
Abstract
Deafness or hearing loss is a major issue in human health. Inner ear hair cells are the main sensory receptors responsible for hearing. Defects in hair cells are one of the major causes of deafness. A combination of induced pluripotent stem cell (iPSC) technology with genome-editing technology may provide an attractive cell-based strategy to regenerate hair cells and treat hereditary deafness in humans. Here, we report the generation of iPSCs from members of a Chinese family carrying MYO15A c.4642G>A and c.8374G>A mutations and the induction of hair cell-like cells from those iPSCs. The compound heterozygous MYO15A mutations resulted in abnormal morphology and dysfunction of the derived hair cell-like cells. We used a CRISPR/Cas9 approach to genetically correct the MYO15A mutation in the iPSCs and rescued the morphology and function of the derived hair cell-like cells. Our data demonstrate the feasibility of generating inner ear hair cells from human iPSCs and the functional rescue of gene mutation-based deafness by using genetic correction.
Similar content being viewed by others

Main
Hearing loss affects ~10% of the global population,1 and ~50% of hearing loss cases have a genetic etiology.2 In particular, mutations in genes such as MYO15A, which are involved in the formation of inner ear sensory hair cells, account for the majority of sensorineural hearing loss cases.3, 4 Owing to the inability to regenerate human hair cells, sensorineural hearing loss is often permanent.5 Human and mouse embryonic stem cells (ESCs), as well as mouse induced pluripotent stem cells (iPSCs), have been used to generate cells with the characteristics of hair cells, that is, hair cell-like cells.6, 7, 8 Mammalian inner ear sensory organs appear to be capable of incorporating transplanted cells.9, 10, 11, 12, 13 These results suggest the potential for the development of a cell-based approach for hearing restoration.
Inner ear hair cells are largely responsible for the sense of hearing.14 Each hair cell contains a dense actin mesh at its apical surface. This mesh is adorned with distinct hair bundles made of F-actin-filled microvilli-derived stereocilia arranged in a staircase pattern.15 The genes that are involved in actin dynamics, including non-muscle unconventional myosin genes, have been identified as being among a limited number of key genes related to deafness.3 MYO15A belongs to the unconventional myosin superfamily and is required for the normal development of stereocilia in hair cells.16, 17, 18, 19 Mutations in MYO15A have been found to be associated with profound, congenital, neurosensory, nonsyndromal deafness in several Asian and European families.20, 21, 22, 23, 24, 25, 26, 27
Since its first development by Yamanaka in 2006, iPSC technology has made rapid development in the field of regenerative medicine.28, 29, 30, 31 CRISPR/Cas9 technology, a gene-editing technique, has offered several further potential advantages including the ease of customization, higher targeting efficiency, and the ability to facilitate multiplex genome editing.32 Combining iPSC technology with CRISPR/Cas9 technology has gradually become an attractive strategy in the study and development of therapies for hereditary human diseases. Here, we report the generation of human iPSCs from members of a Chinese family carrying MYO15A c.4642G>A and c.8374G>A mutations. We then demonstrate the induction of iPSCs into hair cell-like cells and then apply a CRISPR/Cas9-targeting vector and two forms of homologous recombination templates to repair the MYO15A c.4642G>A mutation in the iPSCs. We found that the genetic correction rescued the abnormal phenotypes (F-actin disorganization, abnormally short stereocilia as well as syncytia formation) and dysfunction (lower current density) in the derived hair cell-like cells caused by the MYO15A mutations. Our data demonstrate the feasibility of generating inner ear hair cells from human iPSCs and the functional rescue of gene mutations causing deafness using genetic correction.
Results
Generation and characterization of iPSCs
A customized gene capture panel for 69 known deafness genes and analysis of the mitochondrial genome using next-generation sequencing (Roche NimbleGen, Inc., Madison, WI, USA) were used to identify two novel mutations in the MYO15A gene. These were c.4642G>A causing p.A1548T, and c.8374G>A causing p.V2792M and were identified in a 2-year-old male patient with profound hearing loss (III:1, Figure 1). Sanger sequencing of the entire MYO15A gene in the proband and in the parents showed that the proband carried the compound heterozygous MYO15A c.4642G>A and c.8374G>A mutations. The father and mother were heterozygous for MYO15A c.8374G>A and MYO15A c.4642G>A mutations, respectively. No other mutation was found in the parents or the proband (Figure 1a and b). Dermal fibroblasts isolated from skin punch biopsies of the proband (III:1, deaf, Figure 1c) and the father (II:1, normal hearing, Figure 1c) as well as a female girl (normal hearing) with no MYO15A mutation were designated as M−/−, M+/−, and M+/+, respectively. Four key transcription factor genes OCT4, SOX2, KLF4, and c-MYC were introduced into the three fibroblast lines using a retroviral method (Supplementary Figure S1). Approximately 3 weeks later, human iPSC-like colonies with a high nuclear-cytoplasmic ratio were observed. Single colonies were then selected and separately subcultured (Figure 2a).

