
Abstract
Aggregation of cytosolic proteins is a pathological finding in disease states, including ageing and neurodegenerative diseases. We have previously reported that hypoxia induces protein misfolding in Caenorhabditis elegans mitochondria, and electron micrographs suggested protein aggregates. Here, we seek to determine whether mitochondrial proteins actually aggregate after hypoxia and other cellular stresses. To enrich for mitochondrial proteins that might aggregate, we performed a proteomics analysis on purified C. elegans mitochondria to identify relatively insoluble proteins under normal conditions (110 proteins identified) or after sublethal hypoxia (65 proteins). A GFP-tagged mitochondrial protein (UCR-11 – a complex III electron transport chain protein) in the normally insoluble set was found to form widespread aggregates in mitochondria after hypoxia. Five other GFP-tagged mitochondrial proteins in the normally insoluble set similarly form hypoxia-induced aggregates. Two GFP-tagged mitochondrial proteins from the soluble set as well as a mitochondrial-targeted GFP did not form aggregates. Ageing also resulted in aggregates. The number of hypoxia-induced aggregates was regulated by the mitochondrial unfolded protein response (UPRmt) master transcriptional regulator ATFS-1, which has been shown to be hypoxia protective. An atfs-1(loss-of-function) mutant and RNAi construct reduced the number of aggregates while an atfs-1(gain-of-function) mutant increased aggregates. Our work demonstrates that mitochondrial protein aggregation occurs with hypoxic injury and ageing in C. elegans. The UPRmt regulates aggregation and may protect from hypoxia by promoting aggregation of misfolded proteins.
Similar content being viewed by others

Main
Protein aggregation is one of the most active areas of biological research. Protein aggregation has been linked to disease states including ageing1 and many neurodegenerative diseases.2, 3, 4 It is well established that in these conditions protein aggregation occurs in the cytoplasm where it is a key indicator of pathological progression. In contrast, the presence or pathological consequences of protein aggregation in other cellular compartments, such as mitochondria, remains relatively understudied.
Recent work has established that mitochondrial protein homeostasis is preserved by a mitochondrial specific stress response known as the mitochondrial unfolded protein response (UPRmt).5, 6, 7, 8 The UPRmt is believed to be activated in response to the accumulation of misfolded proteins within mitochondria leading to the upregulation of mitochondrial chaperone levels.6, 7 Most knowledge about the UPRmt has been derived from monitoring levels of mitochondrial chaperones either directly with western blots or through induction of transcriptional reporters of two such chaperones in Caenorhabditis elegans, hsp-6 and hsp-60.6 This has severely limited our understanding of mitochondrial proteostasis as monitoring chaperone levels reflects the response to misfolded proteins rather than a direct readout of mitochondrial protein homeostasis. In addition, chaperone levels do not allow for real-time assessment of the mitochondrial proteostatic environment, which precludes analysis of the dynamics of protein homeostatic changes within mitochondria.
Despite these limitations, a number of studies have identified conditions that are associated with an increase in insoluble mitochondrial protein. Conditions such as hypoxic injury9 and ageing10, 11 have been associated with a rise in detergent-insoluble proteins, which is believed to represent misfolded or aggregated proteins. However, these studies are limited in that they have not established direct evidence for aggregation of proteins within the mitochondria, but rather for protein misfolding, which does not invariably lead to protein aggregation as misfolded proteins may be degraded or misfolded rather than aggregate.12
In this study we identify mitochondrial proteins that misfold following a hypoxic injury and develop real-time reporters of the mitochondrial protein folding environment. Using these reporters we establish that proteins aggregate within mitochondria following both acute stressors such as hypoxic injury and with the chronic stress of ageing. We also establish the dynamics of mitochondrial protein aggregate formation and resolution following hypoxic injury and determine the impact of modulation of the UPRmt on mitochondrial protein aggregate load.
Results
Identification of insoluble mitochondrial proteins
To identify mitochondrial proteins that may aggregate we used an in vivo C. elegans hypoxic injury model13 to produce insoluble proteins, followed by isolation of insoluble mitochondrial proteins and identification by label-free quantitative mass-spectrometry-based proteomic methods. Proteins were isolated after hypoxic incubation, either immediately or following a 24 h recovery period. A total of 1482 proteins were identified in total (Supplementary Table S1). A total of 327 proteins exhibited hypoxia preferential insolubility  either immediately after recovery or after 24 h (Figure 1a). Sixty-five proteins were hypoxia preferentially insoluble at both time points. A total of 330 proteins were enriched in the insoluble fraction in either hypoxia or normoxia-treated samples (
either immediately after recovery or after 24 h (Figure 1a). Sixty-five proteins were hypoxia preferentially insoluble at both time points. A total of 330 proteins were enriched in the insoluble fraction in either hypoxia or normoxia-treated samples ( under either hypoxia or normoxia conditions) (Figure 1b). Of these generally insoluble proteins, 110 were enriched in the insoluble fraction in both the immediate and 24 h recovery samples. Proteins identified in samples from both time points represented hits with highest confidence. Thus, we focused on the 65 hypoxia preferential insoluble proteins and the 110 generally insoluble proteins for further analysis.
under either hypoxia or normoxia conditions) (Figure 1b). Of these generally insoluble proteins, 110 were enriched in the insoluble fraction in both the immediate and 24 h recovery samples. Proteins identified in samples from both time points represented hits with highest confidence. Thus, we focused on the 65 hypoxia preferential insoluble proteins and the 110 generally insoluble proteins for further analysis.

