
Abstract
All current regimens for treating ovarian cancer center around carboplatin as standard first line. The HSP90 inhibitor ganetespib is currently being assessed in advanced clinical oncology trials. Thus, we tested the combined effects of ganetespib and carboplatin on a panel of 15 human ovarian cancer lines. Strikingly, the two drugs strongly synergized in cytotoxicity in tumor cells lacking wild-type p53. Mechanistically, ganetespib and carboplatin in combination, but not individually, induced persistent DNA damage causing massive global chromosome fragmentation. Live-cell microscopy revealed chromosome fragmentation occurring to a dramatic degree when cells condensed their chromatin in preparation for mitosis, followed by cell death in mitosis or upon aberrant exit from mitosis. HSP90 inhibition caused the rapid decay of key components of the Fanconi anemia pathway required for repair of carboplatin-induced interstrand crosslinks (ICLs), as well as of cell cycle checkpoint mediators. Overexpressing FancA rescued the DNA damage induced by the drug combination, indicating that FancA is indeed a key client of Hsp90 that enables cancer cell survival in the presence of ICLs. Conversely, depletion of nuclease DNA2 prevented chromosomal fragmentation, pointing to an imbalance of defective repair in the face of uncontrolled nuclease activity as mechanistic basis for the observed premitotic DNA fragmentation. Importantly, the drug combination induced robust antitumor activity in xenograft models, again accompanied with depletion of FancA. In sum, our findings indicate that ganetespib strongly potentiates the antitumor efficacy of carboplatin by causing combined inhibition of DNA repair and cell cycle control mechanisms, thus triggering global chromosome disruption, aberrant mitosis and cell death.
Similar content being viewed by others


Main
HSP90 inhibitors represent promising agents in cancer treatment. They destabilize a number of HSP90 client proteins required for cancer cell proliferation and survival, and increase proteotoxic stress.1 Among these clients are tumor drivers such as mutant p53 (ref. 2) and MIF,3 but also cell cycle regulators like Wee1(ref. 4) and Chk1,5 as well as DNA repair proteins such as those governing the Fanconi Anemia DNA repair pathway.6 This provides a strong rationale for HSP90 inhibitors in cancer therapy7 and raises the possibility that HSP90 inhibitors can be used in combination with DNA-damaging chemotherapeutics.8
Ovarian cancer is among the most common gynecological tumor types and carries the worst prognosis. HSP90 is highly expressed in advanced ovarian carcinoma9 and thus constitutes a potential therapeutic drug target.10 In support, HSP90 inhibitors were found effective against ovarian cancer in preclinical models,11, 12, 13, 14 alone or in combination with conventional chemotherapy.15, 16
Carboplatin is the key component of all current first-line therapies for ovarian carcinoma. However, while initially often responding with regression, most tumors relapse and develop resistance within less than a year.17, 18 Like other platinum compounds, carboplatin acts by forming crosslinks within DNA.17 Platinum compounds form intrastrand DNA lesions but also interstrand crosslinks (ICLs) by covalently linking opposite DNA strands. Such ICLs represent major obstacles to the DNA replication fork19 and are therefore considered the principal toxic lesion of carboplatin’s mode of action. The ICLs can only be removed by a sophisticated DNA repair system – the Fanconi Anemia pathway (FA) – which consists of numerous protein components acting within complexes. FA-mediated DNA repair operates by orchestrated excision of the crosslinked portion of the DNA. During this process, one template strand of the replicating DNA is temporarily cleaved, leaving a double-strand DNA break (DSB) after completion of the daughter strand synthesis on both sides. Under normal circumstances, the break is subsequently repaired by homologous recombination repair. Identifying ways of targeting the FA pathway may represent a viable sensitization strategy towards ICL-inducing agents such as platinum compounds.
As the HSP90 inhibitor ganetespib destabilizes a number of DNA repair factors and cell cycle regulators, we asked whether it also perturbs the capability of tumor cells to survive ICLs, thereby potentially leading to synergistic tumor cell killing with carboplatin. Our results indicate that carboplatin and ganetespib strongly synergize to reduce the viability of most ovarian cancer cell lines. Strikingly, upon combined treatment, a large fraction of ovarian cancer cells undergo dramatic DNA fragmentation before entering mitosis. We show that degradation of FancA by HSP90 inhibition and the resulting unbalanced action of the nuclease DNA2 are necessary for this phenotype to occur. We propose that the HSP90 inhibitor ganetespib sensitizes cancer cells towards ICLs by preventing DNA repair and by disabling cell cycle checkpoint functions resulting in aberrant mitotic entry and cell death.
Results
Ganetespib and carboplatin strongly synergize to induce death of ovarian cancer cells
A panel of 15 human ovarian cancer cell lines were treated with a clinically relevant range of concentrations of ganetespib or carboplatin followed by luciferase assays to determine cell viability via cellular ATP content. Eight of the 15 lines under study were derived from the high-grade serous subtype of ovarian cancer, representing 70% of all ovarian carcinomas and carrying the worst prognosis.20 After identifying the half-maximum inhibitory concentration (IC50) and the concentration that reduces cell viability by 25% (IC25) for each drug individually, we combined the IC50 and IC25 drug concentrations, respectively, to determine the degree of synergism between ganetespib and carboplatin (Figures 1a and b and Supplementary Figures S1A, S1B). Using the classic algorithm by Chou and Talalay21 to calculate the combination index (CI), we found that 13 out of the 15 lines displayed synergistic effects of the two drugs at both concentrations (CI<1), while the remaining 2 lines showed additive drug cooperation at IC50 (CI=1) and true synergism at IC25. Importantly, combined treatment induced cytotoxic effects in 60–80% of all tumor cells as shown by the ‘Fa’ value (fraction affected; Figure 1a). Moreover, the synergistic effect of ganetespib and carboplatin caused increased induction of apoptosis compared with single drug treatment, shown by Annexin V/7-AAD staining using flow cytometry (Figure 1c) and immunoblot analysis of caspase-mediated PARP cleavage (Figure 1d). Contrasting results were obtained with two non-transformed cell lines (RPE-Tert and MCF10a cells). Here, the two drugs were antagonistic (Figures 1a and b), and unable to induce detectable apoptosis in RPE-Tert cells (Figure 1d). Similar results were obtained when ovarian cancer cell lines were treated with cisplatin, the original platinum drug, in combination with ganetespib (Supplementary Figures S1C and S1D). The cell proliferation rates failed to correlate positively with the CIs, (Supplementary Figure S1E), thus largely excluding a possible bias from slowly replicating cells to explain chemoresistance. Of note, however, p53-proficiency did correlate with resistance towards the drug combination (Supplementary Figure S1F). Lack of drug synergy in p53-proficient cell lines could not be explained by p53-dependent differences in cell proliferation (Supplementary Figures S1G and S1K). Instead, we hypothesized that p53 might protect cells through a p21-mediated cell cycle checkpoint. And indeed, loss of p53 as well as loss of its downstream target p21 re-established drug synergism in (colon cancer-derived) HCT116 cells22 (Supplementary Figure S1H), accompanied by increased γH2AX levels and accumulation of mitotically arrested cells (Supplementary Figures S1I and S1J). Similarly, shRNA-mediated depletion of wild-type p53 in the ovarian cancer-derived cell line A2780 led to increased levels of the G2 checkpoint regulator cyclin B1,23 as well as decreased levels of carboplatin-induced DDB2 (Supplementary Figure S1L), a p53-inducible gene product involved in mediating carboplatin resistance.24 In conclusion, ganetespib and platinum act in a synergistic manner to induce death in the majority of ovarian carcinoma cells but not in non-transformed and/or p53-proficient cells.

