
Abstract
Isthmin (ISM) is a 60 kDa secreted-angiogenesis inhibitor that suppresses tumor growth in mouse and disrupts vessel patterning in zebrafish embryos. It selectively binds to alphavbeta5 (αvβ5) integrin on the surface of endothelial cells (ECs), but the mechanism of its antiangiogenic action remains unknown. In this work, we establish that soluble ISM suppresses in vitro angiogenesis and induces EC apoptosis by interacting with its cell surface receptor αvβ5 integrin through a novel ‘RKD’ motif localized within its adhesion-associated domain in MUC4 and other proteins domain. ISM induces EC apoptosis through integrin-mediated death (IMD) by direct recruitment and activation of caspase-8 without causing anoikis. On the other hand, immobilized ISM loses its antiangiogenic function and instead promotes EC adhesion, survival and migration through αvβ5 integrin by activating focal adhesion kinase (FAK). ISM unexpectedly has both a pro-survival and death-promoting effect on ECs depending on its physical state. This dual function of a single antiangiogenic protein may impact its antiangiogenic efficacy in vivo.
Similar content being viewed by others


Main
Angiogenesis, formation of new blood vessels from pre-existing capillaries, is critical for embryonic development, wound healing, reproduction, as well as tumor progression and metastasis.1 Angiogenesis inhibitors hence offer new promises as therapeutic agents for cancer therapy. In vivo, angiogenesis is believed to be regulated by a balance of pro and antiangiogenic factors. Although a number of endogenous angiogenesis inhibitors such as thrombospondin-1 (TSP-1) and endostatin have been discovered, the full spectrum of angiogenesis inhibitors in vivo and how they regulate angiogenesis remain to be fully elucidated.2
Isthmin (ISM), a 60 kDa secreted matricellular protein, was originally identified as a gene highly expressed in the midbrain-hindbrain organizer in Xenopus embryos.3 It contains a centrally located thrombospondin type 1 repeat (TSR) domain and a C-terminal domain called adhesion-associated domain in MUC4 and other proteins (AMOP). We recently reported that ISM is a novel endogenous angiogenesis inhibitor with functions in both physiological and pathological angiogenesis.4 It suppresses multiple aspects of in vitro angiogenesis including endothelial cell (EC) capillary network formation on Matrigel and EC proliferation. It also induces EC apoptosis. In mice, overexpression of ISM in B16 melanoma cells inhibited tumor growth and angiogenesis. Knockdown of ISM in zebrafish embryos led to abnormal intersegmental vessel (ISV) formation in the trunk.4 Although ISM was shown to selectively bind to alphavbeta5 (αvβ5) integrin, the mechanism of its antiangiogenic action is still largely unclear.
To decipher ISM's mechanism of action, we addressed the following questions in this work: (1) Does αvβ5 integrin mediate ISM's antiangiogenic activities, such as inducing apoptosis? (2) How does ISM induce EC apoptosis? (3) Would ISM generate similar anti-endothelial effects in both soluble and anchored conditions? (4) Would ISM function as a cell adhesion molecule? If yes, how does it compare with other cell adhesion molecule in the extracellular matrix (ECM)? Using both neutralizing antibody as well as RNAi gene expression knockdown, we demonstrated that suppressing integrin αvβ5 could stifle apoptosis induced by ISM and relieve ISM's inhibition of EC proliferation. We further established that ISM induces EC apoptosis through integrin-mediated death (IMD) by activating caspase-8 without causing anoikis. Although soluble ISM potently induces apoptosis, immobilized ISM loses the proapoptotic activity and instead promotes EC adhesion, survival and haptotactic migration. These results suggest that the matricellular protein ISM in different physical state can trigger different intracellular signaling pathways and generate different biological effects through the same integrin receptor. It can function as an integrin agonist as well as an antagonist. This is different from classical ECM integrin ligands such as vitronectin (VN) or fibronectin (FN).
Results
Soluble ISM inhibits angiogenesis and induces EC apoptosis through αvβ5 integrin
We have previously reported that soluble ISM produced from E. coli functions as an angiogenesis inhibitor, inhibiting EC tube formation, inducing EC apoptosis and suppressing vascular endothelial growth factor (VEGF)-stimulated EC proliferation in a dose-dependent manner.4 We also found that ISM selectively binds to αvβ5, but not other integrins expressed in ECs.4 We show here that recombinant ISM protein produced from cultured mammalian cells (HEK293 T) has similar antiangiogenic and apoptosis-induction activity (Supplementary Figure S1). Because of cost-effective production, recombinant ISM made from bacteria is used in this study.