Mitochondrial-associated insoluble proteins. (a) Proteins identified as preferentially hypoxia insoluble  immediately (green) or 24 h (purple) post hypoxia. (b) Proteins identified as generally insoluble (
immediately (green) or 24 h (purple) post hypoxia. (b) Proteins identified as generally insoluble ( for hypoxia and normoxia) immediately (green) or 24 h (purple) post exposure. (c) Gene ontology classes identified as significantly over or under represented by PANTHER overrepresentation test comparing proteins identified as generally insoluble with all detected proteins. (d) The number of proteins with predicted transmembrane domains in the total, generally insoluble, and hypoxia-induced Insoluble sets was estimated by TMHMM 2.0. *P<0.001, **P<0.0001 (hypergeometric probability test)
for hypoxia and normoxia) immediately (green) or 24 h (purple) post exposure. (c) Gene ontology classes identified as significantly over or under represented by PANTHER overrepresentation test comparing proteins identified as generally insoluble with all detected proteins. (d) The number of proteins with predicted transmembrane domains in the total, generally insoluble, and hypoxia-induced Insoluble sets was estimated by TMHMM 2.0. *P<0.001, **P<0.0001 (hypergeometric probability test)
Inconsistent with a stochastic mechanism of insolubility, certain classes of proteins were enriched in the generally insoluble fraction (Supplementary Table S2). Proteins with conserved domains falling in the mitochondrial carrier and acyl-coA dehydrogenase superfamilies were significantly (P<0.01) enriched in the generally insoluble set. In the hypoxia insoluble set, thioredoxin-like and GST-C superfamily containing proteins were highly enriched (P<0.0001). Surprisingly, predicted secondary structure did not correlate with insolubility (Supplementary Figure S1a and b), in contrast to other diseases of protein aggregation.14, 15 We also examined whether transmembrane proteins were present in the insoluble sets.16 In all, 38 out of the 110 generally insoluble proteins were predicted to have at least one transmembrane domain compared with only 316 of the 1482 total set of proteins (P<0.001) (Figure 1c). However, 0 proteins in the hypoxia insoluble set were predicted to have transmembrane domains, a highly statistically significantly difference from both the total and generally insoluble sets (P<0.00001). This suggests that hypoxia-induced insolubility and perhaps aggregation may primarily affect non-transmembrane proteins.
Mitochondrial proteins aggregate with hypoxic stress
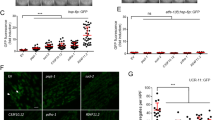
We made use of the proteins in the insoluble sets to develop GFP-tagged fusion reporters that might demonstrate aggregation (Supplementary Table S3). First, we obtained an existing GFP fusion construct for expression of UCR-11,17 a component of complex III of the electron transport chain identified as generally insoluble. Under normoxia, UCR-11::GFP was uniformly expressed throughout mitochondria, but following hypoxia, UCR-11::GFP aggregates were readily apparent in most mitochondria (Figures 2a). To determine if the GFP tag alone could drive aggregation, we tested a strain that expresses an unfused GFP in body wall muscle mitochondria.18 This control strain did not exhibit any GFP aggregates within mitochondria (Figures 2c and d). Next, we constructed two strains expressing GFP-tagged copies of MDH-2 and CRI-3, which were found by proteomics to remain soluble ( under all conditions). Neither of these strains formed any visible aggregates within mitochondria (Figures 2e). From the generally insoluble proteins we generated five additional GFP fusion constructs (Supplementary Table S3). After hypoxia, all of these strains exhibited aggregates within mitochondria (Figures 2h) in all tissues examined, with minimal aggregation under normoxia (Figures 2g), indicating that although these proteins were relatively insoluble under normoxic conditions in our biochemical assay, a stressor is required for visible aggregation. Expression levels alone did not explain the aggregation phenotype, as the unfused GFP control was expressed at much higher levels than transgenes with aggregation (Figure 2q). Notably, these hypoxia-induced aggregates were consistent in size, location, and morphology with those seen previously9 and here by electron microscopy (EM) following hypoxia (Figures 2k and l), suggesting that the GFP-tagged protein aggregates faithfully model the native aggregates.
under all conditions). Neither of these strains formed any visible aggregates within mitochondria (Figures 2e). From the generally insoluble proteins we generated five additional GFP fusion constructs (Supplementary Table S3). After hypoxia, all of these strains exhibited aggregates within mitochondria (Figures 2h) in all tissues examined, with minimal aggregation under normoxia (Figures 2g), indicating that although these proteins were relatively insoluble under normoxic conditions in our biochemical assay, a stressor is required for visible aggregation. Expression levels alone did not explain the aggregation phenotype, as the unfused GFP control was expressed at much higher levels than transgenes with aggregation (Figure 2q). Notably, these hypoxia-induced aggregates were consistent in size, location, and morphology with those seen previously9 and here by electron microscopy (EM) following hypoxia (Figures 2k and l), suggesting that the GFP-tagged protein aggregates faithfully model the native aggregates.

