
Abstract
A recent report from the Laboratory of Heidi McBride (McGill University) presents a role for mitochondria in the de novo biogenesis of peroxisomes in mammalian cells. Peroxisomes are essential organelles responsible for a wide variety of biochemical functions, from the generation of bile to plasmalogen synthesis, reduction of peroxides, and the oxidation of very-long-chain fatty acids. Like mitochondria, peroxisomes proliferate primarily through growth and division of pre-existing peroxisomes. However, unlike mitochondria, peroxisomes do not fuse; further, and perhaps most importantly, they can also be born de novo, a process thought to occur through the generation of pre-peroxisomal vesicles that originate from the endoplasmic reticulum. De novo peroxisome biogenesis has been extensively studied in yeast, with a major focus on the role of the ER in this process; however, in the mammalian system this field is much less explored. By exploiting patient cells lacking mature peroxisomes, the McBride laboratory now assigns a role to ER and mitochondria in de novo mammalian peroxisome biogenesis by showing that the formation of immature pre-peroxisomes occurs through the fusion of Pex3-/Pex14-containing mitochondria-derived vesicles with Pex16-containing ER-derived vesicles.
Similar content being viewed by others

Peroxisome biogenesis in yeast and mammals, version 1.0
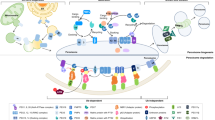
Since their discovery in the early 1950s, the origin and formation of peroxisomes has been extensively studied and hotly debated. It is now widely accepted that peroxisomes are semi-autonomous organelles, which multiply by growth and division, but depend on the ER for supply of other essential components such as lipids.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 There is also general agreement that the ER contributes to the de novo formation of peroxisomes, in particular in cells without pre-existing peroxisomes. Current models derived from studies in different yeast species suppose that the core peroxisomal import machinery is first targeted to the ER (or structures close to the ER), and enriched in pre-peroxisomal vesicles, which fully assemble the peroxisomal import machinery, thereby allowing continued growth and division.8, 13, 14 This model is not without controversy, and differences exist between fungal models, including H ansenula polymorpha, Y arrowia lipolytica, S accharomyces cerevisiae, and S chizosaccharomyces pombe.4, 15, 16, 17, 18 Importantly, the mechanisms identified within the fungal model systems may not fully apply to mammalian cells, given the major evolutionary distance. For example, an essential component of the mammalian import machinery for peroxisomal membrane proteins (PMPs), Pex16,19 is absent in baker’s yeast.20 In addition, although both yeast and mammalian cells contain Pex3, another essential PMP import receptor,21, 22 in the absence of peroxisomes the fungal and mammalian forms of Pex3 target different organelles: in yeast, Pex3 seems to be routed to the ER, where it can function to facilitate de novo biogenesis; instead, in mammals Pex3 targets mitochondria.8, 23 In fact, in cells lacking peroxisomes, it is not just Pex3 that targets mitochondria, but also other PMPs such as Pex14.24 The mis-targeting of peroxisomal proteins to the mitochondria was largely assumed to be an artifact due to loss of PMP import and/or overexpression of PMPs; after all, it was hard to reconcile the function of a peroxisomal protein with its localization on a different organelle. Thus, the field focused mostly on the ER, possibly because Pex16 in mammalian cells is routed to the ER when peroxisomes are absent, a finding that was more 'consistent' with the yeast Pex3 insertion into the ER (reviewed in Hua and Kim9); that said, the focus on an ER-resident protein like Pex16, rather than on the 'partially mitochondrial' Pex3, made sense also in light of the fact that there was no known mechanism that could make a bona fide PMP exit from the mitochondrion and be selectively sorted and delivered to a nascent pre-peroxisome. In contrast, the ER, being a professional sorting station, made it straightforward to envisage a model where peroxisomal proteins like Pex16 initiate peroxisomal biogenesis solely from the ER. However, in 2008 Neuspiel et al.25 (from the McBride Laboratory) identified a novel vesicular transport route between the mitochondria and a subpopulation of peroxisomes within mammalian cells, making it theoretically possible that other types of vesicles might exist to carry cargo to other organelles, such as pre-peroxisomes.1 It is from this starting point that Sugiura et al. revisited the molecular mechanisms of de novo peroxisomal biogenesis in mammalian cells and identified a possible pathway for mitochondria-derived vesicles (MDV) to carry mitochondrial Pex3 to pre-peroxisomes (Figure 1).