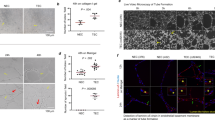
As αvβ5 integrin is a cell surface receptor of ISM, we wish to determine whether ISM inhibits angiogenesis and induces apoptosis through this integrin. In ECs (human umbilical vein endothelial cells, HUVEC) used in this study, integrin β3 or β5 subunit only pairs with αv subunit, with αvβ3 expressed at a much higher level than αvβ5.5, 6 Knockdown of β3 or β5 integrin subunit expression in ECs had no effect on cell adhesion on gelatin, FN or collagen I (data not shown), possibly due to the presence of β1 integrins such as α5β1 in these cells. It is known that β1 integrin mediate cell adhesion to gelatin, FN and collagen I.7 Hence the effect of knockdown of β3 or β5 integrin subunit expression on ISM-mediated inhibition of EC tube formation and induction of apoptosis was carried out in cells seeded on gelatin. As shown in Figure 1a, introduction of siRNA significantly reduced β3 and β5-integrin expression up to 70% in ECs. However, only knockdown of β5-integrin expression alleviated ISM-mediated inhibition of tube formation (Figure 1b). Similarly, ISM-induced EC apoptosis was selectively suppressed by β5, but not β3-knockdown (Figure 2a). In the absence of ISM, knockdown of these integrins did not induce apoptosis. Similarly, only knockdown of β5-integrin expression significantly rescued ISM-mediated inhibition of VEGF-induced EC proliferation (Figure 2b). These results demonstrate that αvβ5 integrin has an essential role in ISM-mediated angiogenesis inhibition and apoptosis.

ISM inhibits in vitro angiogenesis through αvβ5 integrin. (a) Transient transfection of β3 and β5 siRNA effectively knocked down their expression in ECs. Same amount of corresponding scrambled siRNA are transfected as control. (b) Only knockdown of β5-integrin expression abolished the anti-tube formation activity of ISM

ISM induces EC apoptosis through αvβ5 integrin. (a) Knockdown of β5, but not β3 integrin, suppressed ISM-induced apoptosis. (b) Knockdown of β5 integrin rescued ISM-suppressed proliferation. (c) More free αvβ5 integrins are available in ECs growing on FN-coated surface. VN, vitronectin; ISM, isthmin; FN, fibronectin. (d) ISM induces more extensive apoptosis in ECs grown on FN-coated surface
As αvβ5 is essential for ISM-induced apoptosis, the amount of free αvβ5 available for ISM binding should correspond to the extent of EC apoptosis. In ECs attached on VN- or ISM-coated surface, both ligands of αvβ5 integrin, the free αvβ5 available for soluble ISM binding are reduced, comparing with ECs attached on FN-coated surface (Figure 2c), most likely due to αvβ5 occupancy by VN or ISM. Correspondingly, soluble ISM induced only marginal apoptosis in ECs attached on VN and ISM, compared with significant apoptosis with ECs attached on FN (Figure 2d).
Together, these results demonstrate that soluble ISM suppresses angiogenesis and induces EC apoptosis through interacting with αvβ5 integrin on EC surface without causing anoikis. The extent of ISM-induced apoptosis depends on the number of free αvβ5 integrin on EC surface that are available for soluble ISM binding.
Soluble ISM induces EC apoptosis through IMD
It is known that certain cyclic RGD peptides8 or non-peptide RGD mimetics9 induce EC detachment from adhered surface (de-adhesion) through binding to integrins, and hence causing apoptosis through anoikis. To determine whether ISM induces EC apoptosis through anoikis, we examined whether ISM induces detachment of adherent ECs. The adhesion of ECs on different immobilized ECM proteins is mediated by distinct integrins. For example, EC adhesion to VN is mediated primarily by αvβ3 and αvβ5 integrins, whereas EC adhesion to FN occurred mainly through α5β1 integrin.5 As shown in supplementary (Supplementary Figure S2), ISM added into the culture media did not induce de-adhesion of ECs plated on FN- or VN-coated surface, regardless of whether its receptor αvβ5 integrin is bound by a ligand (VN) or not (FN). Under the same condition, ISM potently induced EC apoptosis (Xiang et al.4 and Figure 2a).
We previously established that ISM induces EC apoptosis through a caspase-dependent pathway.4 To further decipher the mechanism of ISM-induced apoptosis, we used specific inhibitors for caspase-3, -8 and -9 to block the extrinsic and intrinsic apoptosis pathways. As shown in Figure 3a, caspase-3 and -8 inhibitors effectively prevented ISM-induced apoptosis, whereas caspase-9-specific inhibitor had no effect.

ISM induces apoptosis through IMD. (A) ISM induced apoptosis through caspase-8-mediated extrinsic pathway, but not caspase-9-mediated intrinsic pathway. (B) Kinetics of ISM-induced apoptosis. Activated caspase-8 can first be detected 4 h after ISM treatment. (C) Knockdown of β5-integrin subunit by siRNA suppressed caspase-8 and caspase-3 activation, as well as cleavage of PARP. (D) ISM induces caspase-8 association with αvβ5 integrin and activation. (E) Soluble ISM induces αvβ5-integrin clustering. Co-localization of β5 integrin (red) with ISM (green) is indicated by yellow signals. DAPI (blue) stains the nucleus. Scale bar: 20 μm
A gradual activation of caspase-8 and caspase-3 under ISM treatment was observed (Figure 3b). The active form of caspase-8 (20 kDa) was first detected at 4 h post ISM treatment, whereas activated caspase-3 was first detected after 16 h ISM treatment. No cleaved form of caspase-9 was detected even after 24 h treatment. As a hallmark indicator of apoptosis, the cleaved form of poly ADP ribose polymerase (PARP, 87 kDa) was also found to gradually increase (Figure 3b). Activation of caspase-8 and caspase-3 by ISM depends on αvβ5 integrin as knockdown of β5 integrin by siRNA-suppressed ISM-induced caspase activations and PARP cleavage (Figure 3c). These results established that ISM induces apoptosis via the extrinsic apoptosis pathway through caspase-8 activation.