Identification of a Chinese family carrying MYO15A mutations. (a) The genogram outlining the inheritance of two MYO15A mutations, c.4642G>A (p.A1548T) and c.8374G>A (p.V2792M) was identified by gene screening of a panel of 69 genes. Family members marked by a * were donors for the next-generation sequencing of deafness genes, Sanger validation, and hearing tests. The 2-year-old male child (III:1) carries two MYO15A mutations. The inheritance of the MYO15A mutations in the family indicated that the child carries compound heterozygous MYO15A mutations. (b) Sanger sequencing confirmed the MYO15A mutations identified in the family members. Double peaks represent a heterozygous mutation. (c) Audiograms of the family carrying MYO15A mutations. Audiograms of the 2-year-old male carrying the compound heterozygous MYO15A mutations (III:1), the parents each carrying a single MYO15A mutation (II:1 and II:2), and the paternal grandparents with the grandfather carrying a single MYO15A mutation (I:1 and I:2). Note that the parents and grandmother have normal hearing. The grandfather has normal hearing below 3 KHz and normal conversational hearing with peak hearing loss at frequencies between 4–6 KHz. An X represents the audition of the left ear and a circle represents the audition of right ear

Generation and characterization of iPSCs from human dermal fibroblasts. (a) Phase contrast microscopy of dermal fibroblasts and derived iPSCs and AP staining of the four putative iPSCs. (b) Immunostaining for pluripotent markers OCT4, SOX2, NANOG, SSEA4, TRA-1–60, and TRA-1-81 in the four putative iPSCs. Nuclei were stained with DAPI (blue). (c) Embryoid bodies formed after 8 days of suspension culture. (d) Teratomas formed from the NOD-SCID mice and stained with hematoxylin and eosin. Tissue structure characteristics of the three germ layers were observed (gut epithelium for endoderm, cartilage for mesoderm, as well as neural rosettes and retinal pigment epithelium for ectoderm). Scale: 50 μm in NANOG images; 100 μm in the other images
Multiple assays were performed to fully characterize the generated putative iPSC lines. Alkaline phosphatase (AP) staining revealed that all three putative iPSC lines were positive for AP (Figure 2a). Immunocytochemistry showed that all three putative iPSC lines expressed iPSC markers OCT4, SOX2, NANOG, SSEA4, TRA-1-60, and TRA-1-81 (Figure 2b). Three clones from each putative iPSC line were picked out to examine the expression of iPSC-specific endogenous and exogenous transgenes. iPSC endogenous marker genes were expressed in all three putative iPSC lines, whereas most exogenous transgenes were silenced (Supplementary Figure S2a). The karyotypes of the three putative iPSC lines were analyzed and confirmed to be normal (Supplementary Figure S2b).
Putative iPSC clones with silenced exogenous transgenes, including Clone 3 (C3) from M+/+, C4 from M+/−, and C7 from M−/− (Supplementary Figure S2a) were subjected to totipotency analysis. Embryoid bodies (EBs) were formed after 8-day suspension culture of these putative iPSCs (Figure 2c). Specific marker genes for each of the three germ layers (AFP and SOX17 for the endoderm, Brachyury and MSX1 for the mesoderm, and PAX6 and MAP2 for the ectoderm) were detected and quantified after a subsequent 8-day adherent culture (Supplementary Figures S2c and e). In addition, teratoma formation was utilized to test the in vivo differentiation totipotency and tridermogenesis of the putative iPSC lines. Each teratoma contained tissue structures characteristic of the three germ layers. More specifically, these included tissues of the gut epithelium, cartilage, and neural rosettes and retinal pigment epithelium of endodermal mesodermal, and ectodermal origin (Figure 2d). These results indicated that we had successfully generated three iPSC lines (M+/+iPSCs, M+/−iPSCs, and M−/−iPSCs) with full characterizations.
Potentials of M+/+iPSCs, M+/−iPSCs, and M−/−iPSCs for differentiation into inner ear hair cells
We subsequently tested the potential of these iPSCs to differentiate into inner ear hair cells (Supplementary Figure S1). We used the monolayer induction method to induce the differentiation of the three iPSC lines toward the otic progenitors.8 After 12 days of induction, cells induced from all three iPSC lines expressed early otic markers, including PAX2, PAX8, SOX2, and NES (Figure 3a and c). The expression of other early otic marker genes, including GATA3, EYA1, SIX1, and DLX5, was also detected (Figure 3d). No significant difference in otic gene expression was observed between the three otic progenitors (Figure 3e, P>0.05).

Induction of human iPSCs toward otic progenitor differentiation. (a–c) Human iPSCs were induced toward otic progenitors and stained for the early otic markers, PAX8+NES (a), PAX8+PAX2 (b), and PAX8+SOX2 (c). Nuclei were stained with DAPI (blue). Scale: 30 μm. (d) RT-PCR analysis of the expression of early otic marker genes. Expression of otic progenitor marker genes, PAX2, PAX8, GATA3, EYA1, SIX1, and DLX5, was detected in all four iPSCs induced toward the otic lineage. (e) The relative gene expression levels were quantified and plotted. The housekeeping gene, GAPDH, was used as an internal reference. Error bars represent the S.D. (n=3)
The otic epithelial progenitors generated from the human iPSCs were isolated and further induced to differentiate into inner ear hair cells.6, 8 The otic epithelial progenitors were cultured for 3 weeks on mitotically inactivated chicken embryonic utricle stromal cells (CUSCs). Immunocytochemistry and Western blot assays were then performed to examine the expression of hair cell marker proteins in the induced hair cell-like cells. The results showed that the hair cell markers, ATOH1,33, 34 and BRN3C,35 the early hair cell structural protein, MYO7A,17 as well as the protein required for hair bundle development, ESPN,36 were all expressed (Figure 4a and e). Gene expression analysis also revealed the expression of hair cell marker genes, including ATOH1, BRN3C, MYO7A, and ESPN (Figure 4f). In addition, transcripts from MYO15A were at similar levels in the induced hair cell-like cells derived from all the three iPSC lines (Figure 4g).