Insolubility predicts aggregation of tagged proteins. (a,b) UCR-11::GFP does not aggregate with normoxia (a) but aggregates following hypoxia independent of reoxygenation (b). Unfused GFP (zcIs14) (c,d) and MDH-2::GFP (gcEx173) (e,f) did not aggregate following normoxia (c,e) or hypoxia (d,f). Aggregation of PHB-1::GFP (gcEx169) (g,h) and MRPL-17::GFP (gcEx167) (i,j) following hypoxia (h,j) but not normoxia (g,i). After hypoxia, a UCR-11::GFP aggregate (arrow) in a single mitochondrion (k) appears similar in relative size and shape to that observed by EM (arrow) (l). (m) Quantification of intra-mitochondrial aggregates. (n) Increased duration of hypoxia but not normoxia resulted in an increase of UCR-11::GFP aggregates. Increased recovery periods decreased aggregation. CRI-3::GFP did not aggregate following normoxia (o) or hypoxia (p). (q) Overexpression alone does not explain aggregation. GFP fluorescence (log10 in arbitrary units, A.U.) of non-aggregating strains (unfused GFP=SJ4103, MDH-2::GFP=MC789) and aggregating strains (UCR-11::GFP=MC776, MRPL-17=MC784). Unfused GFP fluorescence greater than other constructs, P<0.0001, unpaired t-test. Scale bar in (a) =2 μm and applies to (a,b). Scale bar in (c) =2 μm and applies to (c–j) Scale bar in (k) =0.5 μm and applies to (k,l). Scale bar in (o) =2 μm and applies to (o,p). Arrows point to only a portion of the aggregates except for the single aggregate in (k) and (l). *P<0.05, **P<0.01, ***P<0.001, by unpaired t-test with Holm-Šídák correction
To study the dynamics of mitochondrial protein aggregate formation, we varied both the duration of hypoxia and the recovery period and observed the level of UCR-11::GFP aggregation. We found that the number of aggregates per high powered field (HPF) increased with increasing length of hypoxia and decreased with increasing periods of recovery (Figure 2n). Interestingly, the aggregates were present under conditions where the worms were not reoxygenated prior to imaging (Figure 2b, immediate condition in Figure 2n). We also assayed for aggregate formation with exposure to the chemical hypoxia mimetics sodium azide and sodium cyanide. Continuous incubation with both chemicals led to a time dependent increase in the number of aggregates (Supplementary Figure S2) indicating that aggregation can occur rapidly and is not related to lack of environmental oxygen per se, but is instead related to the cellular effects of decreased oxygen utilization. We also asked whether reducing translation rate might reduce aggregate level. This question derives from the hypothesis that newly synthesized proteins being transported into the mitochondria may be particularly prone to aggregation. Further, translational suppression has been shown to confer resistance to hypoxic death and therefore if mitochondrial aggregates were deleterious, cycloheximide might be associated with reduced aggregates.19, 20 However, cycloheximide at concentrations previously shown to inhibit translation to 35% (1 mg/ml) and 20% (3 mg/ml) of baseline did not reduce aggregates load;19 rather, it dose-dependently increased it (Supplementary Figure S3). Our findings demonstrate that aggregation increases with worsening hypoxic stress, that the aggregate load appears to be, in part, reversible, and that cytoplasmic translational inhibition at a level previously shown to protect from hypoxic injury actually increases aggregation.
To test whether protein aggregation generally results from impairment of mitochondrial function or fragmentation, we tested two drugs that block ATP generation by mitochondria, the uncoupling agent carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP)21 and the ATP synthase inhibitor oligomycin A.22 Following an exposure to FCCP sufficient to depolarize and fragment mitochondria, no increase in mitochondrial protein aggregation was observed (Figures 3a–c). Similarly, treatment with oligomycin A produced mitochondrial fragmentation and severe mitochondrial swelling but did not result in mitochondrial protein aggregation (Figures 3d–f).

