
Abstract
Many viruses express proteins which prevent the host cell death that their infection would otherwise provoke. Some insect viruses suppress host apoptosis through the expression of caspase inhibitors belonging to the P35 superfamily. Although a number of P35 relatives have been identified, Autographa californica (Ac) P35 and Spodoptera littoralis (Spli) P49 have been the most extensively characterized. AcP35 was found to inhibit caspases via a suicide substrate mechanism: the caspase cleaves AcP35 within its ‘reactive site loop’ then becomes trapped, irreversibly bound to the cleaved inhibitor. The Maruca vitrata multiple nucleopolyhedrovirus encodes a P35 family member (MaviP35) that exhibits 81% identity to AcP35. We found that this relative shared with AcP35 the ability to inhibit mammalian and insect cell death. Caspase-mediated cleavage within the MaviP35 reactive site loop occurred at a sequence distinct from that in AcP35, and the inhibitory profiles of the two P35 relatives differed. MaviP35 potently inhibited human caspases 2 and 3, DCP-1, DRICE and CED-3 in vitro, but (in contrast to AcP35) only weakly suppressed the proteolytic activity of the initiator human caspases 8, 9 and 10. Although MaviP35 inhibited the AcP35-resistant caspase DRONC in yeast, and was sensitive to cleavage by DRONC in vitro, MaviP35 failed to inhibit the proteolytic activity of bacterially produced DRONC in vitro.
Similar content being viewed by others


Main
Cell death is essential for normal animal development and to destroy pre-cancerous and auto-immune cells, but it has been postulated that apoptosis originally evolved to defend primitive multicellular organisms against intracellular pathogens such as viruses.1 Over evolutionary time, an ‘arms race’ has developed between viruses and their hosts. Cellular machineries detect infection and activate self-destruction pathways, limiting the ability of the virus to replicate and spread to other cells. Viruses, in turn, have evolved ways to suppress their hosts’ apoptotic machineries during the early phase of infection. Targeting caspases is one approach adopted by viruses to block their host cells’ suicidal reaction to infection.2 The P35 family is a group of caspase inhibitors encoded by viruses that infect insects. Almost all of the viruses that possess P35 relatives are baculoviruses:3 the sole exception known to date is the Amsacta moorei entomopoxvirus.4 No cellular P35 homologs have been described as yet, although as baculoviruses usually derive their genes from their hosts,5 it seems likely that P35 genes did evolve from a cellular ancestor.
The best-studied P35 family member is AcP35, encoded by the baculovirus Autographa californica multi nucleopolyhedrovirus (AcMNPV).6 It inhibits caspases via a substrate trap mechanism.7, 8, 9 The caspase cleaves AcP35 within the reactive site loop. This cleavage provokes a conformational change within the inhibitor, targeting its amino terminus to the caspase's active site, preventing hydrolysis of a thioester adduct between the inhibitor and the protease, and thus locking the caspase in an inactive, P35-bound form.7 Of the many mammalian, insect and nematode caspases tested, very few were found to be insensitive to AcP35. The Drosophila initiator caspase DRONC was shown to be resistant to inhibition by AcP35.10, 11 Processing of downstream Spodoptera caspases proceeded in the presence of AcP35,12 implying that a Spodoptera DRONC ortholog (denoted ‘Sf-caspase-X’) is also resistant to AcP35 inhibition. AcP35 could inhibit the enzymatic activity of recombinant caspase 9 (DRONC's mammalian counterpart), however extremely high concentrations of AcP35 were required to prevent apoptosome-activated caspase 9 from cleaving its physiological substrate, caspase 3.13 This suggests that AcP35 cannot efficiently interfere with the function of naturally activated caspase 9.
Bombyx mori nucleopolyhedrovirus (BmNPV) encodes a protein (BmP35), which shares 91% of its amino-acid sequence with AcP35. BmP35 displayed only weak anti-apoptotic activity14 and, unlike AcP35, BmP35 was dispensable for normal viral propagation.15, 16 Extracts from mammalian cells expressing BmP35 were less potent than lysates from AcP35-expressing cells at inhibiting recombinant caspase 3, although lower BmP35 expression levels may have contributed to this difference.13 No quantitative data have been published regarding the caspase inhibitory potency or specificity of BmP35, and no other close relatives of AcP35 have been functionally or biochemically investigated to date.
Some baculoviruses encode distant relatives of AcP35, which constitute the P49 subfamily. Spodoptera littoralis (Spli) NPV-P49 is the best-studied member of this subfamily. Like AcP35, SpliP49 is a broad-spectrum caspase inhibitor that could suppress insect17, 18, 19, 20 and mammalian21 cell death. Unlike AcP35, SpliP49 could inhibit DRONC-mediated yeast lethality,21 but it was incapable of preventing DRICE processing in Drosophila cells.19 SpliP49 could, however, prevent processing of executioner Spodoptera caspases,18, 20 implying that it can inhibit the proposed Sf-caspase-X. AcP35 contains the cleavage sequence DQMD’G within its reactive site loop, but SpliP49 instead possesses the sequence TVTD’G at this position. This sequence is required for SpliP49 to inhibit the distal insect caspase Sf-caspase-X, but its insertion into the AcP35 reactive site loop failed to confer this capability,20 indicating that other regions of the SpliP49 protein, not shared by AcP35, are critical for its ability to inhibit insect initiator caspases. The caspase inhibitor AMVP33 from Amsacta moorei entomopoxvirus is the least homologous member of the P35 superfamily, exhibiting only 25% amino acid identity to AcP35.4
The baculovirus Maruca vitrata (Mavi) MNPV infects the legume pod borer Maruca vitrata, and may offer a biological means of controlling this important pest of legume crops.22 The recent sequencing of the MaviMNPV genome23 revealed the presence of a P35 ortholog (MaviP35). The predicted MaviP35 protein is highly homologous to AcP35, but its predicted reactive site loop possesses a distinct caspase cleavage sequence. Here, we report our characterization of the apoptosis and caspase inhibitory properties of MaviP35.