A hypothetical model depicting some of the steps that might be involved into the formation of ER-derived and mitochondria-derived vesicles; their fusion generates a pre-peroxisome, which then matures into a peroxisome
Mammalian peroxisome biogenesis, version 2.0
Mammalian peroxisome biogenesis can be studied using a genetic approach that is based on utilizing fibroblast cell lines derived from patients suffering from Zellweger syndrome, a rare autosomal-recessive disease characterized by a lack of functional peroxisomes and early neonatal lethality.26, 27 In their study, Sugiura et al. used Zellweger patient’s fibroblasts that lacked peroxisomes due to the loss of either Pex3 or Pex16, two essential components required for the import of PMPs and membrane biogenesis.28, 29 Here, the authors showed that infection of these cells with adenovirus overexpressing the missing peroxin (Pex3 or Pex16) reconstitutes the formation of peroxisomes. Using confocal imaging and immunogold electron microscopy, Sugiura et al. provides evidence that Pex3 is first inserted into the mitochondrial outer membrane, and then exits the mitochondrion within vesicular structures. The study also shows that endogenous Pex14, an integral membrane protein for luminal peroxisomal matrix import, targets mitochondria in these cells, and is enriched within Pex3-positive vesicles that bud from mitochondria. As mitochondrial vesicles can carry cargo to the lysosome,30 Sugiura et al. tested whether Pex3 and Pex14 within vesicular carriers may be destined for degradation. However, this was not the case as inhibition of lysosomal transport with bafilomycin did not affect the rescue of peroxisomal biogenesis, and did not lead to an accumulation of Pex3-positive vesicles or protein. Instead, the Pex3-positive vesicles stained with endogenous peroxisomal markers such as PMP70 and catalase, indicating that the Pex3-containing vesicles released from mitochondria matured into import-competent peroxisomes. Interestingly, de novo peroxisome biogenesis was shown to involve a second class of vesicles, this time emerging from the ER, carrying Pex16, the partner of Pex3 for membrane protein import. Video microscopy and immunogold electron microscopy data showed that Pex16 vesicles, derived from the ER, partially targeted the mitochondrial surface, where they appeared to have the ability to fuse either directly with mitochondria at sites where Pex3 was highly enriched, or with Pex3/Pex14-positive vesicles. The compartmentalization of Pex16 within the ER and Pex3/Pex14 within the mitochondria may ensure that the parental organelles will not gain import competence for peroxisomal matrix proteins, thereby maintaining their functional identity. Such 'double organellar origin' of the peroxisome is an exciting finding that establishes a new paradigm in organellar biogenesis. In this context, it will be very important to follow the transit of endogenous Pex3 during de novo peroxisome biogenesis; other interesting aspects to investigate include the mechanism of fusion between Pex16 and Pex3/Pex14 vesicles, as well as the process of maturation into peroxisomes.