Induction of otic epithelial progenitors toward inner ear hair cell differentiation. (a–c) Generation of inner ear hair cell-like cells from otic epithelial progenitors as revealed by immunostaining with antibodies for hair cell markers BRN3C+ATOH1 (a), BRN3C+MYO7A (b), and BRN3C+ESPN (c). Nuclei were stained with DAPI (blue). Scale: 100 μm in MYO7A images, 30 μm in ATOH1 images and 3 μm in ESPN images. (d) Western blot analysis of the expression of hair cell marker genes. (e) The relative expression levels of hair cell marker proteins were quantified and plotted. The housekeeping gene, GAPDH, was used as an internal reference. Error bars represent the S.D. (n=3). (f) RT-PCR analysis of the expression of hair cell marker genes. Expression of inner ear hair cell marker genes such as BRN3C, ATOH1, MYO7A, as well as genes encoding stereocilia proteins, ESPN and MYO15A, were detected. CUSC represents chicken embryonic utricle stromal cells. (g) The relative expression levels of hair cell marker genes were quantified and plotted. The housekeeping gene, GAPDH, was used as an internal reference. Error bars represent the S.D. (n=3)
We further assessed the functional characteristics of these induced cells using FM1-43 uptake experiments7 and electrophysiological recording of voltage-dependent currents.6, 8 Live cells stained with FM1-43 were sorted by fluorescence-activating cell sorting (FACS) (Figure 5a and b). Gene expression analysis revealed that the sorted cells expressed genes specific for hair cell mechanotransduction, including TMC1/2,37, 38, 39, 40 TMHS,41 and TMIE42, 43 (Figure 5c and d). Electrophysiological recordings showed that these cells exhibited hair cell-like voltage-dependent K+ currents, Ik and Ik1 (Figure 5e and f). Hair cell-like voltage-dependent inward Ca2+ currents, ICa, were also detected in these cells (Figure 5g). Consistent with the electrophysiological recording of hair cell features, the induced cells also developed microvilli-based stereocilia-like structures, as revealed by scanning electronic microscopy (SEM) (Figure 6b).

Hair cell-like cells from iPSCs have hair cell-like electrophysiological properties. (a) Cells stained with FM1-43 (green). Nuclei were stained with DAPI (blue). Cells with FM1-43-positive staining were sorted by FACS for electrophysiological recordings. Scale: 100 μm. (b) The percentage of FM1-43 positive cells in the total number of induced cells. Error bars represent the S.E.M. (n=8), * represents statistically significant difference (P<0.05). (c) RT-PCR analysis of the expression of genes specific for hair cell mechanotransduction, including TMC1/2, TMHS, and TMIE. (d) The relative expression levels of genes specific for hair cell mechanotransduction were quantified and plotted. The housekeeping gene, GAPDH, was used as an internal reference. Error bars represent the S.D. (n=3). (e–f) Hair cell-like cells displayed voltage-dependent K+ currents, outward IK (c) and inward IK1 (d). (g) Voltage-dependent inward ICa was recorded from hair cell-like cells. Currents were elicited by 10 voltage steps from the holding potential (c: –104 mV; d: –64 mV; e: −84), the peak current voltages are also indicated. (h) Statistical analysis of average peak current density in the hair cell-like cells induced from M+/+iPSCs, M+/−iPSCs, M−/−iPSCs, and MC/−iPSCs. Average peak current density is equal to the peak current divided by the capacitance. Details are listed in Supplementary Table S1. A two-tailed test was used for pair-wise comparison. ** represents P<0.01 compared with others. Error bars represent the S.E.M. (n=8)

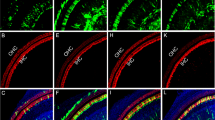
Functional rescue of hair cell-like cells from MYO15A compound heterozygous mutant iPSCs by CRISPR/Cas9 genetic correction. (a) Immunostaining of F-actin in hair cell-like cells. F-actin stress fibers were visualized in hair cell-like cells derived from M+/+iPSCs, M+/−iPSCs and MC/−iPSCs by staining with F-actin dye phalloidin (green). White box: syncytia were observed in hair cell-like cells derived from M−/−iPSCs. Nuclei were stained with DAPI (blue). Arrow: stereocilia-like structure. Scale: 30 μm in the left two columns, 6 μm in the third column. (b) S.E.M. of hair cell-like cells derived from iPSCs. A stereocilia-like structure was observed. (c) The stereocilia length was measured using ImageJ software. The average stereocilia length of hair cell-like cells from M+/+iPSCs, M+/−iPSCs, and MC/−iPSCs was 3.52±0.29 μm, 3.41±0.24 μm, and 3.05±0.23 μm, respectively. The stereocilia length of M−/−iPSCs was significantly shorter at 0.94±0.22 μm. A two-tailed test was used for pair-wise comparison. *** represents a high statistically significant difference (P<0.001). Error bars represent the S.E.M. (n=8)
However, there were significant differences in the morphology and function between the hair cell-like cells differentiated from M−/−iPSCs to those of M+/+iPSCs or M+/−iPSCs. After 7 days of differentiation from otic epithelial progenitors into inner ear hair cells, some of the cells induced from M−/−iPSCs aggregated together and formed syncytia, whereas some other cells floated and finally died. No syncytia were found for cells induced from either M+/+iPSCs or M+/−iPSCs. After 21 days of differentiation from otic epithelial progenitors into inner ear hair cells, F-actin and DAPI staining were performed to examine the effects of MYO15A mutations on F-actin organization. The results revealed that some of the cells induced from M−/−iPSCs formed syncytia with disorganized actin filaments. However, cells induced from M+/+iPSCs or M+/−iPSCs showed no syncytia formation and displayed a normal actin filament structure (Figure 6a). Meanwhile, it is noteworthy that the MYO15A mutation decreased the percentage of FM1-43-positive cells in the total induced cells and that the current density of the hair cell-like cells derived from M−/−iPSCs was significantly lower than that of the hair cell-like cells derived from M+/+iPSCs or M+/−iPSCs (Figure 5h). A comparison of the SEM observations between the three types of induced cells showed that hair cell-like cells induced from M+/+iPSCs or M+/−iPSCs displayed the hair bundle-like structure on the surface of cells where the stereocilia on the hair cell-like cells induced from M−/−iPSCs were significantly shorter than those on the hair cell-like cells induced from M+/+iPSCs and M+/−iPSCs (Figure 6c).