Results
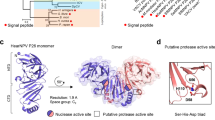
Sequencing of the MaviMNPV genome23 revealed that this virus encoded a P35 ortholog that was 81% identical to the founding member of this family, AcP35 (Figures 1a and b), and modeling suggested the two relatives may adopt similar structures (Figures 1c and d). Residues determined to be essential for the caspase inhibitory activities of AcP35, including C28 and D87,24, 25 were conserved in MaviP35, suggesting it too may function as a caspase inhibitor that could prevent apoptosis. We tested this hypothesis by overexpressing MaviP35 in mammalian and insect cells, and monitoring the transfectants’ sensitivity to apoptosis. MaviP35 inhibited insect cell death and caspase activity triggered by infection with an AcP35-deficient baculovirus, although less efficiently than AcP35 (Figures 2a and b). MaviP35 and AcP35 inhibited insect cell death induced by actinomycin D to a similar extent (Figure 2c), but MaviP35 protected a larger proportion of insect cells than AcP35 against apoptosis induced by UV irradiation (Figure 2d). MaviP35 and AcP35 afforded similar levels of protection to mammalian cells against cisplatin-induced apoptosis (Figure 2e), but MaviP35 was less protective than AcP35 against death induced by TNF-related apoptosis-inducing ligand (TRAIL; Figure 2f).

Relationships between MaviP35 and the other P35 subfamily members. (a) Alignment of baculoviral P35 subfamily proteins. Color intensity reflects the degree of conservation within the family. A pink box shows the P4-P1′ region (based on AcP35) and the caspase cleavage site is marked by an arrow. (b) An average distance tree was calculated using percentage identity from the multiple sequence alignment. The AcP35 structure (c) and predicted structure of MaviP35 (d) are shown. The positions of the AcP35 P1 aspartate residue and the corresponding residue in MaviP35 are indicated by arrows

MaviP35 inhibits mammalian and insect cell death. (a, b) Sf9 cells were transfected with plasmids encoding MaviP35-FLAG, AcP35-FLAG or chloramphenicol acetyl transferase (CAT), then infected with an AcP35-mutant baculovirus. Untransfected cells were subjected to mock infection. (a) Survival was monitored relative to the mock-infected cells. (b) Caspase activity was detected and expressed relative to the DEVDase activity in infected cells expressing the negative control CAT protein. (c and d) Sf21 cells were co-transfected with plasmids encoding GFP and the specified proteins. After 4 h heat shock-mediated induction of transgene expression, the cells were treated with actinomycin D (c) or UV (d). Viability is expressed as the number of GFP-positive cells following exposure to apoptotic stimuli, relative to their number before treatment. (e and f) MEF (e) or LN18 glioblastoma cells (f) were co-transfected with a β-galatosidase expression plasmid and either an empty vector (solid lines), or plasmids encoding MaviP35 (dashed lines) or AcP35 (dotted lines), before treatment with the indicated doses of either cisplatin (e) or crosslinked TRAIL (f). Following 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside staining, the survival of blue (transfected) cells was scored using morphological criteria. Over 80 (e) or 150 (f) blue cells were scored for each condition per experiment. Error bars represent S.E.M. from three separate experiments (a–f)
We have previously exploited yeast-based assays to visualize caspase activity and inhibition, and these were used to provide an indication of the specificity of MaviP35 for various caspases. MaviP35 protected yeast from death induced by mammalian caspases 1, 2, 3, 5, 7 and 8, the Drosophila caspases DCP-1 and DRICE, and CED-3 from Caenorhabditis elegans (Figure 3). In this system, MaviP35 appeared to exhibit similar activity to AcP35, and protected yeast from death induced by caspases 5, 8 and CED-3 better than SpliP49 (Figure 3).

MaviP35 inhibits caspase-dependent yeast death. Yeast were transformed with the indicated expression plasmids. Suspensions containing equivalent concentrations of each transformant were serially diluted and 5 μl of each dilution were spotted onto plates containing galactose (to induce transgene expression) or glucose (to repress transgene expression). Growth on inducing plates indicates survival and proliferation of yeast expressing the transgenes
AcP35 has been shown to inhibit caspases via a pseudosubstrate mechanism, and mutation of the caspase cleavage site abolishes caspase and apoptosis inhibitory activity.7, 8 Comparison of the MaviP35 and AcP35 sequences predicted that caspases may cleave MaviP35 after residue D87 within the site TQFD87′G (Figure 1a). Consistent with this residue being critical for caspase inhibition, a putative P1 mutant failed to prevent DRICE induced yeast death (Figure 4). Lysates from yeast expressing active DRICE exhibited considerable DEVDase activity. Co-expression of untagged or carboxyl terminally FLAG-tagged wild-type MaviP35 or AcP35 abolished this activity, but their cleavage site mutants had negligible impact (Figure 4). Immunoblotting confirmed that the tagged mutants were expressed at least as abundantly as their wild-type counterparts. Interestingly, cleavage products could not be detected in lysates from yeast co-expressing DRICE with either MaviP35-FLAG or AcP35-FLAG. Because DRICE activity in this system is generated through autoactivation, we suspect that only relatively few P35 molecules would be required to block this feedback loop, and immunoblotting may not be sufficiently sensitive to detect this small number of cleaved proteins.

MaviP35 activity requires the P1 aspartate residue but is unaffected by addition of a C-terminal FLAG tag. Yeast were transformed with the indicated expression plasmids. Suspensions containing equivalent concentrations of each transformant were serially diluted, and 5 μl of each dilution were spotted onto inducing and repressing plates. The caspase activities of induced transformants’ lysates were compared using the fluorogenic Ac-DEVD-AFC substrate. Anti-FLAG immunoblotting was performed and loading was visualized by coomassie staining
The yeast system is a sensitive tool for observing caspase inhibition within a naive eukaryotic environment, but only provides limited insight into the strength of inhibition. To gain a quantitative understanding of MaviP35's caspase inhibitory activity, we purified FLAG-tagged MaviP35 and AcP35 and examined their ability to prevent recombinant caspases from cleaving fluorogenic substrates in vitro. When present at 100–1000-fold excess, MaviP35 diminished by at least 90% the activity of caspases 2, 3, DRICE, DCP-1 and CED-3. Weaker inhibition was seen for caspases 1 and 7. MaviP35 only partially reduced the activity of caspases 6, 8 and 9, and negligible inhibition of caspase 10 was observed (Figure 5). A particularly striking difference between AcP35 and MaviP35 related to inhibition of caspase 8. As observed previously,26 AcP35 potently suppressed the proteolytic activity of this enzyme, yet a 100-fold excess of MaviP35 only decreased its activity by about half (Figure 5).