Mitochondrial dysfunction does not necessarily result in aggregation. (a–c), Treatment with the protonophore FCCP (carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone) leads to mitochondrial depolarization but does not lead to an increase in aggregates per high power field. (a), 0 min post addition of FCCP demonstrating a high degree of colocalization of the reporter UCR-11::GFP (green) with the mitochondrial membrane potential sensitive dye tetramethylrhodamine ethyl ester (TMRE) (red). (b) By 90 min of exposure to FCCP mitochondria in the same field are fragmented, and decreased colocalization of UCR-11::GFP and TMRE is observed indicative of mitochondrial depolarization. (c), Treatment with FCCP did not lead to an increase in the amount of aggregates per high power field. (d–f) Overnight treatment with control buffer (d) or the oxidative phosphorylation inhibitor oligomycin A (e) did not induce aggregation of the UCR-11::GFP reporter. (f). Scale bar in (a) =2 μm applies to all images
Ageing promotes mitochondrial protein aggregation
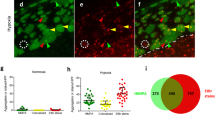
Cytosolic protein aggregation is a hallmark of ageing.1 Further, although controversial,23 induction of the UPRmt has been associated with prolonging lifespan.24 Thus, we hypothesized that mitochondrial protein aggregation may occur with ageing. To test this, we performed EM on wild-type worms at three time points, adult days 1, 5 and 10. Under these conditions the percent of mitochondria with intra-mitochondrial electron-dense material consistent with protein aggregates increased in an age-dependent manner (Figures 4a and c). Next, we measured UCR-11::GFP aggregates with ageing and found that aggregation of the reporter similarly occurred with ageing (Figures 4d–f). Therefore, by both EM and fluorescent reporter, we have established that mitochondrial protein aggregation is a component of the ageing process.

Ageing results in mitochondrial aggregates. Electron micrographs of day 1 adults (a) demonstrated fewer mitochondria with aggregates (arrows) relative to day 10 adults (b). Expanded insets are for better visualization of the mitochondria. (c) Summary of EM data – number of mitochondria scored – day 1=1444, day 5=1609, day 10=227, three animals/age group were scored. (d–f). Day 1 adults with UCR-11::GFP (d) had fewer aggregates (arrows) than day 10 (e). χ2-test (c), Dunnett’s multiple comparison test (f). Inset scale bar in (a) =2 μm and applies to both insets. ns, not significant, **P<0.01, ****P<0.0001
An intact UPRmt is required for mitochondrial aggregates
We and others have previously reported that hypoxia induces the UPRmt and that UPRmt induction can protect from hypoxic cell death.9, 25 Under the assumption that mitochondrial aggregates were deleterious to the cell, we hypothesized that induction of the UPRmt would decrease hypoxia-induced aggregation. To test this hypothesis, we used three RNAi constructs previously shown to activate the UPRmt, (cco-1, letm-1, and F02A9.4).23, 24, 26 We found that pre-hypoxic treatment with the RNAi decreased aggregation and improved hypoxic survival (Figures 5a and b). Some of the proteins targeted by these RNAi constructs were identified as insoluble by proteomics; therefore decreased aggregation may represent a direct effect of decreased protein level rather than actual improved proteostasis via UPRmt activation. To more directly test the role of the UPRmt, we tested the effect on aggregation of an RNAi construct, loss-of-function, and gain-of-function mutations in the UPRmt master transcriptional regulator atfs-1.27, 28 Surprisingly, both atfs-1 RNAi and the loss-of-function mutation atfs-1(tm4919) resulted in a marked reduction of mitochondrial aggregates (Figure 5c). The atfs-1(et18) gain-of-function mutant had a modest but statistically significant increase in aggregates (Figure 5c). We then tested doxycycline, a mitochondrial translation inhibitor and a general inducer of the UPRmt.24 Consistent with the effects of manipulation of atfs-1 activity, treatment with doxycycline also led to increased mitochondrial protein aggregation (Figures 5d–f). Thus, although the data with cco-1, letm-1, and F02A9.4. RNAi constructs are consistent with our original hypothesis, the near abolition of aggregates by atfs-1 loss-of-function and the increase in the atfs-1 gain-of-function mutant and by doxycycline indicate that activation of the UPRmt can promote aggregation.

The UPRmt regulates UCR-11::GFP aggregation following hypoxia. (a,b) RNAi constructs that activate the UPRmt decrease aggregation (a) and increase survival (b) post hypoxia relative to L4440 empty vector control. (c) Aggregates are decreased in the loss-of-function atfs-1(tm4919) mutant and by atfs-1 RNAi and increased in atfs-1(et18) gain-of-function mutant. Treatment with doxycycline (d–f) increased aggregates (arrows in f) relative to control. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 by Dunnett’s multiple comparison test (a,b), unpaired t-test with Holm-Šídák correction (c,d)
Discussion
Hypoxia and ageing induce profound changes in the cellular proteostasis network.11, 19 The role of proteostasis in ageing has been most extensively studied. Aggregation of cytoplasmic proteins is well established to occur with ageing.1, 11 In addition, the cytoplasmic heat shock response pathway is required for both dietary restriction and insulin/IGF-1-like signaling pathway mediated extension of lifespan, and loss-of-function in the heat shock response is associated with premature ageing,29 highlighting the importance of cytoplasmic proteostasis in the ageing process. Although cytoplasmic proteostasis is established as having a fundamental role in ageing, the contribution of mitochondrial proteostasis to the ageing process remains controversial. Previously, activation of the UPRmt by inducing a state of mitonuclear protein imbalance was shown to be protective against ageing,24 however, more recent work23 has found that some conditions of activation of the UPRmt, such as with gain-of-function mutations in the UPRmt regulator atfs-1, do not extend lifespan, but in fact may reduce lifespan. These findings bring into question the role mitochondrial proteostasis plays in the ageing process. In this study, by both EM of wild-type worms and using the UCR-11::GFP reporter, we have established that in fact, aggregation of mitochondrial proteins does occur as part of the normal ageing process.