It should be noted that Pex3-dependent de novo peroxisome biogenesis from mitochondrial membranes has been observed in the yeast S. cerevisiae. Rucktaschel et al.31 generated a mitochondrial Pex3 by fusion with the mitochondrial targeting signal of Tom20p. Expression of mitochondrial Pex3 in Pex3-deficient cells, which lack functional peroxisomes, resulted in the de novo formation of import-competent peroxisomes. These findings confirm that Pex3p-containing mitochondria in yeast and mammalian cells can serve as source for de novo peroxisome biogenesis. Rucktaschel et al. also concluded that natural or artificial targeting of Pex3p to any endomembrane may initiate peroxisome formation. It is thus likely that the specific properties of PMPs allow their targeting to other endomembranes, preferentially ER or mitochondria, when peroxisomal membranes are absent and/or the PMP import machinery is compromised; here, the properties of PMPs may differ between organisms and species, resulting in different affinities for organelle import receptors and chaperones, a possibility that could explain the preferential ER or mitochondrial localization in yeast and mammalian cells. The core import machinery may then exploit these membranes for the generation of pre-peroxisomal structures in order to obtain lipids/membrane for peroxisome formation. It will be interesting to investigate if these processes are linked to quality control mechanisms at organelle membranes to specifically re-localize membrane proteins with altered location.
In peroxisome-containing wild-type fibroblasts, Pex3 localizes exclusively to peroxisomes, and peroxisome biogenesis occurs through growth and division of pre-existing organelles. In their study, Sugiura et al. visualized Pex3 targeting to the mitochondria not just in human fibroblast cells derived by Zellweger patients, but also in control cells under conditions of peroxisome loss. It is known that Pex3 expression can trigger the selective, ubiquitination-linked degradation of peroxisomes (pexophagy).32 Although this was only observed in a small percentage of control cells, Sugiura et al., show that the machinery and processes for Pex3 import into mitochondria exist in wild-type cells and can be triggered upon physiological stress.
This critical finding leads to the important question about the physiological importance and function of de novo peroxisome biogenesis. In yeast cells, which contain few peroxisomes, de novo biogenesis may help to overcome loss of peroxisomes due to the inability to properly distribute peroxisomes to the budding daughter cell.4, 33 In the mammalian system, it is almost completely unknown how, whether, or when de novo peroxisome biogenesis may occur. De novo peroxisome biogenesis may be a rare event because it is coupled with pexophagy, possibly as a mechanism to restore the population of peroxisomes that inhabit a cell. Within such scenario, it would be important to learn whether de novo biogenesis is restricted to select physiological/metabolic states of the cell; also whether de novo peroxisome biogenesis is a constitutive or dominant process only in certain cell types, a possibility that would allow to clarify the etiology of a number of diseases that have dysregulated peroxisome activity.
In searching for the molecular machinery and regulation of Pex3 incorporation into pre-peroxisomal vesicles, Sugiura et al. conducted a number of experiments in which they downregulated candidate proteins either by silencing them or by using a pharmacological approach. These studies revealed a number of important findings. First, the fact that the addition of the proteasomal inhibitor MG132 led to a block in Pex3 exit from mitochondria, and an inhibition of peroxisomal biogenesis, leading the authors to conclude that Pex3 must exit the mitochondria to generate new peroxisomes. Here, addition of MG132 caused a robust accumulation of Pex3, indicating a rapid, ubiquitin- and proteasome-dependent turnover of Pex3 in both the Zellweger and control human fibroblasts; instead, Pex14 remained stably expressed. This implies that Pex3, whether it resides in the mitochondria or in the peroxisome, is subject to regulated ubiquitination, which leads to the retrotranslocation of the single-pass transmembrane protein into the cytosol for degradation by the proteasome. Ubiquitin-dependent turnover of Pex3 was previously observed in the yeast H. polymorpha,34 and ubiquitination of Pex5, a receptor for the import of peroxisomal matrix proteins, is essential for its recycling form the peroxisomal membrane.35 That specific PMPs are selectively targeted for ubiquitination opens a series of new questions, including the identity and regulation of the ligases; further, it highlights the importance of addressing how such rapid turnover on Pex3 differentially impacts on its function as an import protein and as a pexophagy receptor.