MaviP35 inhibits recombinant caspases in vitro. Cleavage of fluorogenic substrates by purified recombinant caspases (at the indicated concentrations) is plotted as the change in RFU per minute, in the presence of 1 μM of FLAG-tagged MaviP35, AcP35 or truncated CED-9 (a cytosolic protein devoid of caspase inhibitory activity). The dotted line indicates 10% activity, relative to the substrate cleavage rate in the presence of the control protein
The alignment of P35 subfamily members revealed that the P4 and P2 residues differed between AcP35 and MaviP35 (P4-DQMD-P1 versus P4-TQFD-P1, respectively). Mutagenesis studies of AcP35 had previously demonstrated that changing its P4 aspartate residue to either alanine or asparagine markedly impaired its ability to inhibit caspases 3 and 8,7 highlighting the importance of the P4 amino acid for caspase inhibition. The cleavage site of MaviP35, containing a P4 threonine residue, was reminiscent of the site at which DRONC auto-processes between its large and small subunit (TQTE)11 and, to a lesser extent, the caspase cleavage site within the SpliP49 reactive site loop (TVTD).17 This prompted us to wonder whether MaviP35 may be the first example of a P35 subfamily member that can inhibit DRONC. Consistent with this notion, expression of MaviP35 completely abolished DRONC-mediated yeast death (Figure 6a) and recombinant DRONC could cleave purified MaviP35, although not as efficiently as DRICE (Figures 6b and c). We therefore sought to determine whether MaviP35 could suppress DRONC activity in insect cells. DRONC can cleave DRICE between its large and small subunits10, 11 and cleavage of DRICE in Drosophila cells has previously been used as a readout of DRONC activity.19, 27 A GFP-tagged active site mutant of DRICE expressed in Drosophila Kc167 cells was completely processed to yield a 39-kDa product following actinomycin D treatment (Figure 6d), as expected from DRONC cleavage between the large and small subunits of DRICE (Figure 6e). Enforced expression of DIAP1 completely inhibited cleavage of DRICEC211A-eGFP in actinomycin D-treated Drosophila cells (Figure 6). Curiously, AcP35 partially inhibited DRICEC211A-eGFP cleavage, with more than half of the protein remaining intact in cells co-expressing AcP35. MaviP35 also partially inhibited this cleavage event, although less potently than AcP35. Purified MaviP35 did not impair the ability of recombinant DRONC to cleave a peptide substrate (Figure 6f). Using a range of substrate and inhibitor concentrations, inhibition by bacterially produced DRONC of MaviP35 was extremely weak (Figure 7). Quantitation of MaviP35's inhibition of other caspases confirmed the data shown in Figure 5: strong inhibition of executioner caspases, but weak to negligible inhibition of initiator caspases (Figure 7c).

MaviP35 is cleaved by DRONC but does not inhibit DRONC activity in vitro. (a) Yeast were transformed with the indicated expression plasmids. Suspensions containing equivalent concentrations of each transformant were serially diluted, and 5 μl of each dilution were spotted onto inducing and repressing plates. A total of 400 ng of FLAG-tagged MaviP35 (b) or AcP35 (c) were incubated with 0, 15, 150 or 1500 nM of DRONC or DRICE, then processing was monitored by anti-FLAG immunoblotting. (d) Drosophila Kc167cells were co-transfected with a plasmid encoding either eGFP or DRICEC211A-eGFP, together with either an empty vector or plasmid directing expresion of AcP35-F, MaviP35-FLAG or DIAP1. Transfectants were incubated in media containing or lacking 1 μM actinomycin D (ActD) fo 12 h. Lysates were subjected to anti-GFP or anti-FLAG immunoblotting, or coomassie staining. (e) The structure of the DRICEC211A–eGFP fusion protein is shown. (f) Cleavage of Ac-TQTD-AFC by purified recombinant DRONC is plotted as the change in RFU per minute, in the presence of 1.6 μM of FLAG-tagged MaviP35, AcP35 or truncated CED-9

MaviP35 is an efficient inhibitor of executioner caspases in vitro. Purified FLAG-tagged MaviP35 and AcP35 were tested in vitro for their ability to inhibit cleavage of fluorogenic peptide substrates by human caspases 3, 8 and 9 and Drosophila DRICE and DRONC. Hydrolysis of the indicated substrates was monitored in the presence of a range of inhibitor concentrations. Graphs show the impact of various concentrations of MaviP35 on DRONC (a) and DRICE (b) activity. (c) Inhibition constants (Ki) were calculated for selected caspases using a competitive model, as described in the Materials and Methods
Discussion
This study describes the apoptosis and caspase inhibitory properties of a new P35 subfamily member: MaviP35. Like AcP35, MaviP35 could inhibit insect and mammalian cell death, was susceptible to caspase cleavage, and could inhibit the proteolytic activity of caspases. Nevertheless, AcP35 and MaviP35 differed in their specificity profile. MaviP35 inhibited executioner apoptotic caspases with similar potency to AcP35. However, MaviP35 was substantially less potent than AcP35 at inhibiting mammalian caspases 8 and 10. This may explain the weaker protection afforded by MaviP35 relative to AcP35 against TRAIL-induced cell death. MaviP35 was also a weaker inhibitor of recombinant caspase 9 than AcP35. However, it is important to note that published data suggest that the susceptibility of recombinant caspase 9 to AcP35 in vitro is not mirrored by apoptosome-activated caspase 9 within cell lysates,13 so it is possible that AcP35 and MaviP35 are both incapable of interfering with the activity of naturally activated caspase 9 in vivo.