Genetic correction rescues morphology and function of the derived hair cell-like cells
We tried to correct the MYO15A c.4642G>A mutation in the M−/−iPSC line and to examine the effects of genetic correction on the morphological and functional recovery of the induced hair cell-like cells. CRISPR/Cas9 technology was applied for genetic correction (Supplementary Figure S1). A CRISPR/Cas9 vector pX330-maxGFP-sgRNA4 specifically targeting the MYO15A c.4642G>A locus, a 130-bp single-stranded donor oligonucleotide (ssODN) and a donor plasmid pUC19-MYO15Asynonymous for homologous recombination were designed and constructed for nucleofection (Supplementary Figures S3 and S4a and c). After nucleofection, cells were sorted. 5% of the cells in ssODN nucleofection and 7.9% in donor plasmid nucleofection were GFP positive (Supplementary Figure S4d). GFP-positive cells were collected and seeded onto the mitotically inactivated MEFs. After 10 days of culture, 96 monoclones (48 from ssODN nucleofection and 48 from donor plasmid nucleofection) were picked and amplified from the GFP-positive cells. Restriction fragment length polymorphism analysis showed that eight clones obtained by ssODN nucleofection and five clones obtained by donor plasmid nucleofection had a single 365 bp band, but no 733 bp bands (Supplementary Figures S5a and b). This may indicate that these clones have been genetically corrected. Furthermore, Sanger sequencing results revealed that the mutation at the MYO15A c.4642G>A locus of these 13 clones had been corrected (sequencing results also revealed a double-peak at the PAM synonymous mutation site in clones obtained by donor plasmid nucleofection) as shown in Supplementary Figure S5c. Off-target detection revealed that there were no double peaks adjacent to the top 12 suspected off-target sites in the 13 iPSC clones (Supplementary Figure S6a). In other words, no off-targeting was found. In addition, several InDel mutations in iPSC clones, which had not been corrected by homology directed repair (HDR), but targeted by the CRISPR/Cas9 plasmid, were also detected (Supplementary Figure S6b).
Subsequently, the expression of pluripotent marker genes and proteins in positive iPSC clones with the genotype of MYO15ACorrection/− (MC/−iPSC) was examined. MC/−iPSCs expressed marker proteins characteristic of hiPSC, including AP, OCT4, SOX2, NANOG, SSEA4, TRA-1-60, and TRA-1-81 (Figure 2a and b), as well as iPSC endogenous marker genes, such as OCT4, SOX2, and NANOG. All exogenous transgenes were silenced in MC/−iPSCs (Supplementary Figure S2a). MC/−iPSCs also formed EBs and teratomas and had the potential to form cells of the three germ layers in vitro and in vivo (Figure 2c and d). Karyotype assays of the MC/−iPSCs showed that the cells displayed a normal karyotype of 46XY (Supplementary Figure S2b). These results showed that the mutation at the MYO15A c.4642G>A locus in M−/−iPSCs had been corrected successfully and that a new iPSC line with a genotype of MYO15ACorrection/− (MC/−iPSCs) had been generated.
To understand the effects of the genetic correction of MYO15A mutations on the morphology and function of the derived hair cell-like cells, we induced the differentiation of the MC/−iPSCs into otic progenitors and further into inner ear hair cell-like cells. After the induction of the differentiation of MC/−iPSCs into otic progenitors for 12 days, the expression of the early otic markers, including PAX2, PAX8, SOX2, and NES, was examined by immunocytochemistry (Figure 3a and c). Gene expression analysis also revealed that the induced cells expressed early otic marker genes. These included PAX2, PAX8, GATA3, EYA1, SIX1, and DLX5 (Figure 3d). No difference in the expression patterns for the early otic markers was observed between the otic progenitors from M+/+iPSCs, M+/−iPSCs, and MC/−iPSCs (Figure 3e). Furthermore, the differentiation of otic epithelial progenitors from the MC/−iPSCs into inner ear hair cells was induced. After 21 days of induction, immunocytochemistry and western blot assays revealed that the hair cell-like cells induced from MC/−iPSCs expressed hair cell markers, including BRN3C, ATOH1, MYO7A, and ESPN (Figure 4a and d). Gene expression analysis indicated that the induced cells expressed hair cell marker genes, including BRN3C, ATOH1, MYO7A, and ESPN (Figure 4f). In addition, MYO15A was also detected, as shown in Figure 4f. The expression pattern of hair cell markers in the inner hair cell-like cells induced from MC/−iPSCs had no significant difference from that in the inner hair cell-like cells induced from M+/+iPSCs or M+/−iPSCs (Figure 4e and g, P>0.05). Hair cell-like cells induced from MC/−iPSCs took up FM1-43 (Figure 5a) and expressed genes specific for hair cell mechanotransduction, including TMC1/2, TMHS, and TMIE (Figure 5c). Electrophysiological recordings showed that the hair cell-like cells induced from the MC/−iPSCs also contained the hair cell-like voltage-dependent currents and current densities similar to those from the M+/+iPSCs or M+/−iPSCs (Figure 5e and h). During the differentiation of otic epithelial progenitors into inner ear hair cells, no syncytia were formed in the cells induced from MC/−iPSCs. This was consistent with the observation from cells induced from M+/+iPSCs or M+/−iPSCs. After 21 days of induction, the F-actin and DAPI-staining assays also showed no syncytia formation in cells induced from the MC/−iPSCs and the actin filaments had remained well organized in these cells as it had in the cells induced from M+/+iPSCs or M+/−iPSCs (Figure 6a). SEM detection revealed that the stereocilia length of hair cell-like cells induced from the MC/−iPSCs had no difference from that in cells induced from the M+/+iPSCs or M+/−iPSCs (Figure 6c, P>0.05). Using restriction fragment length polymorphism analysis, we found that hair cell-like cells induced from the MC/−iPSCs expressed normal MYO15A mRNA (Supplementary Figure S5d). This gave further evidence of the success of the genetic correction.