Finally, silencing experiments showed that Pex3 exit from mitochondria is independent of the core mitochondrial (and peroxisomal) fission GTPase Drp1, as well as of Vps35. Similarly, the GTPases Vps1p and Dnm1p, two proteins required for peroxisome fission in yeast, were not required for Pex3-dependent de novo peroxisome biogenesis from mitochondrial membranes in the yeast S. cerevisiae.31 These are findings that, on one hand, are consistent with previous studies from the McBride Laboratory in which it was shown that the budding of MDV from the organelle did not require Drp1.25 However, on the other hand, the fact that Vps35 is not required for Pex3 mobilization from the mitochondria is intriguing because they previously showed that the protein, which is a component of the retromer complex, regulates the exit of MAPL (a.k.a. MUL1/MULAN) into vesicles that are targeted to a subpopulation of peroxisomes.36 This suggests that mitochondria might be budding out vesicles via Vps35-dependent and Vps35-independent mechanisms. Consistent with this possibility, Sugiura et al. showed electron microscopy data indicating that Pex3-containing vesicles are structurally different from MAPL-containing ones. More specifically, MAPL-containing structures are strictly 70–100 nm in diameter, and contain both inner and outer mitochondrial membranes; instead, Pex3-containing structures are more pleotropic in size, up to 250 nm in diameter, do not appear to have a spherical shape, are electroluscent, and contain at least the outer mitochondrial membrane (sometime both membranes). This finding highlights the flexibility of mitochondria in segregating different cargoes through distinct molecular mechanisms of vesicle biogenesis, which ultimately results in vesicles that carry out different functions.37 To date MDV are known to carry mitochondrial proteins to peroxisomes,25, 36 oxidized proteins to the lysosome in a PINK1/Parkin-dependent manner,30, 38, 39 mitochondrial antigens to the lysosome in a process inhibited by PINK1/Parkin,38, 40 oxidized mtDNA for cellular release within neutrophils,41 and, finally, the protein MAPL to a subpopulation of peroxisomes in a retromer-dependent manner.25, 36 The McBride Laboratory now extend MDV biology by showing that they generate pre-peroxisomal vesicles containing Pex3.1 Clearly, the extent and function of cargoes that exit mitochondria within vesicular structures is likely to continue to expand, and is poised to have major impact on several fields of cell biology.
Coupling the biology of mitochondria to that of peroxisomes: possible impacts on the disease
The identification of cooperative and essential roles for both the ER and mitochondria in de novo peroxisome biogenesis opens entirely new and exciting avenues for future research. One critical initial step will be the development of experimental paradigms to study de novo peroxisomal biogenesis in complex mammalian physiology. To this end, it is essential to understand the functional links between the peroxisomes, mitochondria, and the ER; mitochondria and peroxisomes cooperate in several metabolic and signaling tasks, and even share common machinery for their division.6, 42 Furthermore, the transcriptional regulation of mitochondria and peroxisomal biogenesis is also shared through the PGC1alpha and the PPAR system.43, 44 Last but not the least, they both rely on the ER for lipid biosynthesis, and they both form membrane contact sites with the ER.45, 46, 47, 48, 49 For deciphering the cellular signals that may trigger de novo peroxisomal biogenesis, one can turn to the cell type-specific biochemical processes that utilize this metabolic triad of organelles. This includes hepatocytes, where ER-derived cholesterol is used to generate bile acids using enzymes found within all three organelles; within this context, it is possible that the rapid expansion of hepatocytes during development or injury may require de novo peroxisome biogenesis. Similarly, in the central nervous system, 70 mol% of the myelin sheets is composed by the ether lipid plasmalogen; this lipid is made by oligodendrocytes, in the peroxisomes, using phosphatidylethanolamine, which is generated in the mitochondria from ER-derived phosphatidyl serine50 at mitochondria–ER contact sites.48, 51, 52, 53, 54 This intimate metabolic cycle requires a unique homeostatic niche that may provide an opportunity to better understand the mechanisms and physiology of de novo peroxisomal biogenesis.