The P4 residue of MaviP35 (threonine) differs from that of AcP35 (aspartate). Mutation of P4 in AcP35 to asparagine reduced its ability to inhibit caspase 3 by 47-fold,7 yet MaviP35 – which contains the slightly larger polar uncharged residue threonine at this position – inhibited caspase 3 with similar efficiency to wild-type AcP35. Presumably differences in other regions of the protein compensate, allowing MaviP35 to efficiently suppress caspase 3 activity. The MaviP35 cleavage site resembles the auto-processing site between the large and small subunits of DRONC, and MaviP35 was sensitive to DRONC proteolysis in vitro. MaviP35 inhibited yeast death triggered by high-level expression of DRONC, suggesting that it could function as a pseudo-substrate inhibitor of DRONC. However, two pieces of evidence argue against this possibility. First, MaviP35 was an extremely weak inhibitor of recombinant DRONC activity in vitro. Its Ki was indistinguishable from that of AcP35, which was incapable of inhibiting DRONC in yeast and in vivo10, 11 and was resistant to DRONC cleavage in vitro. Second, MaviP35 only partially inhibited actinomycin D-induced cleavage of the DRICEC211A–eGFP fusion protein, impeding processing to a lesser extent than AcP35. This result implies that actinomycin D treatment provoked DRONC-mediated (AcP35/MaviP35 resistant) processing of the DRICE-eGFP substrate in Kc167 cells, but also suggested that other proteases, sensitive to AcP35/MaviP35 inhibition, also contributed to this proteolysis. Taken together, these data lead us to postulate that MaviP35 functions as a classical substrate for DRONC, rather than as a suicide substrate inhibitor. It is possible that the ability of MaviP35 to suppress DRONC-mediated yeast lethality reflects substrate competition: DRONC's proteolytic attention may be diverted to MaviP35 processing rather than cleavage of essential yeast proteins.
Despite the high homology between MaviP35 and AcP35, this study has revealed an important difference in their caspase specificity. Both inhibitors could suppress downstream caspases from insects and mammals (and CED-3), but only AcP35 could efficiently block the activities of mammalian caspases-8 and -10. Neither AcP35 nor MaviP35 could significantly inhibit the Drosophila initiator caspase DRONC.
Materials and Methods
Sequence comparisons and structural modeling
Sequences of the P35 subfamily members were aligned using the ‘Multialign’ program28 and formatted using Jalview 2.0.29 Genbank accession numbers for the P35 sequences used were: MaviMNPV: YP_950833, AcMNPV: NP_054165.1, RoMNPV: NP_703122, BmNPV: AAO12972, HycuNPV: AAO17287, SpltNPV: CAA71304 and LeseNPV: AAF78504. Protein Homology/analogY Recognition Engine (Phyre) was used to predict the structure of MaviP35.30 AcP35 and predicted MaviP35 structures were depicted using Jmol: an open-source Java viewer for chemical structures in 3D (http://www.jmol.org/).
Plasmid construction
For yeast experiments, coding DNA sequences were expressed from inducible Gal1/10 promoters.31 Plasmids-expressing caspase 2, caspase 3-lacZ, caspase 5, caspase 753, caspase 8, CED-3, reverse DRICE, DCP-1, DRONC, AcP35, AcP35-F, AcP35D87A-F and SpliP49-F have been described previously.11, 21, 32, 33, 34 Other plasmids were generated as follows: the caspase 1 coding region was amplified using oligonucleotides 1 and 2 from pET21b-Casp-1-His (purchased from Addgene Cambridge, MA, USA). The product was cut with BamHI/XbaI and ligated into pGALL-(LEU2)32 cut with BamHI/XbaI. The MaviP35 coding region was amplified with oligonucleotides 3 and 4 from a plasmid kindly donated by Prof. Chung-Hsiung Wang, cut with BamHI/XhoI and ligated into pGALL-(HIS3)32 cut with BamHI/XhoI. MaviP35 was also amplified with oligonucleotides 3 and 5 and cloned the same way into pGALL-(HIS3), to incorporate a carboxyl terminal FLAG tag. Site-directed mutagenesis to generate MaviP35D87A was performed using PCR. After excising MaviP35 from pGALL-(HIS3)-MaviP35 and inserting it into Bluescript II SK+ via BamHI/XhoI, a PCR was conducted using MaviP35-F as a template (oligonucleotides 6 and 7) to amplify the 3′ portion of the gene incorporating a P1 mutation. This product was cut with BclI/XhoI and inserted into BclI/XhoI cut MaviP35-Bluescript II SK+. The resulting P1-mutated MaviP35-F gene was then excised with BamHI/XhoI and ligated into pGALL-(HIS3). For mammalian and insect cell expression, MaviP35-F was amplified from pGALL-(HIS3)-MaviP35-F with oligonucleotides 8 and 7, then cut with BamHI/XhoI, blunted and inserted into pHSP70PLVI+CAT (chloramphenicol acetyl transferase)35 cut with BglII/EcoRI blunted or pEF36 cut with BamHI/XbaI blunted. Caspase 3 was amplified using primers 9 and 10, then digested with NdeI/XhoI and ligated into NdeI/XhoI cut pET23a. All amplified fragments were sequenced to verify the absence of any unintentional mutations. The pAct5eGFP vector was kindly provided by Samuel Le Fort and David Vaux. The coding sequencing of DRICEC211A was amplified from pGMR-DRICEC211A (described below) using primers 11 and 12, cut with BglII and cloned into pACT5eGFP cut with BamHI to generate pACT5c-DRICEC211A-eGFP. Restriction analyses and sequencing were performed to determine the orientation of the insert and to check the sequence was correct. pGMR-DRICE was made by amplifying DRICE with primers 13 and 14, then cutting the product with BglII and XbaI and cloning into pGMR.37 pGMR-DRICEC211A was constructed by amplifying the 3′ portion of DRICE encoding the active site with a mutagenic forward primer (15) and wild-type reverse primer (16), digesting the product with NheI and NotI and ligating into pGMR-DRICE cut with NheI and NotI. The DNA encoding eGFP was removed from pAct5eGFP by cutting with BamHI and XbaI, blunting using Klenow polymerase and religating to yield pAct5c. BamHI/XbaI fragments encoding either DIAP1 (obtained by digesting pGALL-(HIS3)-DIAP1) or MaviP35-FLAG (excised from pGALL-(HIS3)-MaviP35-F) were ligated into BamHI/XbaI cut pAct5c-eGFP, replacing the eGFP gene.