Synergistic antitumor activity of carboplatin and ganetespib in ovarian cancer cell lines. (a) Combination Index (CI) plotted against the fraction affected (Fa) for each ovarian carcinoma cell line tested at drug concentrations pre-determined to reduce cell viability by 50% (IC50) or 25% (IC25) after 72 h incubation. For raw data, cf. Supplementary Figure S1. For drug concentrations, cf. Supplementary Table S1. The area marked in red indicates drug synergism (CI<1) along a gradient of cytotoxic efficacy (Fa) up to 100% (x axis). (b) Representative examples of cell viability assays in EFO-21, ES-2, Ovcar-3 and Ovcar-5 cells, as well as the non-transformed cell lines RPE-Tert and MCF10a. The cells were treated with ganetespib or carboplatin alone or in combination for 72 h, followed by the assessment of cell viability by quantification of ATP. In all ovarian carcinoma cell lines, the combination was more effective than the single drugs, ***P<0.001. In contrast, no drug synergism was observed in untransformed cells. NS, not significant. (a–c) Mean of three biological replica ±S.D. (c) Apoptosis induction upon 48 h treatment with ganetespib or carboplatin alone or in combination. The cells were stained with fluorescently labeled Annexin V (early apoptosis) and 7-AAG (late apoptosis or necrosis) and analyzed by flow cytometry. The drug combination was more effective than single drugs with high significance, in cancer cells but not untransformed cells. IC50 drug concentrations were applied as listed in Supplementary Table S1. (d) The extent of PARP cleavage was greatest with the drug combination in ovarian carcinoma cells. The cells were treated as in c and processed for immunoblot analysis. As full-length PARP decreases, a lower molecular weight cleaved band appears, indicating the induction of caspase activity and apoptosis. Notably, the drug combination did not induce PARP cleavage in the untransformed cell line RPE-Tert
The combination of carboplatin with ganetespib leads to accumulation of persistent DNA damage in ovarian cancer cells
Carboplatin, like other DNA-crosslinking agents, exerts its cytotoxic effects mainly through the induction of DNA damage, in particular ICLs. We therefore asked whether ganetespib might enhance this damage and the cellular response to it. Immunostaining of phosphorylated histone H2AX (γH2AX) revealed that this was indeed the case. At concentrations where single agents enhanced γH2AX levels only moderately or not at all, the combination of ganetespib and carboplatin led to a strong increase of γH2AX, as determined by quantitative immunofluorescence analysis (Figures 2a and b) and by immunoblot analysis (Figure 2c). In the latter case, phosphorylated Kap1 (pKap1) mirrored the accumulation of γH2AX upon combined treatment. The global levels of ATM and Chk2 were not increased but rather diminished by ganetespib, at least at 48 h post treatment, arguing that the enhanced phosphorylation of H2AX and Kap1 is due to enhanced signaling activity, but not simply due to the sheer accumulation of kinases within this pathway (Supplementary Figure S2). Moreover, the detection of phosphorylated RPA32 (pRPA32, Figure 2c) suggested the presence of single-stranded DNA (ssDNA) replication intermediates and/or DNA resection. Thus, ganetespib enhances the DNA damage response elicited by carboplatin, suggesting the accumulation of persistent, unrepaired DNA damage.

Combined ganetespib/carboplatin treatment induces the highest DNA damage response. (a) Phosphorylated H2AX level is highest with the drug combination. Immunofluorescence of Ovcar-3 cells treated with the indicated drugs at IC50 (Supplementary Table S1) for 48 h in the presence of 20 μM zVAD to prevent false positive staining due to apoptosis induction. (b) Quantification of the average phosphorylated H2AX signal intensity by high-content microcopy shown in a. In all cases, the damage signal was highest with the drug combination. Significance was determined by the Student's t-test and the P-value is shown by asterisks (***P-value<0.001) (c) Signaling intermediates of the DNA damage response such as phosphorylated Kap1 (pKap1), phosphorylated H2AX (γH2AX) and phosphoryated RPA32 (S4/S8) are highest with the drug combination. pRPA32 marks single-stranded DNA, as occurring during replicative stress. Immunoblot analysis upon treatment for 24 h without zVAD. (b and c) Drug concentrations as in Figure 1c and Supplementary Table S1
HSP90 inhibition depletes G2/M checkpoint kinases and increases the fraction of mitotic cells
Next, we assessed the cell cycle distribution of ovarian cancer cells upon drug treatment. In response to carboplatin alone, the cells first accumulated in G1 and S phase (Supplementary Figure S3A), and later in G2 (Supplementary Figure S3B), but failed to enter mitosis (Figure 3a). However, treatment with the HSP90 inhibitor, alone or in combination with carboplatin, led to the accumulation of cells in mitosis, as indicated by immunostaining of phosphorylated histone 3 (phospho-H3 Ser10; Figure 3a). In contrast, p53-proficient Cov434 cells did not accumulate at G2/M (Supplementary Figure S3B), possibly due to p53-mediated G1 arrest, correlating with their failure to undergo apoptosis upon combined treatment (Supplementary Figure S1E). In addition, the checkpoint kinases Wee1 and Chk1, both required for a functional G2/M checkpoint25, 26, 27, 28 and both dependent on intact HSP90 function,4, 5 nearly disappeared upon HSP90 inhibition (Figure 3b), a phenomenon that might contribute to the observed entry into mitosis despite DNA damage. Indeed, on treatment of ovarian cancer cells with ganetespib alone or in combination, mitotic phospho-H3 positive cells with fully replicated DNA accumulated (Figure 3c; Supplementary Figure S3C). To probe the relevance of ganetespib-mediated destabilization of Wee1 and Chk1, we transiently overexpressed each of these kinases, followed by treatment with ganetespib and/or carboplatin. However, as this did not detectably alter the response to the two drugs (Supplementary Figure S3D–H), we propose that degradation of additional HSP90 clients that regulate the cell cycle, such as Cdc2 (ref. 29) and Cdc25c,30 may also contribute to the combined drug efficacy. In any case, the combined treatment of ovarian cancer cells with carboplatin and HSP90 inhibitor triggers the accumulation of damaged cells in mitosis, apparently by destabilizing a variety of cell cycle regulators.