Despite advances in the prediction and identification of proteins prone to aggregation, the mechanisms of toxicity of misfolded and aggregated cytoplasmic proteins remain poorly understood. In general, aggregation of proteins correlates poorly with markers of toxicity such as cell death.30 In some cases, protein aggregation is believed to be protective11, 30, 31 against toxicity by protein oligomers.2 It is unknown whether aggregation of mitochondrial proteins is toxic or protective. Our findings that ATFS-1 activity correlates with hypoxia-induced mitochondrial protein aggregation and with hypoxia protection is consistent with the mitochondrial protein aggregates being protective. The increased aggregates with the hypoxia protective compound cycloheximide is also supportive of the hypothesis of aggregation as a protective strategy although how suppression of cytoplasmic translation might promote mitochondrial protein aggregation is obscure. Primarily based on the atfs-1 data herein and our previously published work showing the UPRmt is induced by hypoxia and is hypoxia protective,9 we favor the hypothesis that hypoxia and ageing promote mitochondrial protein misfolding and that atfs-1 responds to this stress by activating the UPRmt in a way that promotes the large aggregates observed by EM and with our reporters. Alternatively, the atfs-1-dependent aggregates are unrelated to mitochondrial toxicity, and atfs-1 provides hypoxia protection by some other mechanism. If indeed toxic, potential mechanisms of aggregate toxicity include preventing the normal functioning of mitochondria by sequestering essential proteins for mitochondrial function, or activation of pathways to degrade aggregate-containing mitochondria. Potential mechanisms by which the aggregates might be protective include sequestering toxic misfolded proteins and/or oligomers. Given the overrepresentation of proteins involved in electron transport and oxidative phosphorylation, the aggregates may serve to sequester toxic proteins that generate harmful free radicals.
In summary, in this study we have identified mitochondrial proteins that aggregate under stress conditions most notably hypoxia and ageing. We have also established the dynamics of aggregate appearance and resolution with hypoxia. Finally, we have found that the activity of atfs-1, which is the master regulator of the UPRmt and a hypoxia protectant, is associated with increased mitochondrial protein aggregation. It remains to be determined whether the aggregates are actually an atfs-1-directed protective response to hypoxia-induced protein misfolding.
Materials and methods
Strains
C. elegans strains were cultured as previously described.32 N2 (Bristol) was used as the wild-type strain for all experiments.33 Strain MC776 containing the gcIs46 transgene was generated by UV irradiation of strain DM732417 and was outcrossed with N2 four times prior to use. Strain MC791 containing atfs-1(et18);gcIs46 was made by crossing strains QC118 (ref. 34) and MC776, with presence of the atfs-1(et18) allele confirmed by sequencing. atfs-1(tm4919);gcIs46 was constructed and confirmed similarly. Proteins were selected for tagging on the basis of known mitochondrial targeting and either were not in operons or were the first gene in an operon. To generate strains with novel extrachromosomal arrays, clones were first generated by using PCR to amplify the following genes including the upstream intergenic region and excluding the stop codon from N2 genomic DNA: F26E4.6 (F: TAAGCACTGCAGGAAAAATGTCGAAATCAGGTAA, R: TAAGCAGGATCCTCTTGGTTGGCCTTTCTGAGTTG), mrpl-17 (F: TAAGCACTGCAGACAAATGAAGTTTTAAGACGGAGAA, R: TAAGCAGGATCCTCTTGAGATTTCTTCGGAGTATCCAGCT), mtx-1 (F: TAAGCAGGATCCCTTTTATCGGTGGAGCGCAC, R: TAAGCAGGTACCCTCTCAGAGATCTCCTCTTCGACTGA), phb-1 (F: TAAGCACTGCAGAGCGGCAAATTTCGATCGAA, R: TAAGCAGGATCCTCGGATTGAAGGTTGAGAAGGGTTTGT), dhod-1 (F: AGAGGAAAACAAAATAACGACTGC, R: ACCGGTACTTTCGTTTTTTCTCTGTGATCAGCT), mdh-2 (F: TAAGCACTGCAGCTTGAGAGTTGACTTGCTGAGA, R: TAAGCAGGTACCCTGTTTCCCTTAACGAAAGCGACT), cri-3 (F: CCCACCCAATTCCCGGTATA, R: ACCGGTACTTTGGAGATGAACTTCTTGATCTTGT). PCR products were cloned in frame into pPD95.75 (F26E4.6, mrpl-17, mtx-1, phb-1, mdh-2) or into pCR-XL-TOPO along with the GFP and the 3’-UTR from pPD95.75 (dhod-1 and cri-3). Strains were then generated by microinjections of the clones along with pRF4[rol-6(su1006)] as a co-injection marker.35, 36
Hypoxic exposure
Hypoxic and normoxic exposures and scoring were performed as previously reported.37 For survival assays, hypoxic exposure length was 18 h, for all other experiments, hypoxic exposure was for 12 h unless otherwise noted. Survival assays were performed with three plates each containing ~50 worms per biological replicate. All animals were 1-day-old adults when placed into the hypoxia chamber.