Discussion
iPSC technology enables the development of study models for many diseases by facilitating the generation of patient-specific stem cells.44 In this study, deafness patient-specific iPSCs were generated from a Chinese boy carrying MYO15A c.4642G>A and c.8374G>A compound heterozygous mutations. These were induced into inner ear hair cell-like cells to examine the effects of the MYO15A mutations on the morphology and function of the derived hair cell-like cells. Our results showed that the MYO15A mutations had no effect on the totipotency of iPSCs. However, the morphology and function of hair cell-like cells induced from the M+/+iPSCs, M+/−iPSCs, and M−/−iPSCs were found to be different. Some of the cells induced from the M−/−iPSCs aggregated together and formed syncytia, whereas no syncytia were found in the cells induced from the M+/+iPSCs or M+/−iPSCs. To our knowledge, no previous studies have yet reported relationships between human MYO15A mutations and syncytia formation. The formation of syncytia may occur through cell fusion or mitotic disorders.45, 46 In this study, some of the induced cells with human MYO15A mutations formed syncytia accompanied with F-actin disorganization. This requires further investigation as to whether syncytia formation of some induced cells can be attributed to a mitotic disorder resulted from a MYO15A mutation-mediated F-actin disorder.
Myo15a localizes to the inner ear sensory cell stereocilia in mouse cochlea and is essential for the formation of a staircase pattern of hair bundles.18 In Myo15ash2 mouse cochlea, the hair cell stereocilia are much shorter compared with those of the wild type.47 In our study, SEM detection showed that although the surface of the cells induced from the M+/+iPSCs, M+/−iPSCs, or M−/−iPSCs displayed the stereocilia-like structure, the stereocilia on the surface of the cells induced from the M−/−iPSCs were significantly shorter than those on cells induced from the M+/+iPSCs and M+/−iPSCs. Actin is fundamental for the structure of stereocilia.48 Previous studies in mice also indicated that Whrn and Eps8 have vital roles in actin regulation in hair cells.16, 49, 50 Myo15a interacts with Whrn and Eps8, and transports these cargos to the tips of the stereocilia.16, 51 This process is considered as the basis of the elongation of stereocilia in hair cells. Based on the phenomenon of F-actin disorganization in the cells induced from M−/−iPSCs, we deduce that human MYO15A mutations may result in F-actin disorganization and consequently cause abnormally shortened stereocilia in the induced hair cell-like cells. Our study confirmed that MYO15A is essential for the elongation of inner ear hair cell stereocilia during the differentiation of otic epithelial progenitors into inner ear hair cells.
CRISPR/Cas9 technology allows us to edit the genome precisely with low probability of changing the cellular genetic background.52 It has been reported that the genetic correction of homozygous mutations does indeed reverse the disease phenotype caused by the homozygous mutation.53, 54 In patients suffering from hereditary diseases, however, homozygous mutations are in the minority, and compound heterozygous mutations are dominant. Although most individuals carry a single allele mutation, no homologous disease symptoms are present. For example, the father of the M−/− deaf patient had a genotype of M+/− with normal hearing. Therefore, we hypothesized that the genetic correction of one locus in the compound heterozygous mutations should be sufficient for the functional reconstruction of M−/−iPSCs. Therefore, we genetically corrected the MYO15A c.4642G>A mutation in the M−/−iPSC line using CRISPR/Cas9 technology and generated a new iPSC line (MC/−iPSC line). The differentiation of MC/−iPSCs into hair cell-like cells was then induced. In this case, the results showed no syncytia formation during differentiation. After 21 days of induction, the immunostaining and electrophysiological recordings showed significant differences in the electrophysiological properties between cells induced from MC/−iPSCs and M−/−iPSCs. SEM observation showed that the length of the stereocilia on the surface of cells induced from MC/−iPSCs was identical to that of cells induced from M+/+iPSCs or M+/−iPSCs and significantly longer than that of stereocilia on cells induced from M−/−iPSCs. These results indicated that the genetic correction of one locus in the compound heterozygous mutation loci was sufficient for the reversal of the abnormal phenotype (F-actin disorganization, abnormally short stereocilia as well as syncytia formation) and dysfunction (current density) caused by the MYO15A mutations.
In summary, our results showed that the rescue of F-actin organization, stereocilia length, and electrophysiological parameters in hair cell-like cells derived from MC/−iPSCs was indeed due to the genetic correction of the MYO15A c.4642G>A mutation in the M−/−iPSCs. Thus, our data indicate that it is feasible to generate iPSCs from human patient tissues to induce the differentiation of these iPSCs into hair cell-like cells and to genetically correct such deafness mutations in human iPSCs. These findings provide a step toward the development of therapeutic interventions for similar cases of hereditary deafness.
Material and Methods
Subjects and clinical evaluations
This study was approved by the Institutional Review Boards of Zhejiang University, Hangzhou, China, and the Ethical Committee of the People’s Hospital of Wenling, Zhejiang, China. Written informed consent was obtained from adult subjects and the parents of minor subjects. The gDNA samples were subjected to a deafness genetic screen using a custom-made capture kit for 69 deafness genes and mitochondrial DNA (Roche NimbleGen, Inc.).
Generation of iPSCs
Human epidermic tissues were obtained from donors’ arms with a 6-mm skin punch (Vetlab Supply, Palmetto Bay, FL, USA). Primary fibroblasts were isolated and cultured as previously described.55 Reprogramming was adapted from Takahashi et al.28 Small molecule compounds were added into the iPSC medium to enhance the induction efficiency. Human iPSCs were cultured on mitomycin C-inactivated MEFs.