HSP90 inhibition abrogates the G2/M checkpoint and leads to the accumulation of mitotic cells with 4N DNA content. (a) Average signal intensity for mitotic marker phospho-H3 (pH3), quantified by high-content microcopy after treatment as in Figure 2a. In all cases, the mitotic index was increased with ganetespib, alone or in combination with carboplatin. Right: examples of pH3 staining in Ovcar-3 cells. Significance was determined by the Student's t-test and the P-value is shown by asterisks (***P-value<0.001) (b) Hsp90 inhibition degrades the G2/M checkpoint kinases Wee1 and Chk1. The cells were treated with 200 nM ganetespib and 75 μM carboplatin for 24 h followed by immunoblot analysis. Carboplatin treatment leads to the activation of Chk1, indicated by phospho—Chk1. Ganetespib treatment alone or in combination with carboplatin rapidly depletes both total and phosphorylated levels of Chk1 and Wee1. (c) Cell cycle analysis of propidium iodide/phospho-H3 double-labeled Ovcar-5 cells, treated as in (a). The y-axis shows the green fluorescence of phospho-H3 positive cells and the x-axis represents the DNA content of stained by red fluorescent propidium iodide. The blue circle shows pH3-positive cells with 2N DNA content, the red circle depicts pH3-positive cells with 4N DNA content
Ganetespib in combination with carboplatin induces massive accumulation of small condensed chromosomal fragments
To better understand the cell fate that follows DNA damage in response to combined treatment, we performed chromosome analysis by metaphase spreads. In response to ganetespib/carboplatin treatment, but not upon single drug treatment, dramatic fragmentation of chromosomes was observed (Supplementary Figures S4A, S4B). Caspase inhibition by additional treatment with the caspase inhibitor zVAD did not alleviate this fragmentation, indicating that it is not a result of apoptosis induction (Figures 4a and b). Of note, the numbers of visible crosslinks and radials induced by carboplatin were not increased by ganetespib. Importantly, this fragmentation was observed in all the chromosomes within a cell and in more than 90% of all metaphases (Figure 4c; Supplementary Figure S4C).

Ganetespib in combination with carboplatin induces massive accumulation of small condensed chromosomal fragments. (a) Ovcar-5 cells were treated with ganetespib (20 nM) and/or carboplatin (90 μM) for 48 h in the presence of 20 μM zVAD to exclude apoptosis-driven DNA fragmentation. To enrich the mitotic cell population, 150 nM paclitaxel was added during the last 5 h. The chromosomes were spread on glass slides followed by Giemsa staining. Representative images of metaphase spreads are shown. The extent of chromosome fragmentation per cell was analyzed by counting the number of fragments in 10 randomly chosen metaphases. For quantification of crosslinks and radials, and of intact chromosomes, 10 random metaphases were chosen and visible interstrand crosslinks/radials were counted. Significance was determined by the Student's t-test and the P-value is shown by asterisks (***P-value<0.001) (b) Metaphase spreads of ES-2 cells that were treated with ganetespib (30 nM) and/or carboplatin (45 μM) in the presence of 20 μM zVAD. Sample generation and analysis as in (a). (c) The percentage of metaphases with fragmented DNA was quantified in 100 random cells per treatment group to show that fragmentation occurred with the drug combination at a far higher frequency than with single drugs or control treatment
DNA fragmentation precedes chromatin condensation upon combined ganetespib and carboplatin
To assess whether the DNA fragmentation occurs before or after entry into mitosis, the cells were treated with the two drugs alone or in combination, and TUNEL staining performed in the presence of zVAD, to detect DNA breaks that were not caused by apoptosis. This revealed the appearance of DNA breaks in the majority of cells only on drug combination (Figure 5a). Live cell imaging provided a more detailed insight into the fate of cells that undergo chromosome fragmentation. Specifically, treated cells entered mitosis and condensed their chromosomes. However, owing to persistent DNA breaks, the chromosomes showed massive fragmentation and were scattered throughout the cell, thereby preventing proper chromosome alignment and faithful execution of mitosis. Subsequently, a minor fraction of cells died in mitosis, whereas the majority of cells de-condensed their chromatin and exited from mitosis without undergoing cell division. Those cells were characterized by irregularly shaped nuclei and micronuclei that were formed on the unscheduled exit from mitosis. Ultimately, most of these cells died in the subsequent interphase (Figures 5b and c; Supplementary Videos S1–S3). Thus, the synergistic effect of the two drugs can be explained, at least in part, by severe, persistent and unrepaired damage to tumor cell DNA. This in turn leads to premitotic DNA fragmentation followed either by immediate death or by uncontrolled transition to mitosis and subsequent cell death.

DNA fragmentation precedes chromatin condensation in response to combined treatment with ganetespib and carboplatin. (a) The drug combination induces apoptosis-independent DNA fragmentation during interphase. Ovcar-5 cells were treated as in Figure 4a in the presence of 20 μM zVAD for 48 h, followed by TUNEL and Hoechst staining. Representative images are shown. (b) Ovcar-5 cells were transiently transfected with GFP-tagged H2B. Live-cell imaging of ganetespib- and/or carboplatin-treated cells (15 nM ganetespib, 90 μM carboplatin) was performed between 24 and 40 h after drug treatment. Representative images were taken from time-lapse microscopy, Supplementary Videos S1–S3. (c) All cells observed within the time frame of live-cell imaging (b) were classified according to the time of their death, either during interphase or during mitosis. Significance was determined by the Student's t-test and the P-value is shown by asterisks (***P-value<0.001)
Ganetespib synergizes with other ICL-inducing agents and induces rapid degradation of Fanconi Anemia pathway components
Next, we found that key components of the ICL repair machinery and Fanconi Anemia (FA) pathway, such as FancA, BLM and ubiquitinated FancD2 were rapidly degraded in response to ganetespib treatment (Figure 6a; Supplementary Figure S6A). Even as little as 1 h of treatment with ganetespib was sufficient to induce FancA degradation, and when combined with 48 h of carboplatin treatment led to the increased accumulation of γH2AX and cleaved PARP1 in Ovcar-5 cells (Figure 6b). In the case of FancD2, carboplatin led to the appearance of a second protein species with higher molecular weight (‘FancD2-L’; Figure 6a), corresponding to monoubiquitinated FancD2 that is induced by the upstream components of the pathway upon activation.31 When carboplatin was combined with ganetespib, FancD2 was not only reduced in its total levels, but the upper band corresponding to ubiquitinated FancD2 disappeared completely. Thus, both the key components and key functionality of the FA pathway are compromised by HSP90 inhibition. Caspase inhibition did not detectably affect the ganetespib-induced loss of FancA and FancD2 levels (Supplementary Figure S6B). This was in line with unchanged PARP1 levels (Supplementary Figure S6B). Mitomycin C, a chemically unrelated ICL-inducing agent, also synergized with ganetespib in its toxicity towards ovarian cancer cells (Figure 6c), similar to carboplatin (Figure 1a). In contrast, combination of ganetespib with the double-strand break-inducing agent neocarzinostatin (Figure 6d) or with the mitotic spindle inhibitor paclitaxel (Figure 6e) showed little if any synergistic effects. Moreover, ovarian cancer cells treated with mitomycin C and ganetespib again displayed intense chromosome fragmentation (Figure 6f, Supplementary Figures S6C and S6D), similar to the combination of carboplatin and ganetespib.