It has been previously reported that unligated or antagonized integrin can cluster on cell surface and subsequently recruit caspase-8 to plasma membrane, where it becomes activated in a death receptor-independent manner, a phenomenon termed IMD.10 To determine if ISM initiates apoptosis through IMD, co-immunoprecipitation was performed to examine the interaction between αvβ5 integrin and caspase-8. As shown in Figure 3d, ISM induced the association of procaspase-8 (55 kDa) and activated caspase-8 (20 kDa) with αvβ5 integrin. A Fas antagonist did not prevent ECs from ISM-induced apoptosis, indicating that Fas is not involved in ISM-induced cell death (Supplementary Figure S3). Correspondingly, Fas-associated protein with death domain (FADD) was not detected in ISM-induced co-immunoprecipitate sample (data not shown), indicating that FADD is not involved. We therefore conclude that soluble ISM act as a αvβ5-integrin antagonist and induce EC apoptosis through IMD.
We found that soluble ISM induced integrin clustering (Figure 3E), with EC bound ISM (green) and integrin β5-subunit (red) co-localized in cell periphery in clusters. The clustered integrin may be responsible for the recruitment and activation of caspase-8.
Immobilized ISM promotes EC survival and migration
After its secretion, ISM could potentially exist in different physical states in vivo, such as immobilized in the ECM of a tissue or soluble in circulation. Indeed, we detected ISM in the mouse plasma (98 nM) and the ECM (in Matrigel, 91.5 nM). Interestingly, when ISM was mixed with Matrigel, it lost the ability to inhibit EC tube formation. Soluble ISM at 1 μM in culture medium completely abolished EC tube formation on Matrigel, whereas immobilized ISM in Matrigel had no effect on tube formation even at 3 μM (Figure 4).

Immobilization of ISM by ECM abolished its anti-tube formation activity. Recombinant ISM was added into culture medium (left) or mixed into Matrigel (right). The concentrations of ISM were labeled in the panels. Scale bar represents ? μm. Control indicates culture medium (containing 2% FBS)
Integrin-mediated cell attachment on immobilized ECM ligands such as FN or VN results in cell spreading, focal adhesion and actin stress fiber formation, as well as activation of focal adhesion kinase (FAK).11 To determine if immobilized ISM could also function as a cell adhesion molecule and support EC survival, we plated ECs onto ISM-coated surface. As shown in Figure 5a, immobilized ISM supported EC adhesion in a dose-dependent and saturable manner. ECs spread on ISM-coated surface in a similar fashion to VN-coated surface (Figure 5B, panels a and b). The adhesion and spreading was completely abolished in the presence of 10 mM EDTA, consistent with the fact that divalent cations are essential for integrin-mediated cell adhesion11 (Figure 5B, panels c and d). ECs attached on ISM-coated surface proliferate at similar rate, compared with ECs attached on VN or gelatin (data not shown). Immobilized ISM also induced actin stress fiber formation and integrin clustering, similar to VN (Figure 5B, panels e–h). Therefore, immobilized ISM supports EC adhesion and survival as a cell adhesion molecule.

Immobilized ISM promotes EC adhesion through αvβ5 integrin. (A) ECs attach to ISM-coated surface in a dose-dependent and saturable manner. (B) Similar to VN, ISM supports EC adhesion and spreading, induces integrin clustering and actin stress fiber formation. Cell spreading in the absence or presence of EDTA are shown in phase contrast micrographs (panels A–D, scale bar: 40 μm). Actin stress fibers (red) and β5 integrin clustering (green) are shown in panels E–H (scale bar: 20 μm). (C) ISM mediates EC adhesion through αvβ5 integrin. (D) Knockdown of β3 and β5 integrin suppresses EC adhesion to VN. (E) Knockdown of β5 integrin suppresses EC adhesion to ISM
We further demonstrated that ECs adhere to ISM-coated surface through αvβ5 integrin, as αvβ5 blocking antibody blocked 85% of this adhesion, whereas control IgG or αvβ3 blocking antibody had no blocking activity (Figure 5c). Anti-αv or anti-β5-subunit antibody also blocked EC adhesion to ISM-coated surface by 56 and 61%, respectively, whereas anti-β1 and anti-β3 antibody had no effect. Collectively, these results confirmed that αvβ5 integrins mediate EC adhesion to ISM and serves as receptor for immobilized ISM. In comparison, EC attachment to collagen I was inhibited by anti-β1(50%), or attachment to VN was inhibited by anti-αvβ3(51%) and anti-αvβ5(29%), respectively, consistent with the ligand-receptor specificity (Supplementary Figure S4, A and B).