Mitochondrial isolation
Mitochondria were isolated from C. elegans raised on HGM plates following 12 h of hypoxia/normoxia without recovery (immediate) or after 24 h of recovery at 20 °C (24 h recovery), using Mitochondria Isolation Kit for Tissues (MitoSciences, Eugene, OR, USA) according to manufacturer’s instructions. For isolation of insoluble protein, mitochondria were resuspended in 2% NP-40, 1 mM DTT, 50 mM Tris pH 7.4, 1 mM EDTA, with Roche cOmplete Mini-Protease Inhibitor Tablets,38, 39 sonicated and centrifuged at 20 000 × g and 4 °C for 30 min twice. Aggregates were then washed in 50 mM Tris, 150 mM NaCl and centrifuged at 20 000 × g and 4 °C for 40 min.
Proteome sample preparations
Proteins were extracted from mitochondrial pellets with buffer (50 mM NH4HCO3, 8 M urea). Protein concentrations were quantified with Bradford assays. Equal amounts of proteins were input for proteomics analysis. Protein extracts were diluted with buffer (50 mM NH4HCO3, 5 % acetonitrile) to urea concentration <1 M, reduced with 10 mM DTT, alkylated with 10 mM iodoacetamide and digested with trypsin in 1:50 ratio (Promega Corporation, Madison, WI, USA) overnight. Tryptic peptides were purified with C18 Sep-Pak columns (Waters), dried, and resuspended with 0.1% formic acid for LC-MS/MS analysis.
LC-MS/MS analysis
Proteome samples were analyzed with a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) coupled to an EASY-nLC 1000 system (Thermo Fisher Scientific). Reversed-phase separation was performed with a 3 cm trap column (packed in-house with 200 Å C18 magic bead, 5 μm) (Bruker) and a 65 cm analytical column (100 Å C18 magic bead, 5 μm), using mobile phase of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). A linear separation gradient with 2–10% solvent B for 1 min, 10–30% solvent B for 89 min, and 30–80% solvent B for 1 min was used. The flow rate was set to 300 nl/min. Data-dependent acquisition analysis with Q Exactive Plus included MS1 analysis with scan range 400–2000 m/z, scan resolution 70 000 and automatic gain control (AGC) target value 1 × 106, and MS/MS analysis for the top 20 most abundant precursor ions detected in MS1 scans. MS/MS analysis included isolation window 1.6 m/z, fragmentation energy 25 NCE, scan resolution 17 500, AGC target value 5 × 104. Charge exclusion for 1+ and unassigned ions was enabled. Dynamic exclusion of 30 s was used.
Proteome quantitation analysis
The raw data was converted to mzXML files with ReAdW (version 4.2.1). The mzXML files were searched with Comet40 (version 2015.01 rev.02) against the concatenated database containing forward and reverse protein sequences of C. elegans N2 strain (total of 52 506 entries, downloaded from Uniprot on 21 April 2015). Search parameters included 20 ppm precursor mass tolerance, 13C offsets (−1/0/1/2/3) enabled, fragment bin tolerance 0.1, fragment bin offset 0, variable modifications of methionine oxidation (15.9949 Da), fixed modification of cysteine carbamidomethylation (57.021464 Da). Only fully tryptic peptide sequences were considered, and up to two missed cleavages were allowed. The false discovery rate for peptide identification was determined with the target-decoy approach,41 and was controlled at 1%. Label-free quantitation for MS1 precursor ion intensity was performed with XPRESS.42 Peptide chromatograms (time, intensity) were used to determine peptide ratios. Only peptides that were quantified in at least two technical replicates, and with Coefficient of variation (CV) of chromatogram intensity <80% in technical replicates were used for quantitative comparison with other samples. The CV filtering process reduced the noise in peptide quantitation. Peptide ratios quantified with different charge states were averaged to represent the peptide ratios. The log2 of the peptide ratios were averaged to determine the log2 of protein ratios. The protein fold changes were obtained with the anti-log2 transformation of log2 protein ratios. Only proteins with two or more unique peptides in each biological replicate were included in further analysis.
Gene ontology
Gene list analysis, overrepresentation test, gene ontology classification, and Figure 1c were generated by PANTHER43, 44, 45 (http://pantherdb.org).
Prediction of conserved domains
Conserved domain analysis was performed using NCBI’s Batch CD-Search Tool (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi).46
Prediction of secondary structure
Secondary structure predictions were performed using Jpred4 (ref. 47) (http://www.compbio.dundee.ac.uk/jpred/).
Prediction of transmembrane domains
Prediction of transmembrane helicies was performed using TMHMM Server v. 2.0 (ref. 16, 48) (http://www.cbs.dtu.dk/services/TMHMM/).