Ganetespib induces rapid degradation of Fanconi Anemia pathway components and synergizes with other ICL-inducing agents. (a) The Fanconi Anemia (FA) pathway components FancD2, FancA and BLM are depleted by ganetespib, regardless of carboplatin co-treatment. The cells were treated with ganetespib and/or carboplatin for 48 h (drug concentrations, cf. Supplementary Table S1), followed by immunoblot analysis. Note that carboplatin alone induces a FancD2 band with a slightly higher molecular weight (FancD2-L) corresponding to FancD2 monoubiquitination, a marker of activation of the FA pathway.45 Ubiquitinated FancD2 is no longer observed in cells treated with the drug combination, indicating that the pathway upstream of FancD2 was rendered inactive. (b) When treated with carboplatin for 48 h, as little as a 1 h co-treatment with ganetespib is sufficient to repress FancA levels leading to the accumulation of DNA damage (γH2AX) and induction of apoptosis (PARP cleavage). Ovcar-5 cells were treated as indicated in the time line below and analyzed by immunoblot. (c) Synergism between ganetespib and another crosslinking compound, mitomycin C. Combination Index (CI) plotted against the fraction affected (Fa) for each cell line at drug concentrations pre-determined to reduce cell viability by 50% (IC50) after 72 h incubation. The area marked in red indicates drug synergism (CI<1) along a gradient of cytotoxic efficacy (fraction affected, Fa) up to 100% (x axis). (d and e) Lack of synergism between ganetespib and a double-strand break-inducing radiomimetic, neocarzinostatin (d) or the mitotic spindle drug paclitaxel (e) in the majority of ovarian cancer cell lines. Treatment and processing as in (c). (f) Ganetespib combined with Mitomycin C also induces massive accumulation of small condensed chromosomal fragments. Metaphase spreads of Ovcar-5 cells treated with 500 nM Mitomycin C alone or in combination with 10 nM ganetespib for 48 h in the presence of 20 μM caspase inhibitor zVAD. The extent of chromosome fragmentation per cell was analyzed by counting the number of fragments in 10 random metaphases
Ganetespib and carboplatin strongly synergize in the inhibition of tumor xenografts in vivo
Next, we tested whether the synergy of carboplatin and ganetespib also holds true in vivo. Ovcar-5 (Figures 7a and b) or BG-1 cells (Figures 7c and d) were subcutaneously injected into immunocompromised SCID SHO mice to grow tumors. Notably, although tumor growth was partially inhibited in mice treated with carboplatin or ganetespib alone, compared with vehicle treatment, the combined treatment led to complete tumor growth stagnation or even tumor regression (Figures 7a and c). Importantly, a single dose of carboplatin combined with ganetespib was sufficient to induce complete depletion of FancA, FancD2 and BLM in the tumor xenografts, accompanied by γH2AX accumulation and PARP1 cleavage (Figures 7b and d). We cannot exclude though that H2AX phosphorylation may partially result from caspase activity, mediated by caspase-activated DNase (CAD) and DNA damage.32

Antitumor synergism of ganetespib and carboplatin in xenograft models. (a and c) SCID SHO mice were inoculated with 106 Ovcar-5 (a) or BG-1 (c) cells (with 1/5 volume Geltrex) into the right and left flanks. (a) Twelve days after inoculation, the mice were treated on day 1 (=12 days after inoculation), day 8 and day 11 with 40 mg/kg carboplatin i.p. and 50 mg/kg ganetespib i.v. (c) Twenty-one days after inoculation, mice were treated every 4–5 days with 30 mg/kg carboplatin and 75 mg/kg ganetespib. (a and c) Tumor volumes were determined before each injection. The drug combination essentially prevented tumor growth and was significantly more effective than the single drugs. Representative tumor explants from each treatment group from day 11 (a) or day 24 (c) are shown below. Significance was determined by the Student's t-test and the P-value is shown by asterisks (*P-value<0.05, ***P-value<0.001) (b and d) Immunoblot analysis of Ovcar-5 (b) and BG-1 (d) tumors. The mice were killed 24 h after treatment with a single dose of carboplatin (40 mg/kg, i.p.) and ganetespib (50 mg/kg, i.v.) alone or in combination. As already seen in cultured cells, FancA, FancD2 and BLM levels were reduced in tumors on ganetespib treatment. Conversely, DNA damage (γH2AX) and apoptosis were highest with the drug combination
Chromosome fragmentation and cell death induced by combined drug treatment is rescued by ectopic FancA expression
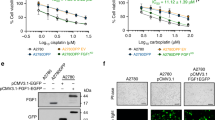
The drop in FancA levels upon ganetespib treatment (Figures 6a and b) suggests that loss of FancA might be causal for the synergistic effect of ganetespib with carboplatin. In support, FancA overexpression rendered Ovcar-5 cells considerably more resistant to both drugs, and this protective effect was most pronounced when ganetespib and carboplatin were used in combination (Figure 8a). Moreover, the synergism between the two drugs was no longer observed upon FancA overexpression. The levels of overexpressed FancA could not be abrogated by ganetespib and/or carboplatin treatment, presumably because such high amounts of FancA no longer depend on HSP90 (Figure 8b). The accumulation of phospho-H3 and γH2AX on treatment with drug combination was reduced, suggesting restored DNA repair capability in these cells (Figure 8b). Most impressively, FancA overexpression in Ovcar-5 and EFO-21 cells largely prevented the massive accumulation of chromosome fragments during mitosis (Figure 8c). In contrast, the amount of detectable crosslinked chromosomes was moderately decreased by FancA in carboplatin-treated cells. However, this was not seen in cells treated by the drug combination (Supplementary Figures S8A and B), presumably because some ICLs remained unrepaired but no longer gave rise to DNA breaks. Taken together, these results indicate that FancA is a crucial HSP90 client in the context of ovarian cancer cell survival after carboplatin treatment.