Knockdown of either β3 or β5-integrin subunit expression by 50-70% through siRNA significantly suppressed cell adhesion on VN-coated surface (Figure 5d), with β3-knockdown having more potent suppression (Figure 5d). This is consistent with the much higher level of β3 integrin expression in HUVECs and the fact that αvβ3 is the main VN receptor.6, 12 In contrast, only knockdown of β5 integrin reduced cell adhesion to ISM-coated surface, consistent with ISM as a αvβ5 specific ligand (Figure 5e). Correspondingly, knockdown of β3 or β5 integrin subunits had no effect on EC adhesion to type I collagen, FN and gelatin (data not shown).
FAK is an important mediator of cell survival, proliferation and migration.13 Integrin clustering leads to FAK phosphorylation and subsequent recruitment to focal adhesions.14 The most crucial site for FAK activation is Y397, which is an Src-binding site. Comparing with cells in suspension, immobilized ISM induced FAK Y397 phosphorylation, similar to immobilized VN (Figure 6a). Notably, immobilized ISM no longer induces EC apoptosis and present similar basal level apoptosis as immobilized VN, suggesting that immobilized ISM functions as an integrin agonist and transmit positive intracellular signals (Supplementary Figure S5).

Immobilized ISM activates FAK and promotes EC haptotactic migration. (a) Immobilized ISM activates FAK Y397 phosphorylation in similar fashion as immobilized VN. S, cells in suspension. (b) ISM dose-dependently promotes EC haptotactic migration with similar potency as FN. (c) ISM promotes EC haptotactic migration through αvβ5 integrin
ECs migrate towards a higher concentration of anchored cell adhesion molecule, a phenomenon termed haptotaxis. Immobilized ISM induced EC haptotactic migration in a concentration-dependent manner with similar potency as FN (Figure 6b). This haptotactic migration is blocked by anti-αv, anti-β5 and anti-αvβ5 antibodies, but not by anti-β1, anti-β3 or anti-αvβ3 antibodies (Figure 6c), indicating αvβ5 as the mediator of this function.
Altogether, the above results revealed that immobilized ISM no longer induces apoptosis, but serves as an adhesion molecule for ECs and an agonist of αvβ5 integrin. It functions in similar fashion as VN or FN, inducing αvβ5 integrin clustering and actin stress fiber formation, promotes EC adhesion, survival, haptotactic migration and stimulates FAK activation.
ISM binds to αvβ5 integrin through a novel ‘RKD’ motif
Using a solid-phase ELISA-based binding assay, we determined that ISM binds αvβ5 integrin with a Kd around 40 μM, in the range of integrin ECM ligand binding affinities (Supplementary Figure S6). We have previously determined that ISM binds to αvβ5 integrin through its C-terminal AMOP domain, and the AMOP domain alone can inhibit EC tube formation in vitro.4 However, there is no ‘RGD’ sequence within the AMOP domain of ISM. To determine how ISM binds to αvβ5 integrin, we produced recombinant proteins, corresponding to overlapping N- and C-terminal truncations of AMOP domain (Figure 7a). ECs attached to AMOP-N coated surface with similar efficacy as the intact AMOP domain, whereas AMOP-C could not support EC attachment at all (Figure 7b). It has been reported that substitution of G for K in the RGD motif of a 12 residue synthetic hepatitis-A peptide did not change the physiochemical properties of the peptide.15 However, it is not known if ‘RKD’ can function as an integrin binding motif. In AMOP-N, there is an ‘RKD’ motif at amino acids 315–317. We mutated the ‘KD at 316–317 position to AA’ and produced the mutant AMOPKD316AA protein in the intact AMOP background (Figure 7a). As shown in Figure 7b, AMOPKD316AA completely lost the ability to support EC adhesion. Furthermore, AMOPKD316AA also lost the ability to inhibit EC tube formation on Matrigel (Figure 7c). These results demonstrate that ISM binds to EC surface αvβ5 integrin through a novel ‘RKD’ motif located in its C-terminal AMOP domain. This motif has a critical role in the antiangiogenic as well as pro-survival functions of ISM. ‘RKD’ is therefore a novel integrin-binding motif, which binds αvβ5, but not αvβ3 integrin.

Identification of the αvβ5 integrin-binding site in ISM. (a) Schematic diagrams showing the truncated ISM protein fragments produced as recombinant proteins in E. coli. Numbers refer to the amino acid position in the full-length secreted ISM protein. The positon of KD316AA mutation is indicated by the arrowhead. The black triangle at the right end of each truncated protein represent His-tag. (b) EC adhesion to various ISM protein fragments compared with 0.2% gelatin. 3% BSA was used as the negative control. (c) KD316AA mutation abolished the anti-tube formation activity of ISM
Discussion
In this work, we demonstrated that the recently identified matricellular angiogenic inhibitor ISM functions through its cell surface receptor αvβ5 integrin. Although soluble ISM suppresses EC tube formation and induces EC apoptosis as an integrin antagonist, immobilized ISM functions as an integrin agonist and promotes cell survival and migration. The antiangiogenic activity of ISM is lost when ISM is immobilized in ECM. We demonstrated that mouse ISM is present in soluble (plasma) and immobilized form (extracellular matrix), thus its dual activity may have an important role in vivo.