Generation of graphs
Graphs were generated using Graphpad Prism 6 or Microsoft Excel. Venn diagrams were generated using Venn Diagram Plotter provided by Pacific Northwest National Laboratory (http://omics.pnl.gov/software/venn-diagram-plotter). Error bars in all graphs represent standard error of the mean.
Statistics
All experiments were performed with a minimum of three independent biological replicates. Statistics were performed using Graphpad Prism 6 or Microsoft Excel. All statistical tests were performed as two-sided tests. Statistical tests for overrepresentation using a hypergeometric probability test were performed using he GeneProf calculator (https://www.geneprof.org/GeneProf/tools/hypergeometric.jsp).49
Light microscopy
Confocal microscopy was performed using a Leica TCS SP8 equipped with a HyD detector. Paralysis was produced by mounting worms in a solution of 50 mM levamisole (Sigma-Aldrich Corp., St. Louis, MO, USA) in M9 prior to imaging. Images were acquired from at least four randomly selected worms (eight worms for ageing) at 1024 × 1024 resolution using a 63 × objective with 8 × zoom producing a 23.07 × 23.07 μm image (defined as one high power field, HPF). All images were acquired as a ten slice Z-stack with scan speed of 800-1800 Hz and flattened as a maximum intensity projection prior to analysis. Analysis of images was performed by an observer blinded to condition. To assess aggregation without reoxygenation, worms were mounted on slides in an anaerobic chamber where a coverslip was applied and prior to removal from the chamber for imaging. Mitochondrial depolarization was assessed using TMRE as previously reported.9 For assessment of total fluorescence images were acquired on a Zeiss Axioskop2 microscope with fluorescence intensity quantified using NIH ImageJ.
Electron microscopy
EM was performed as previously reported.9
Drug treatment
All chemicals were acquired from Sigma-Aldrich. Sodium azide and sodium cyanide were dissolved in M9 with 50 mM levamisole at 500 mM and 50 mM concentrations respectively with M9+levamisole as a control and worms were mounted in the solutions and maintained in them throughout imaging. For experiments with doxycycline (50 μM), or oligomycin A (50 μM) worms were treated overnight with drugs or their respective control solutions. For FCCP treatment worms were mounted in 50 mM levamisole in M9 with FCCP (15 μM) or an equivalent amount of DMSO added immediately prior to imaging, and maintained in the solution throughout imaging.
RNAi
Feeding RNAi was performed as previously described.9
References
Labbadia J, Morimoto RI . The biology of proteostasis in aging and disease. Annu Rev Biochem 2015; 84: 435–464.
Ross CA, Poirier MA . Protein aggregation and neurodegenerative disease. Nat Med 2004; 10: S10–S17.
Opattova A, Filipcik P, Cente M, Novak M . Intracellular degradation of misfolded tau protein induced by geldanamycin is associated with activation of proteasome. J Alzheimers Dis 2013; 33: 339–348.
Ciechanover A, Kwon YT . Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 2015; 47: e147.
Zhao Q, Wang J, Levichkin I, Stasinopoulos S, Ryan M, Hoogenraad N . A mitochondrial specific stress response in mammalian cells. EMBO J 2002; 21: 4411–4419.
Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D . Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 2004; 117: 4055–4066.
Haynes CM, Ron D . The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci 2010; 123: 3849–3855.
Jovaisaite V, Mouchiroud L, Auwerx J . The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J Exp Biol 2014; 217: 137–143.
Kaufman DM, Crowder CM . Mitochondrial proteostatic collapse leads to hypoxic injury. Curr Biol 2015; 25: 2171–2176.
David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C . Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol 2010; 8: e1000450.
Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P et al. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 2015; 161: 919–932.
Pulido P, Llamas E, Llorente B, Ventura S, Wright LP, Rodriguez-Concepcion M . Specific Hsp100 chaperones determine the fate of the first enzyme of the plastidial isoprenoid pathway for either refolding or degradation by the stromal clp protease in arabidopsis. PLoS Genet 2016; 12: e1005824.
Scott BA, Avidan MS, Crowder CM . Regulation of hypoxic death in C. elegans by the insulin/IGF receptor homolog DAF-2. Science 2002; 296: 2388–2391.
Fink AL . Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des 1998; 3: R9–23.
Bratko D, Blanch HW . Effect of secondary structure on protein aggregation: a replica exchange simulation study. J Chem Phys 2003; 118: 5185–5194.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL . Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001; 305: 567–580.
Meissner B, Rogalski T, Viveiros R, Warner A, Plastino L, Lorch A et al. Determining the sub-cellular localization of proteins within Caenorhabditis elegans body wall muscle. PLoS One 2011; 6: e19937.
Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D . Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 2006; 174: 229–239.
Scott B, Sun CL, Mao X, Yu C, Vohra BP, Milbrandt J et al. Role of oxygen consumption in hypoxia protection by translation factor depletion. J Exp Biol 2013; 216: 2283–2292.
Anderson LL, Mao X, Scott BA, Crowder CM . Survival from hypoxia in C. elegans by inactivation of aminoacyl-tRNA synthetases. Science 2009; 323: 630–633.
