zeps N, Cregan M, Hartmann P, Martin T . 14-3-3σ (sigma) regulates proliferation and differentiation of multipotent p63-positive cells isolated from human breastmilk. Cell Cycle 2011; 10: 278–284.
Lee GY, Kenny PA, Lee EH, Bissell MJ . Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods 2007; 4: 359–365.
Ossowski L, Biegel D, Reich E . Mammary plasminogen activator: correlation with involution, hormonal modulation and comparison between normal and neoplastic tissue. Cell 1979; 16: 929–940.
Monks J, Rosner D, Geske FJ, Lehman L, Hanson L, Neville MC et al. Epithelial cells as phagocytes: apoptotic epithelial cells are engulfed by mammary alveolar epithelial cells and repress inflammatory mediator release. Cell Death Differ 2005; 12: 107–114.
Monks J, Smith-Steinhart C, Kruk ER, Fadok VA, Henson PM . Epithelial cells remove apoptotic epithelial cells during post-lactation involution of the mouse mammary gland. Biolo Reprod 2008; 78: 586–594.
Stein T, Salomonis N, Gusterson BA . Mammary gland involution as a multi-step process. J Mammary Gland Biol Neoplasia 2007; 12: 25–35.
Tobar N, Guerrero J, Smith PC, Martinez J . NOX4-dependent ROS production by stromal mammary cells modulates epithelial MCF-7 cell migration. Br J Cancer 103 (7): 1040–1047.
Mori K, Shibanuma M, Nose K . Invasive potential induced under long-term oxidative stress in mammary epithelial cells. Cancer Res 2004; 64: 7464–7472.
Visvader JE, Lindeman GJ . Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8: 755–768.
Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010; 7: 403–417.
Lyons TR, Schedin PJ, Borges VF . Pregnancy and breast cancer: when they collide. J Mammary Gland Biol Neoplasia 2009; 14: 87–98.
O’Brien J, Schedin P . Macrophages in breast cancer: do involution macrophages account for the poor prognosis of pregnancy-associated breast cancer? J Mammary Gland Biol Neoplasia 2009; 14: 145–157.
Asselin-Labat ML, Shackleton M, Stingl J, Vaillant F, Forrest NC, Eaves CJ et al. Steroid hormone receptor status of mouse mammary stem cells. J Natl Cancer Inst 2006; 98: 1011–1014.
Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER et al. Control of mammary stem cell function by steroid hormone signalling. Nature 2010; 465: 798–802.
Wadia JS, Chalmers-Redman RM, Ju WJ, Carlile GW, Phillips JL, Fraser AD et al. Mitochondrial membrane potential and nuclear changes in apoptosis caused by serum and nerve growth factor withdrawal: time course and modification by (-)-deprenyl. J Neurosci 1998; 18: 932–947.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Medela AG, Switzerland, financially supports Professor Peter Hartmann's laboratory. The remaining authors declare no conflict of interest.
Additional information
Edited by A Finazzi-Agró
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Thomas, E., Zeps, N., Rigby, P. et al. Reactive oxygen species initiate luminal but not basal cell death in cultured human mammary alveolar structures: a potential regulator of involution. Cell Death Dis 2, e189 (2011). https://doi.org/10.1038/cddis.2011.69
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.69
Keywords
This article is cited by
-
Pistacia lentiscus extract enhances mammary epithelial cells’ productivity by modulating their oxidative status
Scientific Reports (2020)
-
Investigation on the suitability of milk-derived primary bovine mammary epithelial cells grown on permeable membrane supports as an in vitro model for lactation
In Vitro Cellular & Developmental Biology - Animal (2020)
-
Stem cells in human breast milk
Human Cell (2019)
-
A genetic variant in SLC30A2 causes breast dysfunction during lactation by inducing ER stress, oxidative stress and epithelial barrier defects
Scientific Reports (2018)
-
Gene expression in breastmilk cells is associated with maternal and infant characteristics
Scientific Reports (2015)