We also identified a novel αvβ5-integrin-binding site ‘RKD’ located in the AMOP domain of ISM. Loss of αvβ5 binding through point mutations of this ‘RKD’ motif obliterated the antiangiogenic activity of ISM in vitro and abolished the ability of ISM to support EC adhesion. Therefore, integrin αvβ5 has an essential role in the antiangiogenic as well as pro-survival functions of ISM. This dual function of ISM through the same cell surface integrin receptor and the intracellular signaling pathways involved are summarized in Figure 8. As αvβ5 integrin is upregulated in tumor blood vessels, ISM most likely inhibits tumor angiogenesis through this integrin in vivo.

Schematic illustration of the dual functions of ISM. (a) Immobilized ISM serves as integrin agonist to mediate cell survival/migration by activating FAK. (b) Soluble ISM induces cell apoptosis through IMD (direct activation of caspase-8). ‘?’ indicates the unresolved question if additional molecules are involved in the integrin–caspase-8 complex formation
Similar to VN, immobilized ISM promoted EC attachment and spreading, integrin clustering, actin stress fiber formation, FAK activation and EC haptotactic migration (Figures 5 and 6). The clustering of integrins upon ECM engagement is known to transmit positional cues from ECM to intracellular signaling cascades that promote cell survival.14 Both neutralizing antibody to αvβ5 integrin, as well as siRNA knockdown of β5 integrin, abrogated EC adhesion to immobilized ISM, demonstrating that immobilized ISM is an αvβ5 integrin agonist (Figure 5).
Different from VN, soluble ISM inhibits in vitro angiogenesis, induces EC apoptosis and suppresses EC proliferation in an αvβ5 integrin-dependent manner (Figures 1 and 2). Knockdown of β5, but not β3 integrin in ECs rendered ISM inactive in inhibition of tube formation and induction of apoptosis. Correspondingly, ISM induced more extensive apoptosis in ECs cultured on non-αvβ5 ligand-coated surface such as FN (Figure 2d), due to the higher amount of free αvβ5 integrin available for soluble ISM binding in cells adhered to FN comparing with VN or ISM (Figure 2c). As ISM binds to αvβ5 integrin with a low affinity (Kd around 40 μM), it cannot effectively displace bound VN by competitively binding to αvβ5 integrin (Supplementary Figure S6).
We further demonstrated that ISM induces apoptosis through IMD by recruiting caspase-8 to the plasma membrane and activating caspase-8 in a Fas-independent manner. Both the zymogen and active forms of caspase-8 are found in the αvβ5 protein complex upon ISM induction of apoptosis (Figure 3d). Soluble ISM therefore serves as an integrin antagonist and transmits a direct death signal involving caspase-8 and caspase-3. This is similar to IMDs induced by RGD containing αvβ3 integrin antagonist echistatin or overexpression of unligated αvβ3 integrins.10, 16 In contrast to previous report that β5 integrin is non-apoptotic,10 our results provide the first evidence that β5 integrin also mediates IMD.
Both soluble and immobilized ISM induces αvβ5-integrin clustering (Figures 3E and 5B). Although clustering of unligated or antagonized integrins is known to induce apoptosis in adherent cells, clustering of agonized integrin promotes cell survival.17 Whether soluble and immobilized ISM induce different mode of integrin clustering which lead to different interactions with focal adhesion proteins and activate different intracellular signaling pathways warrant further investigation.
The full spectrum of Ism's physiological role is not clear at this moment. Our preliminary investigation indicated that Ism is expressed at a significantly higher level in lung, comparing with other tissues (Supplementary Figure S7). Future investigation by immunohistochemistry is needed to reveal how ISM is presented to ECs in vivo.
The phenomenon that an endogenous antiangiogenic protein in different physical state (soluble versus immobilized) carries out contrasting functions in angiogenesis may impact the in vivo efficacy of the antiangiogenic protein. This dual-function characteristic may also exist in other endogenous antiangiogenic proteins. For example, immobilized endostatin has been reported to support EC adhesion in an integrin-dependent manner, whereas soluble endostatin inhibits integrin-dependent cell functions such as cell migration and induces apoptosis.18 It seems that integrin can promote or suppress apoptosis depending on the physical state of its ligand. This characteristic places integrin as a type of dependence receptors that require immobilized ligands to promote cell survival, whereas soluble ligands act as antagonists inducing IMD.16, 19 In contrast to ISM, ECM adhesion molecules such as VN or FN, which serve as integrin agonists when immobilized, do not function as antagonists to trigger apoptosis in soluble state.
Concluding remarks
In this work, we demonstrated that the endogenous angiogenesis inhibitor ISM could function as either antagonist or agonist of αvβ5 integrin and modulate EC apoptosis and survival. Although immobilized ISM promotes EC survival and haptotactic migration, soluble ISM suppresses EC tube formation and induces EC apoptosis through IMD. This study also identified a novel ‘RKD’ motif within the AMOP domain of ISM, which mediates the specific binding of ISM to αvβ5, but not αvβ3. As other endogenous angiogenesis inhibitor proteins may behave in similar fashion, it is conceivable that an antiangiogenic protein may inhibit or promote angiogenesis depending on its physical state in vivo. This may impact on the in vivo efficacy of the antiangiogenic proteins as anticancer drugs.