Totipotency of human iPSCs
To characterise the totipotency of induced iPSCs, EB formation from iPSCs was performed as previously described.53 For analysis of in vivo differentiation of iPSCs, teratoma formation was performed as previously described.56 Animal care and usage was approved by the Institutional IACUC Committee and the Laboratory Animal Welfare Ethical Committee of Zhejiang University.
Genetic correction of iPSCs carrying the MYO15A c.4642G>A mutation
The genetic correction of iPSCs with the MYO15A c.4642G>A mutation was performed using CRISPR-Cas9 technology (Supplementary Figure S2). For FACS, we added a selectable marker, a CMV-maxGFP-polyA cassette amplified from pmaxGFP (Lonza, Shanghai, China), and cloned it into the plasmid pX330 to construct the plasmid pX330-maxGFP. We constructed a 130-bp ssODN and a donor plasmid pUC19-MYO15Asynonymous for HDR templates. Nucleofection technology was used to transfect the Cas9 plasmid and HDR templates. Positive clones were selected by FACS sorting, restriction fragment length polymorphism analysis and Sanger sequencing.
Induction of iPSCs toward inner ear hair cells
iPSCs were induced toward inner ear hair cells using a slightly modified protocol.8 After 12 days of differentiation, otic epithelial progenitors (OEPs) were isolated and collected for hair cell induction. To induce differentiation of OEPs into hair cells, the cells collected were transferred and seeded at a density of 1000 cell/cm2 onto a Matrigel-coated plate that had been pre-seeded with mitotically inactivated embryonic day 18 CUSCs.6
Gene expression analysis
Total RNA was extracted from cells using TRIzol reagent (Life Technologies, Shanghai, China) including treatment with RNase-Free DNase I (Sigma-Aldrich, Shanghai, China). cDNA synthesis was performed with the M-MLV Reverse Transcriptase Kit (Fermentas, Shanghai, China). Total gDNA was isolated with QuickExtract DNA Extraction Solution (Epicentre Biotechnologies, Beijing, China). The primers and PCR conditions are listed in Supplementary Table S4.
SEM
Cells were fixed in 2.5% glutaraldehyde (Sigma-Aldrich) in PBS at 4 °C overnight. After three PBS washes for 15 min each, cells were post-fixed in 1% OsO4 in PBS for 2 h and then washed three times for 15 min. Cells were then dehydrated with a graded ethanol series for 15 min each. After being dehydrated two times in 100% ethanol, samples were transferred into isoamyl acetate for 1 h, dehydrated with liquid CO2 in a Hitachi Model HCP-2 critical point dryer, coated with gold-palladium in a Hitachi Model E-1010 ion sputter for 5 min, and observed under a Hitachi SU8010 SEM.
Electrophysiological recordings
For cells that had been differentiated for 21 days, a whole-cell patch-clamp technique was used to measure the membrane currents with EPC10 Patch-Clamp Amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). A FM1-43FX (Life Technologies) working staining solution of 5 μg/ml was prepared in PBS without Ca2+ and Mg2+. Cells that took up FM1-43FX exhibited green fluorescence and were sorted for electrophysiological recordings. The data were acquired using the PatchMaster software and a Digidata analog-to-digital converter (Molecular Devices, Sunnyvale, CA, USA) and analyzed using the Clampfit and Origin (OriginLab, Guangzhou, China) software.
Statistics
Experimental results are expressed as±the S.E.M. unless otherwise stated. Statistical analysis of one-way variance (ANOVA) was performed using Origin software. Statistical significance was considered as P<0.05.
Fuller details of material and methods are presented in Supplementary material and methods.
Abbreviations
- iPSC:
-
induced pluripotent stem cell
- ESC:
-
embryonic stem cell
- CRISPR:
-
clustered regularly interspaced short palindromic repeat
- Cas9:
-
CRISPR-associated protein 9
- HDR:
-
homology directed repair
- CUSC:
-
chicken utricle stromal cell
- AP:
-
alkaline phosphatase
- ssODN:
-
single-strand oligonucleotide
- GFP:
-
green fluorescent protein
- FACS:
-
fluorescence-activated cell sorting
- WT:
-
wild type
References
Lentz JJ, Jodelka FM, Hinrich AJ, McCaffrey KE, Farris HE, Spalitta MJ et al. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat Med 2013; 19: 345–350.
Muller U, Barr-Gillespie PG . New treatment options for hearing loss. Nat Rev Drug Discov 2015; 14: 346–365.
Brownstein Z, Abu-Rayyan A, Karfunkel-Doron D, Sirigu S, Davidov B, Shohat M et al. Novel myosin mutations for hereditary hearing loss revealed by targeted genomic capture and massively parallel sequencing. Eur J Hum Genet 2014; 22: 768–775.
Kochhar A, Hildebrand MS, Smith RJ . Clinical aspects of hereditary hearing loss. Genet Med 2007; 9: 393–408.
Nadol JB Jr . Hearing loss. N Engl J Med 1993; 329: 1092–1102.
Oshima K, Shin K, Diensthuber M, Peng AW, Ricci AJ, Heller S . Mechanosensitive hair cell-like cells from embryonic and induced pluripotent stem cells. Cell 2010; 141: 704–716.
Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E . Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature 2013; 500: 217–221.
Chen W, Jongkamonwiwat N, Abbas L, Eshtan SJ, Johnson SL, Kuhn S et al. Restoration of auditory evoked responses by human ES-cell-derived otic progenitors. Nature 2012; 490: 278–282.
Watada Y, Yamashita D, Toyoda M, Tsuchiya K, Hida N, Tanimoto A et al. Magnetic resonance monitoring of superparamagnetic iron oxide SPIO)-labeled stem cells transplanted into the inner ear. Neurosci Res 2015; 95: 21–26.