This and previous studies indicate that mitochondrial function may be directly coupled to that of the peroxisome; by implication, mitochondrial dysfunctions might also impact on peroxisome activity and vice versa.55 We have witnessed a renaissance in mitochondrial cell biology over the last decade, particularly with the established links to diseases such as Parkinson’s,56 Alzheimer’s,57, 58 cancer,59 metabolic syndromes, and a host of rare diseases that, together, afflict millions of people worldwide. In addition, new links between mitochondrial ultrastructure, dynamics, and inter-organellar contacts with changes in cell metabolism have expanded the field. Although it has been known that there is extensive metabolic flux between the mitochondria and peroxisome, this knowledge hasn’t yet led to significant experimental examination of the contribution of peroxisomal dysfunction to these diseases. Peroxisomal disorders like Zellweger syndrome or adrenoleukodystrophy are linked to neurological defects, and it is conceivable that peroxisomal dysfunction may play an important role in neurodegeneration and aging. In this respect, it is not clear, for example, whether there is a specific role for peroxisomes within axons and dendrites. It is commonly believed that there are few peroxisomes in axons, which instead appear to be segregated into dendrites: why is this, and how is it regulated? What does it mean for the metabolism within the axon? Similarly, peroxisomes are central to the function of oligodendrocytes, but have not been actively studied in the process of multiple sclerosis, or during oligodendrocyte degeneration and regeneration processes. Indeed, a primary phenotype of Zellweger patients is a loss of myelination. Even within liver disease peroxisomes are generally seen as a passive generator of bile acids, without a major contribution to the etiology of disease. It has only been a few years since we learned of the molecular importance of mitophagy in disease, and we imagine that the work published by the group of Heidi McBride will open a similar resurgence of interest in peroxisomal biology.
References
Sugiura A, Mattie S, Prudent J, McBride HM . Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 2017; 542: 251–254.
Wanders RJ . Peroxisomes in human health and disease: metabolic pathways, metabolite transport, interplay with other organelles and signal transduction. Subcell Biochem 2013; 69: 23–44.
Lazarow PB, Fujiki Y . Biogenesis of peroxisomes. Annu Rev Cell Biol 1985; 1: 489–530.
Motley AM, Hettema EH . Yeast peroxisomes multiply by growth and division. J Cell Biol 2007; 178: 399–410.
Huybrechts SJ, Van Veldhoven PP, Brees C, Mannaerts GP, Los GV, Fransen M . Peroxisome dynamics in cultured mammalian cells. Traffic 2009; 10: 1722–1733.
Schrader M, Costello JL, Godinho LF, Azadi AS, Islinger M . Proliferation and fission of peroxisomes - an update. Biochim Biophys Acta 2016; 1863: 971–983.
Bonekamp NA, Sampaio P, de Abreu FV, Luers GH, Schrader M . Transient complex interactions of mammalian peroxisomes without exchange of matrix or membrane marker proteins. Traffic 2012; 13: 960–978.
Agrawal G, Subramani S . De novo peroxisome biogenesis: evolving concepts and conundrums. Biochim Biophys Acta 2016; 1863: 892–901.
Hua R, Kim PK . Multiple paths to peroxisomes: mechanism of peroxisome maintenance in mammals. Biochim Biophys Acta 2016; 1863: 881–891.
Kim PK, Mullen RT, Schumann U, Lippincott-Schwartz J . The origin and maintenance of mammalian peroxisomes involves a de novo PEX16-dependent pathway from the ER. J Cell Biol 2006; 173: 521–532.
Delille HK, Agricola B, Guimaraes SC, Borta H, Luers GH, Fransen M et al. Pex11pbeta-mediated growth and division of mammalian peroxisomes follows a maturation pathway. J Cell Sci 2010; 123: 2750–2762.