Materials and Methods
Cell culture and recombinant protein production
Endothelial cells (ECs) used in this work are HUVECs. Fresh umbilical cords were collected from consented maternal ward patients at the National University Hospital according to the protocol (DSRB C/00/553), which is approved by Singapore National Healthcare Group's Domain-Specific Review Board (DSRB) ethics approval committee. ECs were cultured in CSC complete medium (Cell System Corporation, Kirkland, WA, USA). Only cells of passages 4–8 were used in experiments.
Cell attachment assays
The assays were performed as described previously, with minor modifications.20 96-well plates (Nunc, Rochester, NY, USA) were coated with the indicated concentrations of ISM or ECM proteins (Sigma, St. Louis, MO, USA) at 4°C overnight. Nonspecific binding sites were blocked with 1% BSA. Subconfluent ECs were harvested and suspended in CSC basal medium. Cells (2 × 104) were plated to each well and allowed to attach for 60 min at 37°C in CO2 incubator. In some experiments, ECs were pre-incubated for 30 min with integrin-specific antibodies (αv antibody (clone M9), β1 antibody (clone P4C10), β3 antibody (clone 25E11), β5 antibody (clone E19), αvβ3 antibody (clone LM609), αvβ5 antibody (clone P1F6)) (Millipore, Billerica, MA, USA) or control IgG prior to their addition to the wells. After removal of the nonattached cells by washing with PBS, attached cells were fixed with 10% formalin (Sigma) and stained with 0.2% crystal violet. The crystal violet was extracted by 10% acetic acid and cell attachment was quantified by measuring the absorbance of eluted dye at 595 nm with a microplate reader.
Apoptosis assays
ECs (1 × 104 cells per well) in 96-well plate were starved in basal CSC medium containing 2% FBS overnight before treated with 1 μM ISM and 15 ng/ml VEGF for 24 h. Apoptosis was determined by measuring cytosolic oligonucleosome-bound DNA using a Cell Death ELISA kit (Roche Diagnostics Asia Pacific, Singapore). Caspase inhibitors Z-DEVD-FMK (caspase 3 inhibitor), Z-IEDT-FMK (caspase 8 inhibitor), Z-LEHD-FMK (caspase 9 inhibitor) and Kp7-6 (Fas antagonist) (Merck, Singapore) were added to cells 1 h prior to treatment with ISM and VEGF.
Cell proliferation assay
ECs or siRNA transfected ECs (2 × 104 per well) were seeded in a gelatin-coated 96-well plate with CSC complete medium for 24 h at 37°C and 5% CO2. Cells were starved with CSC basal medium supplemented with 2% FBS overnight. Then 1 μM ISM and 15 ng/ml VEGF were added at the beginning of the proliferation assay. ECs were allowed to proliferate for 24 h and proliferation was determined by BrdU labeling (Millipore), according to manufacturer's instruction.
ELISA measurement of free αvβ5 integrin
A total of 96-well plates were coated with VN (50 μg/ml), FN (50 μg/ml) and ISM (1 μM) at 37°C for 2 h. Nonspecific binding sites were blocked with 1% BSA in PBS for 2 h at room temperature. HUVECs (2 × 104 cells) were seeded into each well and allowed to attach overnight. After removal of the nonattached cells by washing with PBS, ECs were fixed and blocked with 3% BSA prior to incubating with anti-αvβ5 antibody (clone P1F76, Santa Cruz, Santa Cruz, CA, USA) for 1 h. Detection of integrin was achieved by anti-mouse IgG-HRP and developed with TMB (3,3′,5,5′-tetramethylbenzidine) (Dako, Glostrup, Denmark) for 15 min. The reaction was stopped with the addition of 50 μl 1 M H2SO4. The absorbance was then determined at 450 nm.
Immunofluorescence staining
ECs were plated for 2 h on 96-well plates coated with 20 μg/ml of either VN or ISM. Cells were rinsed with PBS twice, fixed with 4% paraphormaldehyde in PBS, permeabilized with 0.1% Triton X-100 in PBS for 10 min and blocked with 3% BSA for 1 h at room temperature. Cells were stained with rhodamine-conjugated phalloidine (actin stress fibers) or goat anti-β5 antibody (Santa Cruz) with FITC-labeled rabbit anti-goat IgG (Invitrogen, Carlsbad, CA, USA) and counter-stained with DAPI (Sigma). For ISM binding to ECs, cells were incubated with ISM for 1 h at 37°C and 5% CO2 before they are fixed and stained with mouse anti-His tag (sc-57598, Santa Cruz) and goat anti-β5 (sc-5401, Santa Cruz) in 3% BSA for 1 h. ISM and β5 integrin were then visualized with a mix of Alexa Fluor 488 rabbit anti-mouse IgG and Alexa Fluor 568 rabbit anti-goat IgG (Invitrogen). Fluorescence images were obtained with a Zeiss laser scanning confocal microscope system (Carl Zeiss South East Asia, Singapore).