The sequences of the nucleotides referred to above were as follows:
- 1::
-
5′-CGGGATCCATGGCCGACAAGGTCCTGAAGGAG-3′
- 2::
-
5′-GCTCTAGATTAATGTCCTGGGAAGAGGTAGAAACATC-3′
- 3::
-
5′-GGGATCCCATATGTGTGTAATTTTTCCAGTAG-3′
- 4::
-
5′-GCCTCGAGTTAATCAATGTTTAATATTATATTG-3′
- 5::
-
5′-GCCTCGAGTTACTTGTCATCGTCGTCCTTGTAGTCCATATCAATGTTTAATATTA TATTGTTG-3′
- 6::
-
5′-CAATTTGATCAACTAGAACGCGACCACAGCACTCAATTCGCT GGAGGCC-3′
- 7::
-
5′-CTTTATTATTTTTATTTTATTGAGAGGGTGG-3′
- 8::
-
5′-GCGGATCCGCCATGTGTGTAATTTTTCCAGTAG-3′
- 9::
-
5′-GGAATTCCATATGGAGAACACTGAAAACTCAGTGG-3′
- 10::
-
5′-CCCTCGAGGTGATAAAAATAGAGTTCTTTTGTGAGC-3′
- 11::
-
5′-GTCAGATCTCAAAATGGACGCCACTAACAATGGAG-3′
- 12::
-
5′-GTCAGATCTACCCGTCCGGCTGGAGCCAAC-3′
- 13::
-
5′-CGAGATCTCCGCCATGGACGCCACTAACAATGGAGAATCC-3′
- 14::
-
5′-CGTCTAGACTAAACCCGTCCGGCTGGAGCCAACTGC-3′
- 15::
-
5′-CCTCGCTAGCCGGCAAACCCAAGTTGTTCTTCATACAGGCCGCCCAGGGC-3′
- 16::
-
5′-GCACTAGTGCGGCCGCCTAAACCCGTCCGGCTGGAGCCAACTGC-3′
Apoptosis assays from insect cells
Sf21 cells were plated at 8 × 105 cells per well in six-well plates in TC-100 insect medium (Invitrogen, Carlsbad, CA, USA) plus 10% fetal bovine serum (FBS; Atlanta Biologicals, Atlanta, GA, USA), and allowed to attach overnight at 27°C. Transfections were performed using lipofectin, which was prepared as a 1.5 : 1 mixture of DOTAP ((N-(1-(2,3-Dioleoyloxy)propyl)-N,N,N,-trimethylammonium chloride salt; Avanti Polar Lipids, Alabaster, AL, USA) and DOPE (L-α Phosphatidylethanolamine, dioleoyl; Sigma-Aldrich, St. Louis, MO, USA). For each well, 2.5 μg of the eGFP expression plasmid pHSP70GFPBsu36I38 was mixed with 2.5 μg of either pHSP70PLVI+CAT, pHSP70PLVI+AcP35 or pHSP70PLVI+MaviP35F. This DNA was diluted to 100 μl with TC-100 lacking FBS and incubated for 5 min at room temperature (RT). In a separate tube, 6 μl lipofectin was diluted to 100 μl with TC-100 lacking FBS and incubated for 5 min at RT. The mixtures of DNA and lipofectin were then combined and allowed to incubate for 15 min at RT. During this incubation the Sf21 cells were washed twice with 1 ml of TC-100 lacking FBS. After the last wash, 800 μl of TC-100 lacking FBS was left in each well and the DNA/lipofectin mixture (200 μl) was added to each well and allowed to incubate at 27°C for 4 h. The mixture plus the media were removed and 2 ml of TC-100 plus 10% FBS was added to each well. The cells were heat shocked 24 h post transfection at 42°C for 30 min, to drive expression from the hsp70 promoter. The cells were then induced to undergo apoptosis 4 h post heat shock, by treatment with either UV (by placing the plates on a transilluminator for 10 min) or actinomycin D (Invitrogen; 250 ng/ml). To determine cell viability, the number of GFP-expressing cells was counted in each well both immediately before and 17 h after UV or actinomycin D treatment. Three random fields of view per sample were counted per well, and three separate wells were assayed per treatment.
To assay sensitivity to infection-mediated apoptosis, Sf9 cells (106) were plated in six-well culture dishes for 2 h in the TC-100 medium with 10% FBS. After 2 h, the medium was replaced with Grace's insect unsupplemented medium (Invitrogen). Cells were transiently transfected with pHSP70PLVI+CAT, pHSP70PLVI+AcP35 or pHSP70PLVI+MaviP35F using 3 μg of each plasmid and 6 μl of lipofectin. Transfection mixtures were replaced with TC-100 plus 10% FBS after 5 h incubation with cells. Cells were infected at 24 h post transfection with vAcP35KO-PG39 at a multiplicity of infection of 1 PFU/cell, and then harvested at 48 h post infection for caspase and viability assays. Caspase assays were performed using the substrate Ac-DEVD-AFC (MP Biomedicals, Solon, OH, USA) as described previously.40 To assess viability, three random fields of view were photographed ( × 200 magnification), and viable cells were counted for each well. Cell viability was determined by counting the non-apoptotic cells and comparing to the number of viable cells in a mock-infected control at 0 h post infection, which was set at 100%.
DRICE cleavage assays in Kc167 cells
Two million Kc167 cells (kindly provided by Gary Hime) were transfected with 0.2 μg of either pAct5c-eGFP or pAct5c-DRICE-eGFP plus 1.8 μg of either pAct5c, pAct5c-MaviP35-F or pAct5c-DIAP1, using the Effectene transfection reagent (Qiagen, Doncaster, Victoria, Australia) according to the manufacturer's instructions. After 24 h transfection, the cells were incubated in media containing 0 or 1 μM of actinomycin D for 12 h. Cells were lysed in mammalian lysis buffer (50 mM Tris pH 7.5, 375 mM NaCl, 1 mM ethylenediamine tetra-acetic acid, 1% Triton X-100) containing protease inhibitors (protease inhibitor cocktail set 1; Calbiochem, Darmstadt, Germany) and subjected to 12% SDS-PAGE and either Coomassie stained or immunoblotted using anti-GFP (Roche Applied Science no. 11814460001; Castle Hill, New South Wales, Australia) or anti-FLAGM2 (Sigma no. F3165) and anti-mouse IgG-HRP (Sigma no. A9044).