Okano T, Kelley MW . Stem cell therapy for the inner ear: recent advances and future directions. Trends Amplif 2012; 16: 4–18.
Zhang PZ, He Y, Jiang XW, Chen FQ, Chen Y, Xue T et al. Up-regulation of stromal cell-derived factor-1 enhances migration of transplanted neural stem cells to injury region following degeneration of spiral ganglion neurons in the adult rat inner ear. Neurosci Lett 2013; 534: 101–106.
Tateya I, Nakagawa T, Iguchi F, Kim TS, Endo T, Yamada S et al. Fate of neural stem cells grafted into injured inner ears of mice. Neuroreport 2003; 14: 1677–1681.
Doyle KL, Kazda A, Hort Y, McKay SM, Oleskevich S . Differentiation of adult mouse olfactory precursor cells into hair cells in vitro. Stem Cells 2007; 25: 621–627.
Hudspeth AJ . Integrating the active process of hair cells with cochlear function. Nat Rev Neurosci 2014; 15: 600–614.
Scheffer DI, Zhang DS, Shen J, Indzhykulian A, Karavitaki KD, Xu YJ et al. XIRP2, an actin-binding protein essential for inner ear hair-cell stereocilia. Cell Rep 2015; 10: 1811–1818.
Manor U, Disanza A, Grati M, Andrade L, Lin H, Di Fiore PP et al. Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr Biol 2011; 21: 167–172.
Karolyi IJ, Probst FJ, Beyer L, Odeh H, Dootz G, Cha KB et al. Myo15 function is distinct from Myo6, Myo7a and pirouette genes in development of cochlear stereocilia. Hum Mol Genet 2003; 12: 2797–2805.
Belyantseva IA, Boger ET, Friedman TB . Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc Natl Acad Sci USA 2003; 100: 13958–13963.
Delprat B, Michel V, Goodyear R, Yamasaki Y, Michalski N, El-Amraoui A et al. Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum Mol Genet 2005; 14: 401–410.
Woo HM, Park HJ, Baek JI, Park MH, Kim UK, Sagong B et al. Whole-exome sequencing identifies MYO15A mutations as a cause of autosomal recessive nonsyndromic hearing loss in Korean families. BMC Med Genet 2013; 14: 72.
Gao X, Zhu QY, Song YS, Wang GJ, Yuan YY, Xin F et al. Novel compound heterozygous mutations in the MYO15A gene in autosomal recessive hearing loss identified by whole-exome sequencing. J Transl Med 2013; 11: 284.
Fattahi Z, Shearer AE, Babanejad M, Bazazzadegan N, Almadani SN, Nikzat N et al. Screening for MYO15A gene mutations in autosomal recessive nonsyndromic, GJB2 negative Iranian deaf population. Am J Med Genet A 2012; 158A: 1857–1864.
Chang MY, Kim AR, Kim NK, Lee C, Lee KY, Jeon WS et al. Identification and clinical implications of novel MYO15A mutations in a non-consanguineous Korean family by targeted exome sequencing. Mol Cells 2015; 38: 781–788.
Miyagawa M, Nishio SY, Hattori M, Moteki H, Kobayashi Y, Sato H et al. Mutations in the MYO15A gene are a significant cause of nonsyndromic hearing loss: massively parallel DNA sequencing-based analysis. Ann Otol Rhinol Laryngol 2015; 124 Suppl 1: 158S–168S.
Xia H, Huang X, Guo Y, Hu P, He G, Deng X et al. Identification of a novel MYO15A mutation in a Chinese family with autosomal recessive nonsyndromic hearing loss. PLoS One 2015; 10: e0136306.
Liburd N, Ghosh M, Riazuddin S, Naz S, Khan S, Ahmed Z et al. Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with Smith-Magenis syndrome. Hum Genet 2001; 109: 535–541.
Kalay E, Uzumcu A, Krieger E, Caylan R, Uyguner O, Ulubil-Emiroglu M et al. MYO15A (DFNB3) mutations in Turkish hearing loss families and functional modeling of a novel motor domain mutation. Am J Med Genet A 2007; 143A: 2382–2389.
Takahashi K, Yamanaka S . Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126: 663–676.
Martins AM, Vunjak-Novakovic G, Reis RL . The current status of iPS cells in cardiac research and their potential for tissue engineering and regenerative medicine. Stem Cell Rev 2014; 10: 177–190.
Revilla A, Gonzalez C, Iriondo A, Fernandez B, Prieto C, Marin C et al. Current advances in the generation of human iPS cells: implications in cell-based regenerative medicine. J Tissue Eng Regen Med 2015 (e-pub ahead of print 11 March 2015; doi:10.1002/term.2021).
Okano H . Stem cell research and regenerative medicine in 2014: first year of regenerative medicine in Japan. Stem Cells Dev 2014; 23: 2127–2128.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F . Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8: 2281–2308.
Izumikawa M, Minoda R, Kawamoto K, Abrashkin KA, Swiderski DL, Dolan DF et al. Auditory hair cell replacement and hearing improvement by Atoh1 gene therapy in deaf mammals. Nat Med 2005; 11: 271–276.
Zheng JL, Gao WQ . Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat Neurosci 2000; 3: 580–586.
Weiss S, Gottfried I, Mayrose I, Khare SL, Xiang M, Dawson SJ et al. The DFNA15 deafness mutation affects POU4F3 protein stability, localization, and transcriptional activity. Mol Cell Biol 2003; 23: 7957–7964.
Donaudy F, Zheng L, Ficarella R, Ballana E, Carella M, Melchionda S et al. Espin gene (ESPN) mutations associated with autosomal dominant hearing loss cause defects in microvillar elongation or organisation. J Med Genet 2006; 43: 157–161.
Kurima K, Ebrahim S, Pan B, Sedlacek M, Sengupta P, Millis BA et al. TMC1 and TMC2 localize at the site of mechanotransduction in mammalian inner ear hair cell stereocilia. Cell Rep 2015; 12: 1606–1617.