RNAi mediated gene silencing
siRNAs were purchased from 1st BASE (Singapore): siRNA targeting human β3 integrin subunit mRNA (5′-CAAGCCUGUGUCACCAUAC-dTdT-3′); human β5 subunit integrin mRNA (5′-GCUCGCAGGUCUCAACAUA-dTdT-3′); and a random sequence control siScramble (5′-GACGUGGGACUGAAGGGGU-dTdT-3′) were as described before.21
Determination of caspase activation by immunoblot
Activation of caspases 3, 8, and 9 was determined as a function of procaspase cleavage.22 ECs (1 × 105/ml) were incubated with ISM (1 μM) and VEGF (15 ng/ml) for 24 h. Caspase activation and PARP cleavage were analyzed at 0, 2, 4, 8, 16 and 24 h post ISM-treatment. Cell lysates were separated by SDS-PAGE and probed with antibodies to caspase 3 (R&D systems, Minneapolis, MN, USA), caspase 8, caspase 9, PARP (Millipore).
Co-immunoprecipitation
ECs were incubated with 1 μM ISM for 16 h at 37°C, 5% CO2. After removing the unbound ISM, cells were washed, lysed and cell lysates were incubated with anti-αvβ5 antibody, control IgG and protein A/G Sepharose beads (Santa Cruz) for 2 h at 4°C. The precipitates were resolved and analyzed for the presence of caspase-8 or FADD by immunoblotting.
Cell haptotaxis assay
Haptotactic cell motility was measured by using 8 μm cell culture inserts, as previously described.23 The undersurface of the membrane filter was pre-coated with serial concentration of BSA, ISM or FN. ECs (1 × 106 cells/ml) were added to each insert. The cells were allowed to migrate for 6 h at 37°C and 5% CO2 before being fixed and stained with crystal violet. Cell migration was quantified by measuring the absorbance of eluted crystal violet at 560 nm. Anti-integrin antibodies were used at 10 μg/ml in the antibody-neutralization experiments.
Determination of integrin binding site of ISM
AMOP-N and AMOP-C region were cloned into pET-M vector (as described in reference 4) using the following primers: AMOPF1EcoRl: CCGGAATTCCGGCTGCTTGCGGG AAGTGAGGAGT; AMOPR1Xhol: CCGCTCGAGCGGCTCCAGGGACAGCATAGA GCGGA; AMOPF2EcoRl: CCGGAATTCCGGTGCATCCGCTCTATGCTGTCCCT; AMOPR2Xhol: CCGCTCGAGTCGGTCTGGCTTCTTGGAACTGTTTG. The point mutants denoted AMOPKD316AA was obtained from a pET-M-AMOP plasmid using site-directed mutagenesis procedures (Stratagene, La Jolla, CA, USA). The recombinant proteins were expressed in E.coli (BL21DE3) and purified using Ni-NTA affinity chromatography in 6 M urea, according to the manufactures’ instructions (Promega, Madison, WI, USA). The proteins were then further purified by reverse-phase HPLC. Protein concentration was determined using BioRad Bradford assay reagent (BIORAD, Hercules, CA, USA). Cell attachment assays were then used to determine the ability of various protein fragments to support EC adhesion, as described in the above section.
Statistical analysis
All data values were reported as mean±S.E.M. Statistical differences were determined using the Student's t-test. P-values of 0.05 were considered to be statistically significant.
Abbreviations
- ISM:
-
isthmin
- EC:
-
endothelial cells
- IMD:
-
integrin mediated death; FAK, focal adhesion kinase
- TSP-1:
-
thrombospondin-1
- TSR:
-
thrombospondin type 1 repeat
- AMOP:
-
adhesion-associated domain in MUC4 and other proteins
- ISV:
-
intersegmental vessel
- ECM:
-
extracellular matrix
- VEGF:
-
vascular endothelial growth factor
- HUVEC:
-
human umbilical vein endothelial cells
- FADD:
-
fas-associated protein with death domain
- VN:
-
vitronectin
- FN:
-
fibronectin
References
Carmeliet P . Angiogenesis in life, disease and medicine. Nature 2005; 438: 932–936.
Ribatti D . The discovery of antiangiogenic molecules: a historical review. Curr Pharm Des 2009; 15: 345–352.
Pera EM, Kim JI, Martinez SL, Brechner M, Li SY, Wessely O et al. Isthmin is a novel secreted protein expressed as part of the Fgf-8 synexpression group in the Xenopus midbrain-hindbrain organizer. Mech Dev 2002; 116: 169–172.
Xiang W, Ke Z, Zhang Y, Cheng GH, Irwan ID, Sulochana KN et al. Isthmin is a novel secreted angiogenesis inhibitor that inhibits tumor growth in mice. J Cell Mol Med 2011; 15: 359–374.
Silva R, D’Amico G, Hodivala-Dilke KM, Reynolds LE . Integrins: the keys to unlocking angiogenesis. Arterioscler Thromb Vasc Biol 2008; 28: 1703–1713.