Mammalian apoptosis assays
SV-40 transformed mouse embryonic fibroblasts (MEFs) were co-transfected with 1 μg of CMV-lacZ and either 3 μg of pEF, AcP35-pEF or MaviP35-pEF using FuGENE HD transfection reagent (Roche; Basel, Switzerland). LN18 glioblastoma cells (ATCC; Manassas, VA, USA) were co-transfected with 1 μg of CMV-lacZ and either 2 μg of pEF, AcP35-pEF or MaviP35-pEF using Lipofectamine (Invitrogen). Transfections were performed according to the manufacturers’ instructions. Twenty-four hours after transfection, the medium was removed and the cells were incubated with fresh unsupplemented media or media containing cisplatin (Mayne Pharma, Mulgrave, Victoria, Australia) or Superkiller (crosslinked) TRAIL (Alexis Biochemicals, Lausen, Switzerland). After 24 h, the cells were stained with 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside and the blue cells were scored for viable versus apoptotic morphology, as previously published.36
Yeast transformation and death assays
Saccharomyces cerevisiae yeast strain W303α was transformed31 and analyzed in survival assays21 as described previously.31 Caspase 1 was expressed using the pGALL-(LEU2)-Casp1 vector described above.
Protein assays from yeast
Yeast transformants were grown and transgene expression induced as described previously.11 The lysates were subjected to SDS-PAGE and the gels were then stained with Coomassie brilliant blue (Sigma) to visualize protein loading and immunoblotted as described previously.11 The membranes were probed with an antibody recognizing the FLAG tag (clone M2; Sigma) and anti-mouse-HRP (Sigma). To measure the caspase activity via fluorescence analysis, the yeast were treated as follows: An overnight culture was pelleted, washed twice with 1 ml of TE (Tris HCl 10 mM pH 8, EDTA 1 mM) and induced for 6.5 h in complete media containing 2% galactose. After pelleting the yeast culture, the yeast was weighed and glass beads were added. To lyse the cells, 5 ml of CelLyticY reagent (Sigma) with 10 mM DTT was added per 1 g of yeast cells. After gently shaking the cells for 30 min at RT, the debris was removed by centrifugation at 16 100 × g for 10 min at 4°C. The protein concentration of the supernatant was measured using the Bicinchoninic acid protein assay kit (Sigma). In fluorescence assays, lysate (0.5 mg/ml) was mixed with Ac-DEVD-AFC (100 μM) in DRICE activity buffer (50 mM HEPES pH7.5, 10% sucrose, 0.1% CHAPS, 5 mM DTT, 100 mM NaCl, 1 mM EDTA), and the fluorescence (excitation 410 nm, emission 500 nm) was measured every 30 s for 1 h. The slope of each curve was calculated using Graphpad Prism 5.0 (La Jolla, CA, USA). The slope of each curve determines the concentration of free AFC per minute, which provides a measure of caspase activity.
Protein purification from yeast
Transformants were grown in 5 ml glucose-containing selective medium to stationary phase, then expanded by addition of 195 ml glucose-containing selective medium. After expanding the cells for 16–18 h, the pelleted yeast were washed once with 100 ml TE and resuspended in 1 l of galactose-containing complete media for 6.5 h induction. Cells were harvested at 12 100 × g, 4°C for 15 min then glass beads were added after weighing the pellets. To lyse the cells CelLyticY reagent (Sigma) was used as described above. The supernatant was incubated for 30 min at 4°C with 200 μl Anti-FLAG M2 affinity gel (Sigma), which had been previously washed three times with 12 × resin volume washing buffer (Tris HCl 50 mM pH 7.4, 150 mM NaCl). Incubated beads were pelleted 5 min at 3750 g, RT then washed with 32 resin volumes of washing buffer for 10 min at 4°C. Five elution fractions were collected each using one resin volume of elution buffer (1 mM HEPES pH 7.0, 0.1% PEG, 0.001% CHAPS, 0.1 mM DTT, 200 ng/μl FLAG peptide (Sigma)). For each elution step the beads were incubated for 2 min at 4°C with agitation, and supernatant was collected after a pelleting for 1 min at 16 100 g, 4°C. After subsequent SDS-PAGE analysis and Coomassie brilliant blue (Sigma) staining, the fractions containing pure FLAG-tagged proteins were pooled, and protein concentration was determined using the Bio-Rad Protein Assay (Bio-Rad, Gladesville, New South Wales, Australia).
Recombinant caspases
Caspases 1, 2, 6, 7, 8, 9 and 10 were purchased from Enzo Life Sciences (Farmingdale, NY, USA). BL21-(DE3)-pLysS (Merck, Darmstadt, Germany) bacteria were transformed with caspase 3-pET23a (described above) or the following previously published plasmids: DCP-1-pET23a,32 DRICE-pET23a,32 CED-3-pET23a,34 DRONC-pET23a.11 Caspase 3, DCP-1, DRICE and CED-3 were purified as described previously.34 DRONC purification was carried out as follows: A transformant colony was inoculated into 1.5 ml 2YT-amp/chlor (16 g/l tryptone, 10 g/l yeast extract, 5 g/l NaCl, 100 μg/ml ampicillin and 35 μg/ml chloramphenicol) and grown overnight at 37°C. A volume of 1 ml of this culture was expanded into 50 ml of pre-warmed 2YT-amp/chlor and grown at 37°C for 2 h, 200 r.p.m. A total of 10 ml of this was mixed with 190 ml of pre-warmed 2YT amp/chlor in a 2-l baffled flask and shaken at 37°, 200 r.p.m. until OD600 reached 0.6–0.8. IPTG was added to a final concentration of 1 mM and the culture was shaken at 20°C for 18 h, then pelleted for 10 min at 3000 g, 4°C and then frozen at −80°C. The pellet was thawed then resuspended in a 10-ml Bug Buster Mastermix (Merck) by pipetting, then incubated for 20 min at RT. Insoluble cell debris was removed by centrifugation at 16 100 × g for 20 min at 4°C. Half a milliliter of NiNTA resin (Qiagen) was washed twice in phosphate buffer (50 mM NaHPO4, 300 mM NaCl), then incubated with the induced bacterial lysate for 30 min at 4°C, gently mixing. The beads were washed twice with phosphate buffer containing 5 mM imidazole, then the caspase was eluted with phosphate buffer containing 250 mM imidazole.