Koch J, Pranjic K, Huber A, Ellinger A, Hartig A, Kragler F et al. PEX11 family members are membrane elongation factors that coordinate peroxisome proliferation and maintenance. J Cell Sci 2010; 123: 3389–3400.
Smith JJ, Aitchison JD . Peroxisomes take shape. Nat Rev Mol Cell Biol 2013; 14: 803–817.
Yuan W, Veenhuis M, van der Klei IJ . The birth of yeast peroxisomes. Biochim Biophys Acta 2016; 1863: 902–910.
van der Zand A, Gent J, Braakman I, Tabak HF . Biochemically distinct vesicles from the endoplasmic reticulum fuse to form peroxisomes. Cell 2012; 149: 397–409.
Knoops K, Manivannan S, Cepinska MN, Krikken AM, Kram AM, Veenhuis M et al. Preperoxisomal vesicles can form in the absence of Pex3. J Cell Biol 2014; 204: 659–668.
Motley AM, Galvin PC, Ekal L, Nuttall JM, Hettema EH . Reevaluation of the role of Pex1 and dynamin-related proteins in peroxisome membrane biogenesis. J Cell Biol 2015; 211: 1041–1056.
Hettema EH, Erdmann R, van der Klei I, Veenhuis M . Evolving models for peroxisome biogenesis. Curr Opin Cell Biol 2014; 29: 25–30.
Honsho M, Tamura S, Shimozawa N, Suzuki Y, Kondo N, Fujiki Y . Mutation in PEX16 is causal in the peroxisome-deficient Zellweger syndrome of complementation group D. Am J Hum Genet 1998; 63: 1622–1630.
Kiel JA, Veenhuis M, van der Klei IJ . PEX genes in fungal genomes: common, rare or redundant. Traffic 2006; 7: 1291–1303.
Matsuzaki T, Fujiki Y . The peroxisomal membrane protein import receptor Pex3p is directly transported to peroxisomes by a novel Pex19p- and Pex16p-dependent pathway. J Cell Biol 2008; 183: 1275–1286.
Muntau AC, Mayerhofer PU, Paton BC, Kammerer S, Roscher AA . Defective peroxisome membrane synthesis due to mutations in human PEX3 causes Zellweger syndrome, complementation group G. Am J Hum Genet 2000; 67: 967–975.
Hoepfner D, Schildknegt D, Braakman I, Philippsen P, Tabak HF . Contribution of the endoplasmic reticulum to peroxisome formation. Cell 2005; 122: 85–95.
Sacksteder KA, Jones JM, South ST, Li X, Liu Y, Gould SJ . PEX19 binds multiple peroxisomal membrane proteins, is predominantly cytoplasmic, and is required for peroxisome membrane synthesis. J Cell Biol 2000; 148: 931–944.
Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA et al. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 2008; 18: 102–108.
Goldfischer S, Moore CL, Johnson AB, Spiro AJ, Valsamis MP, Wisniewski HK et al. Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science 1973; 182: 62–64.
Klouwer FC, Berendse K, Ferdinandusse S, Wanders RJ, Engelen M, Poll-The BT . Zellweger spectrum disorders: clinical overview and management approach. Orphanet J Rare Dis 2015; 10: 151.
Gould SJ, Valle D . Peroxisome biogenesis disorders: genetics and cell biology. Trends Genet 2000; 16: 340–345.
Erdmann R . Assembly, maintenance and dynamics of peroxisomes. Biochim Biophys Acta 2016; 1863: 787–789.
Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 2012; 22: 135–141.
Rucktaschel R, Halbach A, Girzalsky W, Rottensteiner H, Erdmann R . De novo synthesis of peroxisomes upon mitochondrial targeting of Pex3p. Eur J Cell Biol 2010; 89: 947–954.
Yamashita S, Abe K, Tatemichi Y, Fujiki Y . The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy 2014; 10: 1549–1564.