Pan B, Geleoc GS, Asai Y, Horwitz GC, Kurima K, Ishikawa K et al. TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron 2013; 79: 504–515.
Kawashima Y, Geleoc GS, Kurima K, Labay V, Lelli A, Asai Y et al. Mechanotransduction in mouse inner ear hair cells requires transmembrane channel-like genes. J Clin Invest 2011; 121: 4796–4809.
Kawashima Y, Kurima K, Pan B, Griffith AJ, Holt JR . Transmembrane channel-like (TMC) genes are required for auditory and vestibular mechanosensation. Pflugers Arch 2015; 467: 85–94.
Xiong W, Grillet N, Elledge HM, Wagner TF, Zhao B, Johnson KR et al. TMHS is an integral component of the mechanotransduction machinery of cochlear hair cells. Cell 2012; 151: 1283–1295.
Zhao B, Wu Z, Grillet N, Yan L, Xiong W, Harkins-Perry S et al. TMIE is an essential component of the mechanotransduction machinery of cochlear hair cells. Neuron 2014; 84: 954–967.
Liedtke W . A precisely defined role for the tip link-associated protein TMIE in the mechanoelectrical transduction channel complex of inner ear hair cells. Neuron 2014; 84: 889–891.
Grskovic M, Javaherian A, Strulovici B, Daley GQ . Induced pluripotent stem cells—opportunities for disease modelling and drug discovery. Nat Rev Drug Discov 2011; 10: 915–929.
Shemer G, Podbilewicz B . Fusomorphogenesis: cell fusion in organ formation. Dev Dyn 2000; 218: 30–51.
Finger FP, White JG . Fusion and fission: membrane trafficking in animal cytokinesis. Cell 2002; 108: 727–730.
Probst FJ, Fridell RA, Raphael Y, Saunders TL, Wang A, Liang Y et al. Correction of deafness in shaker-2 mice by an unconventional myosin in a BAC transgene. Science 1998; 280: 1444–1447.
Tilney LG, Derosier DJ, Mulroy MJ . The organization of actin filaments in the stereocilia of cochlear hair cells. J Cell Biol 1980; 86: 244–259.
Mburu P, Mustapha M, Varela A, Weil D, El-Amraoui A, Holme RH et al. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat Genet 2003; 34: 421–428.
Kikkawa Y, Mburu P, Morse S, Kominami R, Townsend S, Brown SD . Mutant analysis reveals whirlin as a dynamic organizer in the growing hair cell stereocilium. Hum Mol Genet 2005; 14: 391–400.
Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM et al. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol 2005; 7: 148–156.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339: 819–823.
Wang Y, Zheng CG, Jiang Y, Zhang J, Chen J, Yao C et al. Genetic correction of beta-thalassemia patient-specific iPS cells and its use in improving hemoglobin production in irradiated SCID mice. Cell Res 2012; 22: 637–648.
Howden SE, Gore A, Li Z, Fung HL, Nisler BS, Nie J et al. Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy. Proc Natl Acad Sci USA 2011; 108: 6537–6542.
Park IH, Lerou PH, Zhao R, Huo H, Daley GQ . Generation of human-induced pluripotent stem cells. Nat Protoc 2008; 3: 1180–1186.
Prokhorova TA, Harkness LM, Frandsen U, Ditzel N, Schroder HD, Burns JS et al. Teratoma formation by human embryonic stem cells is site dependent and enhanced by the presence of matrigel. Stem Cells Dev 2009; 18: 47–54.
Acknowledgements
We would like to thank Professor Chris Wood in Zhejiang University for critical reading of the manuscript. This study was supported by the grants from National Basic Research Program of China (2012CB967902 and 2014CB541705), National Development Program of Important Scientific Instruments (2013YQ030595), Strategically Guiding Scientific Special Projects from The Chinese Academy of Sciences (XDA04020202-23), Opening Foundation of the State Key Laboratory of Space Medicine Fundamentals and Applications (SMFA12K02), TZ-1 Application Program (KYTZ01-0901-FB-003), and The Chinese National Science Foundation (81570932 and 81330024). Also with grateful thanks for help of Miss Yu-Qin Luo Y.Q.L during karyotype analysis.
Author contributions
JRC, ZHT, JD, XDQ, CZ, JLC, CCW, LL and JZS performed the experiments and contributed to data analysis. JZ, JZC and TSH collected the clinical data. HSS and SKY performed the electrophysiological recordings. JFW, MXG, and PC drafted the conception of the study, designed the experiments, and monitored the project progression, data analysis, and interpretation. JRC prepared the initial draft of the manuscript. JFW prepared the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by RA Knight
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Chen, JR., Tang, ZH., Zheng, J. et al. Effects of genetic correction on the differentiation of hair cell-like cells from iPSCs with MYO15A mutation. Cell Death Differ 23, 1347–1357 (2016). https://doi.org/10.1038/cdd.2016.16
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.16
This article is cited by
-
The applications of CRISPR/Cas-mediated genome editing in genetic hearing loss
Cell & Bioscience (2023)
-
Impaired AIF-CHCHD4 interaction and mitochondrial calcium overload contribute to auditory neuropathy spectrum disorder in patient-iPSC-derived neurons with AIFM1 variant
Cell Death & Disease (2023)
-
Recent Advances in CRISPR/Cas9 Delivery Approaches for Therapeutic Gene Editing of Stem Cells
Stem Cell Reviews and Reports (2023)
-
Application of organoids in otolaryngology: head and neck surgery
European Archives of Oto-Rhino-Laryngology (2023)
-
Analysis of the genotype–phenotype correlation of MYO15A variants in Chinese non-syndromic hearing loss patients
BMC Medical Genomics (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}