In vitro quantitation of caspase inhibition
Caspases 1, 2, 3, 6, 7, 8, 9, 10, DRICE, DCP-1 and CED-3 were pre-activated for 10 min at 37°C in universal caspase citrate buffer (10 mM HEPES pH 7.0, 10% sucrose, 0.1% CHAPS, 10 mM DTT, 100 mM NaCl, 1 mM EDTA, 0.65 M Na-Citrate). After the activation step, the caspase was incubated either with buffer alone, F-CED-91–251, AcP35-F or with MaviP35-F for 1 h at 37°C. The appropriate fluorescent substrate was then added (100 μM): Ac-WEHD-AFC for caspase 1; Ac-VDVAD-AFC for caspase 2; Ac-DEVD-AFC for caspases 3, 7, DRICE, DCP-1 and CED-3; Ac-VEID-AFC for caspase 6; Ac-LEHD-AFC for caspases 8, 9 and 10 and Ac-TQTD-AFC for DRONC (Enzo Life Sciences). Fluorescence (excitation 410 nm, emission 500 nm) was measured every minute for 2 h. The maximal slope of each curve was calculated using Prism 5.0 and graphed.
Determination of inhibition constants
Caspases were pre-activated for 10 min at 37°C in the following buffers: caspase 3: 100 mM HEPES pH 7.0, 10% PEG, 0.1% CHAPS, 10 mM DTT; DRICE: 50 mM HEPES pH 7.5, 10% sucrose, 0.1% CHAPS, 5 mM DTT, 100 mM NaCl, 1 mM EDTA; caspases 8 and 9: 10 mM HEPES pH 7.0, 10% sucrose, 0.1% CHAPS, 10 mM DTT, 100 mM NaCl, 0.1 mM EDTA, 0.65 M Na-Citrate; DRONC: 50 mM Tris pH 7.4, 100 mM NaCl, 0.65 M Na-Citrate. Subsequently, the caspase was incubated with either AcP35-F or MaviP35-F in the appropriate activity buffer for 1 h at 37°C. Substrates were added at concentrations ranging from 0.001 to 1000 μM. Substrates used were: Ac-DEVD-AFC for caspase 3 and DRICE; Ac-LEHD-AFC for caspases 8 and 9 and Ac-VEID-AFC for DRONC (Enzo Life Sciences). Fluorescence (excitation 410 nm, emission 500 nm) was measured every minute for 2 h. The slope of each curve was calculated using Prism 5.0. Inhibition constants were calculated by non-linear regression using Prism 5.0 software, using a competitive inhibition model as described by these equations: KmObs=Km × (1+[I]/Ki) and Y=Vmax × X/(KmObs+X), where [I] is the inhibitor concentration (μM); Ki is the inhibition constant (μM), Vmax is the maximum enzyme velocity (relative fluorescence units (RFU)/min), Km is the Michaelis–Menten constant (μM), X is the concentration of substrate (μM) and Y is the change in fluorescence (RFU/min).
Abbreviations
- Mavi:
-
Maruca vitrata
- MNPV:
-
multiple nucleopolyhedrovirus
- Ac:
-
Autographa californica
- Bm:
-
Bombyx mori
- NPV:
-
nucleopolyhedrovirus
- Spli:
-
Spodoptera littoralis
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- FBS:
-
fetal bovine serum
- MEF:
-
mouse embryonic fibroblast
- Xgal:
-
5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside
- CAT:
-
chloramphenicol acetyl transferase
- RFU:
-
relative fluorescence units
References
Ameisen JC . On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ 2002; 9: 367–393.
Best SM . Viral subversion of apoptotic enzymes: escape from death row. Annu Rev Microbiol 2008; 62: 171–192.
Jabbour AM, Hawkins CJ . The p35 family of apoptosis inhibitors. Curr Genomics 2004; 5: 215–229.
Means JC, Penabaz T, Clem RJ . Identification and functional characterization of AMVp33, a novel homolog of the baculovirus caspase inhibitor p35 found in Amsacta moorei entomopoxvirus. Virology 2007; 358: 436–447.
Herniou EA, Olszewski JA, O’Reilly DR, Cory JS . Ancient coevolution of baculoviruses and their insect hosts. J Virol 2004; 78: 3244–3251.
Clem RJ, Fechheimer M, Miller LK . Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science 1991; 254: 1388–1390.
Xu G, Rich RL, Steegborn C, Min T, Huang Y, Myszka DG et al. Mutational analyses of the p35-caspase interaction. A bowstring kinetic model of caspase inhibition by p35. J Biol Chem 2003; 278: 5455–5461.
Xu G, Cirilli M, Huang Y, Rich RL, Myszka DG, Wu H . Covalent inhibition revealed by the crystal structure of the caspase-8/p35 complex. Nature 2001; 410: 494–497.
Riedl SJ, Renatus M, Snipas SJ, Salvesen GS . Mechanism-based inactivation of caspases by the apoptotic suppressor p35. Biochemistry 2001; 40: 13274–13280.
Meier P, Silke J, Leevers SJ, Evan GI . The Drosophila caspase DRONC is regulated by DIAP1. EMBO J 2000; 19: 598–611.
Hawkins CJ, Yoo SJ, Petersen EP, Wang SL, Vernooy SY, Hay BA . The Drosophila caspase DRONC cleaves following glutamate or aspartate and is regulated by DIAP1, HID, and GRIM. J Biol Chem 2000; 275: 27084–27093.
LaCount DJ, Hanson SF, Schneider CL, Friesen PD . Caspase inhibitor P35 and inhibitor of apoptosis Op-IAP block in vivo proteolytic activation of an effector caspase at different steps. J Biol Chem 2000; 275: 15657–15664.
Vier J, Furmann C, Hacker G . Baculovirus P35 protein does not inhibit caspase-9 in a cell-free system of apoptosis. Biochem Biophys Res Commun 2000; 276: 855–861.
Morishima N, Okano K, Shibata T, Maeda S . Homologous p35 proteins of baculoviruses show distinctive anti-apoptotic activities which correlate with the apoptosis-inducing activity of each virus. FEBS Lett 1998; 427: 144–148.
Kamita SG, Majima K, Maeda S . Identification and characterization of the p35 gene of Bombyx mori nuclear polyhedrosis virus that prevents virus-induced apoptosis. J Virol 1993; 67: 455–463.
Gomi S, Zhou CE, Yih W, Majima K, Maeda S . Deletion analysis of four of eighteen late gene expression factor gene homologues of the baculovirus, BmNPV. Virology 1997; 230: 35–47.
Pei Z, Reske G, Huang Q, Hammock BD, Qi Y, Chejanovsky N . Characterization of the apoptosis suppressor protein P49 from the Spodoptera littoralis nucleopolyhedrovirus. J Biol Chem 2002; 277: 48677–48684.