Overexpressed FancA rescues the cytotoxicity of combined carboplatin and ganetespib. (a) Left: Ovcar-5 cells were transfected with a control plasmid (pcDNA3) or an expression plasmid for FancA (pMMP_FancA), followed by treatment with ganetespib and/or carboplatin. In all cases, FancA overexpression strongly increased cell viability (P<0.001). Right: Combination Index (CI) plotted against the fraction affected (Fa) for control and FancA-overexpressing Ovcar-5 cells. FancA overexpression abolishes the synergistic action of the combined drugs. (b) FancA overexpression overcomes γH2AX and pH3 accumulation in combination-treated cells (compare the last three lanes). Ovcar-5 cells were transfected with a control (pcDNA3) or expression plasmids for 3-myc-tagged FancA (pcDNA_FancA) or non-tagged FancA (pMMP_FancA). Immunoblot analysis performed 24 h after treatment with 20 nM ganetespib and 90 μM carboplatin, alone or in combination. (c) Control and FancA-overexpressing Ovcar-5 and EFO-21 cells were treated for 48 h with the IC50s of ganetespib and/or carboplatin (drug concentrations, cf. Supplementary Table S1) in the presence of 20 μM zVAD. The cells were processed as in Figure 4a and representative images are shown. The extent of chromosome fragmentation per cell was analyzed by counting the fragments in 10 randomly chosen metaphases. Significance was determined by the Student's t-test and the P-value is shown by asterisks (***P-value<0.001)
The nuclease DNA2 is required for chromosome fragmentation upon combined treatment with ganetespib and carboplatin
We showed that the drug combination triggers global chromosome fragmentation at least in large part owing to abrogation of the FA DNA repair pathway. ICL repair by the FA pathway also involves multiple DNA nucleases. Thus, depletion of FA core proteins can trigger uncontrolled resection of DNA.19, 33 In particular, isolated depletion of FancD2 is known to induce over-resection of DNA by the helicase/nuclease DNA2 (albeit not chromosome condensation).33 Thus, we tested whether DNA2 knockdown could overcome chromosome fragmentation in ganetespib/carboplatin-treated ovarian cancer cells. Indeed, stable depletion of DNA2 (Supplementary Figure S9A) led to near-complete recovery of the chromosome structure (but did not abolish the formation of detectable crosslinks), as shown by chromosome spread analyses of Ovcar-3 (Figure 9a, Supplementary Figure S9B) and Ovcar-5 (Figure 9b, Supplementary Figure S9B) cells. This was accompanied by reduced accumulation of γH2AX and of pKap1 in DNA2-depleted cells (Supplementary Figure S9C, compare lanes 4 and 8). Hence, the fragmentation of DNA in response to ganetespib/carboplatin treatment can be explained by loss of FA pathway components leading to the unscheduled persistence of nuclease DNA2 activity.

Chromatin fragmentation is ameliorated by reducing nuclease DNA2. (a and b) Depletion of DNA2 results in a near-complete recovery of the chromosome structure. Upon transfection with control siRNA or siRNA-targeting DNA2, Ovcar-3 (a) and Ovcar-5 (b) cells were treated with the IC50s of ganetespib and/or carboplatin (for drug concentrations, cf. Supplementary Table S1) for 48 h. The cells were processed as in Figure 4a, and representative images are shown (left). The extent of chromosome fragmentation per cell was analyzed by counting the fragments in 10 metaphases in each case (right). The quantification of intact chromosomes and crosslinks is presented in Supplementary Figure S9B. Significance was determined by the Student's t-test and the P-value is shown by asterisks (***P-value<0.001) (c) Proposed model of how HSP90 inhibition synergizes with platinum drugs. On inhibition of HSP90, the premitotic cell cycle checkpoint is compromised, leading to aberrant entry into mitosis despite DNA damage. This checkpoint deficiency is further aggravated by p53-mutations in cancer cells. As an additional consequence of HSP90 inhibition, components of the FA pathway are degraded, thus impairing the repair of bulky adducts formed by platinum compounds. Both effects enhance cell death and thus contribute to synergistic cytotoxicity. The cell cycle scheme was adapted from an image created by Simon Caulton. (https://commons.wikimedia.org/wiki/File:Cell_cycle_simple.png). The platinum-DNA adduct was depicted according to an image provided at https://de.wikipedia.org/wiki/Cisplatin#/media/File:Molek_working_sheet_5.png, both under a creative commons licence 3.0
Discussion
Here we show that the HSP90 inhibitor ganetespib synergizes with carboplatin in the effective destruction of ovarian cancer cells. Mechanistically, this drug combination induces severe DNA damage due to depletion of key elements of the FA repair pathway, causing cells to undergo massive chromosome fragmentation. The unrepaired cells continue unhindered along the cell cycle and aberrantly enter mitosis, followed by cell death (Figure 9c). Thus, interstrand crosslinking of DNA combined with HSP90 inhibition induces an unusual and particularly effective kind of cell death, triggered by premitotic DNA fragmentation, in about 90% of cells in a broad panel of ovarian cancer cell lines, but not in non-transformed p53-proficient cells. Overall, these encouraging preclinical data might endorse the use of this drug combination to treat patients with newly diagnosed ovarian cancer. As carboplatin is already a standard therapy in first line, we propose to combine it with ganetespib as part of a ‘hit hard and early’ strategy, thereby hopefully avoiding or at least substantially delaying the emergence of platinum-resistant cells.
Our results are in agreement with previous reports. Homologous recombination repair of double-stranded DNA breaks, as well as non-homologous end joining, are impaired by HSP90 inhibitors, enhancing radiosensitivity of tumor cells.34, 35, 36, 37 Moreover, HSP90 inhibitors interfere with DNA damage signaling by degrading ATR, Chk1 and Rad51.38, 39, 40 Nonetheless, the phenotype of cancer cells in response to the combined action of HSP90 inhibitors and ICL-inducing agents is rather unique. It combines features of severe and unrepaired chromosome fragmentation with a futile attempt to get through mitosis. Only portions of this dramatic phenotype have previously been reported. For instance, selective inhibition of Chk1 or Wee1 concomitant with DNA damage results in a phenotype termed ‘premature mitosis’.25, 41, 42 In this scenario, the cells enter mitosis despite being far away from having completed DNA replication.43 In contrast, we did not observe premature mitosis upon combined treatment with carboplatin and ganetespib (Figures 3a and c), despite the depletion of Chk1 and Wee1 by the HSP90 inhibitor (Figure 3b). The frequency of ICLs on treatment with a medically relevant concentration of carboplatin is relatively rare, that is, generating less than 1000 ICLs per cell.44 Thus, we assume that the vast majority of cellular DNA can still be replicated despite the futile attempts of repairing the ICLs.