Alghisi GC, Ponsonnet L, Ruegg C . The integrin antagonist cilengitide activates alphaVbeta3, disrupts VE-cadherin localization at cell junctions and enhances permeability in endothelial cells. PLoS One 2009; 4: e4449.
Yamamoto M, Yamato M, Aoyagi M, Yamamoto K . Identification of integrins involved in cell adhesion to native and denatured type I collagens and the phenotypic transition of rabbit arterial smooth muscle cells. Exp Cell Res 1995; 219: 249–256.
Eskens FA, Dumez H, Hoekstra R, Perschl A, Brindley C, Bottcher S et al. Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins alphavbeta3 and alphavbeta5 in patients with advanced solid tumours. Eur J Cancer 2003; 39: 917–926.
Maubant S, Saint-Dizier D, Boutillon M, Perron-Sierra F, Casara PJ, Hickman JA et al. Blockade of alpha v beta3 and alpha v beta5 integrins by RGD mimetics induces anoikis and not integrin-mediated death in human endothelial cells. Blood 2006; 108: 3035–3044.
Stupack DG, Puente XS, Boutsaboualoy S, Storgard CM, Cheresh DA . Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J Cell Biol 2001; 155: 459–470.
Schwartz MA, Schaller MD, Ginsberg MH . Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol 1995; 11: 549–599.
Ruegg C, Yilmaz A, Bieler G, Bamat J, Chaubert P, Lejeune FJ . Evidence for the involvement of endothelial cell integrin alphaVbeta3 in the disruption of the tumor vasculature induced by TNF and IFN-gamma. Nat Med 1998; 4: 408–414.
Golubovskaya VM, Kweh FA, Cance WG . Focal adhesion kinase and cancer. Histol Histopathol 2009; 24: 503–510.
Burridge K, Turner CE, Romer LH . Tyrosine phosphorylation of paxillin and pp125FAK accompanies cell adhesion to extracellular matrix: a role in cytoskeletal assembly. J Cell Biol 1992; 119: 893–903.
Sospedra P, Mestres C, Haro I, Munoz M, Busquets MA . Effect of amino acid sequence change on peptide-membrane interaction. Langmuir 2002; 18: 1231–1237.
Brassard DL, Maxwell E, Malkowski M, Nagabhushan TL, Kumar CC, Armstrong L . Integrin alpha(v)beta(3)-mediated activation of apoptosis. Exp Cell Res 1999; 251: 33–45.
Stupack DG, Cheresh DA . Apoptotic cues from the extracellular matrix: regulators of angiogenesis. Oncogene 2003; 22: 9022–9029.
Rehn M, Veikkola T, Kukk-Valdre E, Nakamura H, Ilmonen M, Lombardo C et al. Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci USA 2001; 98: 1024–1029.
Stupack DG . Integrins as a distinct subtype of dependence receptors. Cell Death Differ 2005; 12: 1021–1030.
Eikesdal HP, Sugimoto H, Birrane G, Maeshima Y, Cooke VG, Kieran M et al. Identification of amino acids essential for the antiangiogenic activity of tumstatin and its use in combination antitumor activity. Proc Natl Acad Sci USA 2008; 105: 15040–15045.
Monferran S, Skuli N, Delmas C, Favre G, Bonnet J, Cohen-Jonathan-Moyal E et al. Alphavbeta3 and alphavbeta5 integrins control glioma cell response to ionising radiation through ILK and RhoB. Int J Cancer 2008; 123: 357–364.
Buckley CD, Pilling D, Henriquez NV, Parsonage G, Threlfall K, Scheel-Toellner D et al. RGD peptides induce apoptosis by direct caspase-3 activation. Nature 1999; 397: 534–539.
McCarthy JB, Furcht LT . Laminin and fibronectin promote the haptotactic migration of B16 mouse melanoma cells in vitro. J Cell Biol 1984; 98: 1474–1480.
Acknowledgements
This work is supported by a grant from the Singapore Biomedical Research Council (BMRC07/1/21/19/493) to Ruowen Ge.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by P Salomoni
Supplementary Information accompanies the paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Zhang, Y., Chen, M., Venugopal, S. et al. Isthmin exerts pro-survival and death-promoting effect on endothelial cells through alphavbeta5 integrin depending on its physical state. Cell Death Dis 2, e153 (2011). https://doi.org/10.1038/cddis.2011.37
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.37
Keywords
This article is cited by
-
Isthmin-1 attenuates allergic Asthma by stimulating adiponectin expression and alveolar macrophage efferocytosis in mice
Respiratory Research (2023)
-
Isthmin-1 (Ism1) modulates renal branching morphogenesis and mesenchyme condensation during early kidney development
Nature Communications (2023)
-
Advances in research of biological functions of Isthmin-1
Journal of Cell Communication and Signaling (2023)
-
ISM1 suppresses LPS-induced acute lung injury and post-injury lung fibrosis in mice
Molecular Medicine (2022)
-
Identification of Isthmin1 in the small annual fish, Nothobranchius guentheri, as a novel biomarker of aging and its potential rejuvenation activity
Biogerontology (2022)