Zoog SJ, Schiller JJ, Wetter JA, Chejanovsky N, Friesen PD . Baculovirus apoptotic suppressor P49 is a substrate inhibitor of initiator caspases resistant to P35 in vivo. EMBO J 2002; 21: 5130–5140.
Lannan E, Vandergaast R, Friesen PD . Baculovirus caspase inhibitors P49 and P35 block virus-induced apoptosis downstream of effector caspase DrICE activation in Drosophila melanogaster cells. J Virol 2007; 81: 9319–9330.
Guy MP, Friesen PD . Reactive-site cleavage residues confer target specificity to baculovirus P49, a dimeric member of the P35 family of caspase inhibitors. J Virol 2008; 82: 7504–7514.
Jabbour AM, Ekert PG, Coulson EJ, Knight MJ, Ashley DM, Hawkins CJ . The p35 relative, p49, inhibits mammalian and Drosophila caspases including DRONC and protects against apoptosis. Cell Death Differ 2002; 9: 1311–1320.
Lee S-T, Srinivasan R, Lo Y-J, Talekar NS . Identification, characterization and bioassays of Maruca vitrata multiple nucleopolyhedrovirus (MaviNPV) against Maruca vitrata (Lepidoptera, Pyralidae). BioControl 2007; 52: 801–809.
Chen YR, Wu CY, Lee ST, Wu YJ, Lo CF, Tsai MF et al. Genomic and host range studies of Maruca vitrata nucleopolyhedrovirus. J Gen Virol 2008; 89: 2315–2330.
Fisher AJ, Cruz W, Zoog SJ, Schneider CL, Friesen PD . Crystal structure of baculovirus P35: role of a novel reactive site loop in apoptotic caspase inhibition. EMBO J 1999; 18: 2031–2039.
Bertin J, Mendrysa SM, Lacount DJ, Gaur S, Krebs JF, Armstrong RC et al. Apoptotic suppression by baculovirus p35 involves cleavage by and inhibition of a virus-induced CED-3/ICE-like protease. J Virol 1996; 70: 6251–6259.
Zhou Q, Krebs J, Snipas S, Price A, Alnemri E, Tomaselli K et al. Interaction of the baculovirus anti-apoptotic protein p35 with caspases. Specificity, kinetics, and characterization of the caspase-p35 complex. Biochemistry 1998; 37: 10757–10765.
Dorstyn L, Read S, Cakouros D, Huh JR, Hay BA, Kumar S . The role of cytochrome c in caspase activation in Drosophila melanogaster cells. J Cell Biol 2002; 156: 1089–1098.
Corpet F . Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 1988; 16: 10881–10890.
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ . Jalview Version 2 – a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009; 25: 1189–1191.
Kelley LA, Sternberg MJ . Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 2009; 4: 363–371.
Hawkins CJ, Wang SL, Hay BA . Monitoring activity of caspases and their regulators in yeast Saccharomyces cerevisiae. Methods Enzymol 2000; 322: 162–174.
Hawkins CJ, Wang SL, Hay BA . A cloning method to identify caspases and their regulators in yeast: identification of Drosophila IAP1 as an inhibitor of the Drosophila caspase DCP-1. Proc Natl Acad Sci 1999; 96: 2885–2890.
Hawkins CJ, Silke J, Verhagen AM, Foster R, Ekert PG, Ashley DM . Analysis of candidate antagonists of IAP-mediated caspase inhibition using yeast reconstituted with the mammalian Apaf-1-activated apoptosis mechanism. Apoptosis 2001; 6: 331–338.
Jabbour AM, Ho PK, Puryer MA, Ashley DM, Ekert PG, Hawkins CJ . The Caenorhabditis elegans CED-9 protein does not directly inhibit the caspase CED-3, in vitro nor in yeast. Cell Death Differ 2004; 11: 1309–1316.
Clem RJ, Miller LK . Control of programmed cell death by the baculovirus genes p35 and iap. Mol Cell Biol 1994; 14: 5212–5222.
Hawkins CJ, Uren AG, Hacker G, Medcalf RL, Vaux DL . Inhibition of interleukin 1-beta-converting enzyme-mediated apoptosis of mammalian cells by baculovirus IAP. Proc Natl Acad Sci USA 1996; 93: 13786–13790.
Hay BA, Wolff T, Rubin GM . Expression of baculovirus P35 prevents cell death in Drosophila. Development 1994; 120: 2121–2129.
Clarke TE, Clem RJ . Lack of involvement of haemocytes in the establishment and spread of infection in Spodoptera frugiperda larvae infected with the baculovirus Autographa californica M nucleopolyhedrovirus by intrahaemocoelic injection. J Gen Virol 2002; 83: 1565–1572.
Huang N, Wu W, Yang K, Passarelli AL, Rohrmann GF, Clem RJ . Baculovirus infection induces a DNA damage response that is required for efficient viral replication. J Virol 2011; 85: 12547–12556.
Wang H, Blair CD, Olson KE, Clem RJ . Effects of inducing or inhibiting apoptosis on Sindbis virus replication in mosquito cells. J Gen Virol 2008; 89: 2651–2661.
Acknowledgements
We thank Chung-Hsiung Wang for providing the plasmid bearing the MaviP35 gene, Sam Le Fort and David Vaux for the pAct5c-eGFP plasmid, Gary Hime for the Kc167 cells and Anissa Jabbour and Paul Ekert for the MEF cells. This work was funded by the National Health and Medical Research Council project grant to CJH and RJC (#602525), an Australian Research Council Future Fellowship to CJH (#FT0991464), scholarships to ILB and MML from the La Trobe University and a scholarship to ILB from the Cooperative Research Centre for Biomarker Translation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by P Salomoni
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Brand, I., Green, M., Civciristov, S. et al. Functional and biochemical characterization of the baculovirus caspase inhibitor MaviP35. Cell Death Dis 2, e242 (2011). https://doi.org/10.1038/cddis.2011.127
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.127
Keywords
This article is cited by
-
Reconstitution of human pyroptotic cell death in Saccharomyces cerevisiae
Scientific Reports (2023)
-
The response of newly established cell lines of Spodoptera littoralis to group I and group II baculoviruses
Cytotechnology (2019)
-
Yeast techniques for modeling drugs targeting Bcl-2 and caspase family members
Cell Death & Disease (2013)