Heytler PG, Prichard WW . A new class of uncoupling agents—carbonyl cyanide phenylhydrazones. Biochem Biophys Res Commun 1962; 7: 272–275.
Lardy HA, Connelly JL, Johnson D . Antibiotic studies. II. inhibition of phosphoryl transfer in mitochondria by oligomycin and aurovertinN. Biochemistry 1964; 3: 1961–1968.
Bennett CF, Vander Wende H, Simko M, Klum S, Barfield S, Choi H et al. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat Commun 2014; 5: 3483.
Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013; 497: 451–457.
Pena S, Sherman T, Brookes PS, Nehrke K . The mitochondrial unfolded protein response protects against anoxia in Caenorhabditis elegans. PLoS One 2016; 11: e0159989.
Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG et al. Rates of behavior and aging specified by mitochondrial function during development. Science 2002; 298: 2398–2401.
Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM . Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell 2015; 58: 123–133.
Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM . Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012; 337: 587–590.
Taylor RC, Dillin A . Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol 2011; 3: a004440.
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S . Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004; 431: 805–810.
Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G et al. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 2009; 139: 1157–1169.
Sun CL, Kim E, Crowder CM . Delayed innocent bystander cell death following hypoxia in Caenorhabditis elegans. Cell Death Differ 2014; 21: 557–567.
Brenner S . The genetics of Caenorhabditis elegans. Genetics 1974; 77: 71–94.
Rauthan M, Ranji P, Aguilera Pradenas N, Pitot C, Pilon M . The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. Proc Natl Acad Sci USA 2013; 110: 5981–5986.
Mello C, Fire A . DNA transformation. Methods Cell Biol 1995; 48: 451–482.
Mello CC, Kramer JM, Stinchcomb D, Ambros V . Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. Embo J 1991; 10: 3959–3970.
Mao XR, Crowder CM . Protein misfolding induces hypoxic preconditioning via a subset of the unfolded protein response machinery. Mol Cell Biol 2010; 30: 5033–5042.
Tomoyasu T, Mogk A, Langen H, Goloubinoff P, Bukau B . Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol Microbiol 2001; 40: 397–413.
Pimenta de Castro I, Costa AC, Lam D, Tufi R, Fedele V, Moisoi N et al. Genetic analysis of mitochondrial protein misfolding in Drosophila melanogaster. Cell Death Differ 2012; 19: 1308–1316.
Eng JK, Jahan TA, Hoopmann MR . Comet: an open-source MS/MS sequence database search tool. Proteomics 2013; 13: 22–24.
Kall L, Storey JD, MacCoss MJ, Noble WS . Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J Proteome Res 2008; 7: 29–34.
Deutsch EW, Mendoza L, Shteynberg D, Farrah T, Lam H, Tasman N et al. A guided tour of the Trans-Proteomic Pipeline. Proteomics 2010; 10: 1150–1159.
Mi H, Muruganujan A, Thomas PD . PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 2013; 41: D377–D386.
Mi H, Muruganujan A, Casagrande JT, Thomas PD . Large-scale gene function analysis with the PANTHER classification system. Nat Protoc 2013; 8: 1551–1566.
Thomas PD, Kejariwal A, Guo N, Mi H, Campbell MJ, Muruganujan A et al. Applications for protein sequence–function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res 2006; 34: W645–W650.
Marchler-Bauer A, Bryant SH . CD-Search: protein domain annotations on the fly. Nucleic Acids Res 2004; 32: W327–W331.
Drozdetskiy A, Cole C, Procter J, Barton GJ . JPred4: a protein secondary structure prediction server. Nucleic Acids Res 2015; 43: W389–W394.
Sonnhammer EL, von Heijne G, Krogh A . A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 1998; 6: 175–182.
Halbritter F, Vaidya HJ, Tomlinson SR . GeneProf: analysis of high-throughput sequencing experiments. Nat Methods 2011; 9: 7–8.
Acknowledgements
This work was supported by R01-NS045905, R21-NS084360-02, T32-GM007200, 5R01AI101307, and the University of Washington's Proteomics Resource UWPR95794. O.A.I. is supported by a postdoctoral award from the AHA. pPD95.75 was a gift from Andrew Fire. Some strains were provided by the CGC, which is funded by P40 OD010440.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Baehrecke
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
About this article

Cite this article
Kaufman, D., Wu, X., Scott, B. et al. Ageing and hypoxia cause protein aggregation in mitochondria. Cell Death Differ 24, 1730–1738 (2017). https://doi.org/10.1038/cdd.2017.101
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.101
This article is cited by
-
Hypoxia-induced mitochondrial stress granules
Cell Death & Disease (2023)
-
Mitochondrial dysfunction, UPRmt signaling, and targeted therapy in metastasis tumor
Cell & Bioscience (2021)
-
Effect of the mitochondrial unfolded protein response on hypoxic death and mitochondrial protein aggregation
Cell Death & Disease (2021)
-
Protein Aggregation in the Pathogenesis of Ischemic Stroke
Cellular and Molecular Neurobiology (2021)

{kind=link}
{kind=link}
{kind=link}