Knoblach B, Rachubinski RA . How peroxisomes partition between cells. A story of yeast, mammals and filamentous fungi. Curr Opin Cell Biol 2016; 41: 73–80.
Williams C, van der Klei IJ . Pexophagy-linked degradation of the peroxisomal membrane protein Pex3p involves the ubiquitin-proteasome system. Biochem Biophys Res Commun 2013; 438: 395–401.
Platta HW, El Magraoui F, Schlee D, Grunau S, Girzalsky W, Erdmann R . Ubiquitination of the peroxisomal import receptor Pex5p is required for its recycling. J Cell Biol 2007; 177: 197–204.
Braschi E, Goyon V, Zunino R, Mohanty A, Xu L, McBride HM . Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr Biol 2010; 20: 1310–1315.
Sugiura A, McLelland GL, Fon EA, McBride HM . A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 2014; 33: 2142–2156.
McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA . Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 2014; 33: 282–295.
McLelland GL, Lee SA, McBride HM, Fon EA . Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol 2016; 214: 275–291.
Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M et al. Parkinson's disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell 2016; 166: 314–327.
Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 2016; 213: 697–713.
Schrader M, Yoon Y . Mitochondria and peroxisomes: are the 'big brother' and the 'little sister' closer than assumed? Bioessays 2007; 29: 1105–1114.
Bagattin A, Hugendubler L, Mueller E . Transcriptional coactivator PGC-1alpha promotes peroxisomal remodeling and biogenesis. Proc Natl Acad Sci USA 2010; 107: 20376–20381.
Berger J, Moller DE . The mechanisms of action of PPARs. Annu Rev Med 2002; 53: 409–435.
Costello JL, Castro IG, Hacker C, Schrader TA, Metz J, Zeuschner D et al. ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J Cell Biol 2017; 216: 331–342.
Hua R, Cheng D, Coyaud E, Freeman S, Di Pietro E, Wang Y et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J Cell Biol 2017; 216: 367–377.
Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau KF et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun 2014; 5: 3996.
Giacomello M, Pellegrini L . The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ 2016; 23: 1417–1427.
Sood A, Jeyaraju DV, Prudent J, Caron A, Lemieux P, McBride HM et al. A mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc Natl Acad Sci USA 2014; 111: 16017–16022.
Braverman NE, Moser AB . Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta 2012; 1822: 1442–1452.
Herrera-Cruz MS, Simmen T . Of yeast, mice and men: MAMs come in two flavors. Biol Direct 2017; 12: 3.
Vance JE . Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem 1990; 265: 7248–7256.
Vance JE . Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J Biol Chem 1991; 266: 89–97.
Vance JE . MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta 2014; 1841: 595–609.
Schrader M, Costello J, Godinho LF, Islinger M . Peroxisome-mitochondria interplay and disease. J Inherit Metab Dis 2015; 38: 681–702.
Pickrell AM, Youle RJ . The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 2015; 85: 257–273.
Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J 2012; 31: 4106–4123.
Area-Gomez E, Schon EA . Mitochondria-associated ER membranes and Alzheimer disease. Curr Opin Genet Dev 2016; 38: 90–96.
Vyas S, Zaganjor E, Haigis MC . Mitochondria and Cancer. Cell 2016; 166: 555–566.
Acknowledgements
We are grateful to Emma Clara Pellegrini for drawing the artwork shown in Figure 1.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Rights and permissions
About this article

Cite this article
Schrader, M., Pellegrini, L. The making of a mammalian peroxisome, version 2.0: mitochondria get into the mix. Cell Death Differ 24, 1148–1152 (2017). https://doi.org/10.1038/cdd.2017.23
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.23
This article is cited by
-
Organelle-targeted therapies: a comprehensive review on system design for enabling precision oncology
Signal Transduction and Targeted Therapy (2022)
-
The peroxisome: an update on mysteries 2.0
Histochemistry and Cell Biology (2018)