The absence of a functional FA pathway has long been known to sensitize ovarian cancer cells towards crosslinking agents.45 Pharmacological inhibition of FA components prolongs the persistence of phosphorylated H2AX,46 suggesting accumulation of DNA breaks. Importantly, however, condensed chromatin fragments were not described in this scenario, likely owing to intact G2/M checkpoint function. Thus, HSP90 inhibition may be even more effective in sensitizing cells towards platinum compounds.
DNA damage normally prevents the onset of mitosis. This is mediated at least in part through p53 activation and G2 arrest (G2/M checkpoint). It is therefore significant that up to 70% of all ovarian cancers47 and over 90% of high-grade serous ovarian cancers48 carry mutations in p53, which is also true for most of the cell lines studied here. Thus, the futile attempt to undergo mitosis may contribute to the selectivity of the carboplatin/ganetespib drug combination for cancer versus normal cells. In support, the p53-proficient cells did not undergo apoptosis in response to the combined treatment, whereas targeted deletion of p53 restored treatment efficacy (Supplementary Figure S1).
The dramatic chromosome fragmentation observed with the drug combination requires DNA2 activity. It appears that ganetespib induces an imbalance between DNA synthesis in the context of repair and the removal of damaged DNA by nucleases. Thus, DNA2 nuclease activity and its independence of HSP90 contribute to the catastrophic response to the drug combination. In general, the cells require DNA2 for repression of replicative stress. DNA2 can trigger proper intra-S-phase checkpoints,49 prevent replication fork reversal,50 and restart replication when forks have already undergone reversal.51 However, in response to ICL-inducing agents, DNA2 contributes to over-resection of DNA due to an imbalanced hyperactivation of its nuclease activity in response to loss of FancD2 and/or to impairment of the replication fork stabilizer BOD1L.33, 52 These considerations are in line with our observation of phospho-RPA32 accumulation (Figure 2c), a marker for ssDNA intermediates and DNA resection, as phospho-RPA recruits DNA2 to chromatin.53, 54
Interfering with DNA repair as a means of tumor cell chemosensitization has long been proposed as a promising anticancer strategy.55, 56 However, the development of specific inhibitors of DNA repair factors is far from being trivial. Moreover, targeted inhibitors always bear the risk of resistance development, either by compensatory mutations within the target itself, or by activation of alternative repair pathways. The advantage of HSP90 inhibitors may lie precisely in their pleiotropic activity.7 The dual inactivation of ICL repair and cell cycle checkpoints, but not nucleases, by HSP90 inhibition results in a highly effective chemosensitization. Overall, this paper represents strong preclinical evidence to open the door towards designing first-line clinical trials with the combination.
Materials and Methods
Cell culture and treatment
Human EFO-21, Ovcar-3 and BG-1 cells were a gift from Carsten Gründker (UMG, Göttingen). Ovcar-5 cells that contain an RFP expression cassette were purchased from Cell Biolabs. Skov-6, Hoc-7, Skov-3 and A2780 cells were obtained from the German Collection of Cell lines (DSMZ, Braunschweig, Germany). Kuramochi, Cov318 and Snu-119 cells were obtained from Hagen Kulbe (Charité, Berlin, Germany); Cov434 and Cov362 cells were purchased from Sigma-Aldrich (Taufkirchen, Germany); ES-2 cells were a gift from Hermann Lage (Charité Berlin); and EFO-27 cells were purchased from the DSMZ. RPE-Tert and MCF10a cells were purchased from ATCC, (Virginia, USA). Ovcar-3, A2780, Snu-119 and Kuramochi cells were maintained in RPMI medium. All the other cell lines were maintained in Dulbecco’s modified Eagle’s medium. Cell culture media were supplemented with 10% fetal bovine serum and antibiotics. The cells were maintained at 37 °C in a humidified atmosphere with 5% CO2. For the treatment of cells, ganetespib (50 mM, Synta Pharmaceuticals, Lexington, MA, USA), zVAD (Z-VAD-FMK, 40 μM in DMSO, Enzo, Lausen, Switzerland), neocarzinostatin (NCS, 0.5 mg/ml, Sigma-Aldrich), paclitaxel (10 mM in DMSOSigma-Aldrich), mitomycin C (2 mM in DMSO, Sigma-Aldrich), cisplatin (1 mg/ml, Teva GmbH, Ulm, Germany) or carboplatin (10 mg/ml, Actavis, Luxembourg) were diluted in pre-warmed medium and added to the cells for the indicated periods of time.
Stable transduction and transfection
For stable knockdown of DNA2 in cell lines, HEK293T cells were co-transfected with lentiviral packaging vectors (pMD2.G from Addgene, Teddington, UK and pCMV-R8.91 from Plasmid Factory, Bielefeld, Germany) and the GIPZ-shDNA2 plasmid (V2LHS90523) or a GIPZ control plasmid (RHS4348; both from Dharmacon, Lafayette, CO, USA). Ovcar-3 and Ovcar-5 cells were transduced with the generated lentivirus in the presence of 8 μg/ml polybrene. To generate knockdown cells that contain shRNA, the plasmids plko-p53kd_1 (TRCN0000003753), plko-p53kd_2 (TRCN0000003755) or a plko control plasmid containing shRNA against luciferase (all plasmids from Dharmacon) were co-transfected with packaging vectors into HEK293T cells to generate vector-containing supernatant. A2780 cells were transduced with these vectors in the presence of polybrene, followed by selection with 1 μg/ml puromycin. For transient overexpression of FancA, 5 × 104 cells per six wells were transfected with 5 μl Lipofectamine 2000 (Life Technologies, Darmstadt, Germany) and 2 μg of the plasmids pcDNA_FancA or pMMP_FancA (kindly provided by Hyungjin Kim, Stony Brook University and Gary Kupfer, Yale University), or the respective pMMP and pcDNA3.1 control plasmids. Transient overexpression of Chk1 and Wee1 was achieved by transfection of 12 × 104 cells with 2 μg pcDNA4-Chk1-flag (gift from Aziz Sancar, Addgene #22894), pCMV-Sport6-Wee1 (IRAT collection, Geneservice, Cambridge, UK) or 2 μg control pcDNA3.1 (V79020, Invitrogen, Carlsbad, CA, USA) or pCMV-Sport6 (IRAT collection, Geneservice) using 5 μl Lipofectamine 2000.
Quantitative mRNA analysis by qRT-PCR
Total RNA was isolated using TRIzol (Invitrogen), followed by DNase I digest (Thermo Fisher Scientific, Darmstadt, Germany) and reverse transcription with Moloney Murine Leukemia Virus reverse transcriptase and random hexamer primers (Thermo Fisher Scientific). Maxima SYBR Green master mix (Thermo Fisher Scientific) was used for real-time PCR. The primer sequences for DNA2 were DNA2forward: 5′-ACCCAACAATCCTGTTTGTTTCC-3′ and DNA2reverse: 5′-TCCTCCTACATCATCCCTAAGC-3′. PCR conditions were as follows: initial denaturation 15 min at 95 °C, followed by 40 cycles of 95 °C for 15 s and 60 °C for 45 s. Gene expression levels were normalized to ActinB as reference gene (actin_forward: 5′-CGACAGGCTGCAGAAGGAG-3′, actin_reverse: 5′-GTACTTGCGCTCAAGAGGAG-3′) and calculated using the 2ΔΔCt method. Depicted is the mean ratio, normalized to control knockdown cells.
Immunoblot analysis
The cells were harvested in protein lysis buffer (20 mM TRIS-HCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1 mM beta-glycerophosphate, 2 M urea, protease inhibitor cocktail, Roche, Mannheim, Germany). After 10 min lysis on ice, the samples were briefly sonicated to disrupt DNA–protein complexes. Total protein concentration was measured using a Pierce BCA Protein assay kit (Thermo Fisher Scientific). After boiling the samples in Laemmli buffer at 95 °C for 5 min, equal amounts of protein samples were separated by SDS-PAGE, transferred onto nitrocellulose, and visualized with the following antibodies: PARP1 (9542, Cell Signalling, Frankfurt, Germany), phospho-Kap1/pKap1 (Ser824; ab70369, Abcam, Cambridge, UK), γH2AX (S139; 9718, Cell Signalling), phospho-RPA32/pRPA32 (Ser4/Ser8; A300-246A, Bethyl, Montgomery, USA), Hsc70 (sc-7298, Santa Cruz, Heidelberg, Germany), FancD2 (Santa Cruz), FancA, (A301-980A, Bethyl), BLM (A300-120A, Bethyl), β-actin (ab8227 Abcam), Wee1 (4936, Cell Signalling), phospho-Wee1/pWee1 (Ser642; 4910, Cell Signalling), Chk1 (2360, Cell Signalling), phospho-Chk1/pChk1 (Ser17; 2344, Cell Signalling), phospho-H3 (Ser10; 3377, Cell Signalling), p21 (2947, Cell Signalling), phospho-ATM (S1981; 4526, Cell Signalling), ATM (2873, Cell Signalling), ATR (2790, Cell Signalling), Chk2 (sc-5278, Santa Cruz), cyclin B1 (ab7260, Abcam) and DDB2 (sc-81256, Santa Cruz).
Determination of drug synergism
For determination of drug synergism, cell viability was assessed using the Cell TiterGlo Kit (Promega, Madison, WI, USA). Briefly, 1000–2000 cells were seeded 24 h before treatment. To determine the individual IC50 drug concentrations of carboplatin, ganetespib, neocarzinostatin, paclitaxel and mitomycin C, rising drug concentrations were applied for 72 h and relative cell viability was assessed by luminometry. Vehicle-treated control cells were set to 100% cell viability and the relative cell viability on drug treatment was calculated from three independent biological replicates for each drug concentration.
Drug synergism was assessed by treating cells with the single drugs at their IC25 and IC50 side by side with the combination of two drugs used at the same concentrations. Vehicle-treated cells were set to 100%, and the fraction affected (Fa) of each treatment was calculated by subtracting the relative remaining cell viability from DMSO control-treated cells. For calculating the Combination Index (CI), Fa values from single and combination treatments served as input using the Compusyn software.21 CI values >1 describe antagonistic or non-synergistic effects of two drugs, CI=1 indicates additive effects and CI values <1 correspond to synergistic effects of combined drug treatment.
Cell cycle analysis and mitotic index
For cell cycle analysis and determining the percentage of cells arrested in mitosis, the cells were collected after 24 and 48 h drug treatment (in the presence of 20 μM zVAD in case of 48 h treatment). The samples were fixed in ethanol and stained with propidium iodide and/or anti-phospho-H3 (Ser10). Cell cycle profiles were obtained by flow cytometry using the Guava system (Millipore, Darmstadt, Germany). For each condition, three independent biological replicates were processed, and all the samples were analyzed using the same gate settings.
Determination of apoptosis
Annexin V/7-AAD staining was conducted on living cells using the Nexin reagent (Millipore). The percentage of apoptotic cells were determined by flow cytometry using the Guava system (Millipore). For quantification of Annexin V-positive, or Annexin V/7-AAD double-positive cells, the same gate settings were applied for all the samples. For each experiment, at least three independent biological replicates were analyzed. Significance was calculated using Student’s t-test.
Immunofluoresence, TUNEL staining and high-content microscopy
For immunofluorescence analysis, the cells were fixed with 4% paraformaldehyde for 30 min followed by permeabilization with 0.5% Triton X-100/PBS. After 4 h incubation with antibodies against γH2AX (05-636, Millipore) or phospho-H3 (pH3, 3377, Cell Signalling) and extensive washing, the cells were stained with respective secondary antibodies (AlexaFluor488 and AlexaFluor546, Invitrogen) and nuclei counterstained with Hoechst 3342 (Roche). For single-cell analysis, the slides were assessed with a Zeiss AxioVert microscope. For quantification of γH2AX intensity and the number of pH3-positive cells, automated high-content microscopy was done using the Pathway HT Cell Imaging System and the AttoVision image acquisition software (Becton Dickinson, Heidelberg, Germany). Analysis was performed as previously described.57 Briefly, fluorescence signals were normalized to Hoechst staining intensity, and the relative γH2AX signal and the percentage of pH3-positive cells were calculated. For each treatment group, at least three independent biological replica were analyzed. Significance was calculated using Student’s t-test. For the assessment of cells with fragmented DNA, TUNEL staining (click-it TUNEL AlexaFluor488 imaging kit, Thermo Fisher Scientific) was applied on cells that had been treated with drugs for 48 h in the presence of the caspase inhibitor zVAD. The cells were co-stained with Hoechst solution and assessed using a Zeiss AxioVert microscope and × 20 objective.
Analysis of metaphase spreads
For analysis of chromosome structures, the cells were incubated for 48 h with the indicated drug concentrations in the presence or absence of 20 μM zVAD. Five hours before collection, 150 nM paclitaxel was added to enrich the population in mitotically arrested cells with condensed chromosomes. The cells were collected, swollen in a hypotonic solution (40% RPMI, 60% deionized H2O) for 20 min and then fixed using ice-cold Carnoy’s fixative (3 : 1 acetic acid: methanol). After extensive washing with Carnoy’s fixative, the cells were resuspended in 100% waterfree acetic acid and subsequently spread on slides, followed by staining with Giemsa solution (Carl Roth, Karlsruhe, Germany). Analysis was performed with a × 100 objective in oil on a Zeiss AxioVert microscope. For quantification of undamaged chromosomes, crosslinks or radials and chromosome fragments, 10 randomly chosen cells were analyzed per treatment group; for quantification of total cell numbers showing fragmented chromosome structures, 100 random cells per treatment group were counted.
Live-cell imaging
For live-cell analyses, Ovcar-5 cells were transiently transfected with H2B-GFP expressing plasmids using Lipofectamine 2000 and seeded on glass bottom dishes with four compartments (Greiner Bio-One, Germany). Twenty-four hours later, the cells were treated with ganetespib (20 nM) and/or carboplatin (50 μM) for 30 h. Subsequently, the cells were observed for 18 h by time-lapse microscopy at 37 °C and 5% CO2 using a DeltaVision ELITE microscope (GE Healthcare, NW, USA). Image stacks were recorded every 5 min using a PCO Edge sCMOS camera (PCO, Stuttgart, Germany) with a Z-optical spacing of 4 μm. The images were deconvoluted and further processed using the SoftWorx 6.0 (Applied Precision, Mississauga, Canada) software.
Xenograft experiments
Mouse experiments were conducted in full agreement with the Göttingen University Animal Care Committee and the Institutional Guidelines for Human Use of Animals in Research. Ovcar-5 and BG-1 cells (1 × 106 each) were mixed with Geltrex (1 : 5) and subcutaneously injected into the left and right flank of SCID SHO mice. Upon reaching a tumor size of 12–25 mm3, treatment started. The drugs were administered every 4–5 days until tumor sizes reached end point (150 mm3). The tumor size was monitored using a caliper and tumor volumes calculated as ellipsoid (V=abc × 4/3π). Mice with Ovcar-5 xenografts were treated with 40 mg/kg carboplatin i.p. and 50 mg/kg ganetespib i.v. (dissolved in 10% DMSO, 18% Cremophor RH40, 3,6% Dextrose solution), while BG-1 xenografts received 30 mg/kg carboplatin and 75 mg/kg ganetespib. At the end point, the mice were killed and the tumors dissected for analysis. For analysis of the acute drug response in xenografts, the mice were treated once with single or combined drugs, killed 24 h later, their tumors dissected and tumor lysates were prepared for immunoblot analysis.
Assessment of cell proliferation and doubling time
The cells were seeded at a density of 105 cells/well in 12-well plates. Twenty-four hours later, the cells were treated with ganetespib and/or carboplatin at the indicated concentrations. Subsequently, the percentage of cell confluence was determined every 24 h by bright-field microscopy using a Celigo Adherent Cell Cytometer (Brooks, Chelmsford, MA, USA). Cell proliferation and doubling time was calculated from the increase in plate confluence using the Celigo software and evaluating three biological replica at each time point.
Abbreviations
- 7-AAD:
-
7-aminoactinomycin D
- CAD:
-
caspase-activated Dnase
- CI:
-
combination index
- del.:
-
deletion
- DSB:
-
double-strand breaks
- Fa:
-
Fraction affected
- FA:
-
Fanconi Anemia
- ICL:
-
interstrand crosslinks
- IC50/IC25:
-
inhibitory concentration 50/25
- mt:
-
mutant
- NCS:
-
neocarzinostatin
- wt:
-
wild type
- zVAD:
-
Z-VAD-FMK
References
Taipale M, Jarosz DF, Lindquist S . HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 2010; 11: 515–528.
Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015; 523: 352–356.
Schulz R, Marchenko ND, Holembowski L, Fingerle-Rowson G, Pesic M, Zender L et al. Inhibiting the HSP90 chaperone destabilizes macrophage migration inhibitory factor and thereby inhibits breast tumor progression. J Exp Med 2012; 209: 275–289.
Aligue R, Akhavan-Niak H, Russell P . A role for Hsp90 in cell cycle control: Wee1 tyrosine kinase activity requires interaction with Hsp90. EMBO J 1994; 13: 6099–6106.
Arlander SJ, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM . Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. J Biol Chem 2003; 278: 52572–52577.
Oda T, Hayano T, Miyaso H, Takahashi N, Yamashita T . Hsp90 regulates the Fanconi anemia DNA damage response pathway. Blood 2007; 109: 5016–5026.
Dobbelstein M, Moll U . Targeting tumour-supportive cellular machineries in anticancer drug development. Nat Rev Drug Discov 2014; 13: 179–196.
Proia DA, Bates RC . Ganetespib and HSP90: translating preclinical hypotheses into clinical promise. Cancer Res 2014; 74: 1294–1300.
Elstrand MB, Stavnes HT, Trope CG, Davidson B . Heat shock protein 90 is a putative therapeutic target in patients with recurrent advanced-stage ovarian carcinoma with serous effusions. Hum Pathol 2012; 43: 529–535.
Stope MB, Koensgen D, Burchardt M, Concin N, Zygmunt M, Mustea A . Jump in the fire - heat shock proteins and their impact on ovarian cancer therapy. Crit Rev Oncol Hematol 2016; 97: 152–156.
Huntoon CJ, Nye MD, Geng L, Peterson KL, Flatten KS, Haluska P et al. Heat shock protein 90 inhibition depletes LATS1 and LATS2, two regulators of the mammalian hippo tumor suppressor pathway. Cancer Res 2010; 70: 8642–8650.
Jiao Y, Ou W, Meng F, Zhou H, Wang A . Targeting HSP90 in ovarian cancers with multiple receptor tyrosine kinase coactivation. Mol Cancer 2011; 10: 125.
Kim YJ, Lee SA, Myung SC, Kim W, Lee CS . Radicicol, an inhibitor of Hsp90, enhances TRAIL-induced apoptosis in human epithelial ovarian carcinoma cells by promoting activation of apoptosis-related proteins. Mol Cell Biochem 2012; 359: 33–43.
Liu H, Xiao F, Serebriiskii IG, O'Brien SW, Maglaty MA, Astsaturov I et al. Network analysis identifies an HSP90-central hub susceptible in ovarian cancer. Clin Cancer Res 2013; 19: 5053–5067.
Banerji U, Sain N, Sharp SY, Valenti M, Asad Y, Ruddle R et al. An in vitro and in vivo study of the combination of the heat shock protein inhibitor 17-allylamino-17-demethoxygeldanamycin and carboplatin in human ovarian cancer models. Cancer Chem Pharmacol 2008; 62: 769–778.
Zhang Z, Xie Z, Sun G, Yang P, Li J, Yang H et al. Reversing drug resistance of cisplatin by hsp90 inhibitors in human ovarian cancer cells. Int J Clin Exp Med 2015; 8: 6687–6701.
Kelland L . The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 2007; 7: 573–584.
Agarwal R, Kaye SB . Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer 2003; 3: 502–516.
Deans AJ, West SC . DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011; 11: 467–480.
Domcke S, Sinha R, Levine DA, Sander C, Schultz N . Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 2013; 4: 2126.
Chou TC . Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010; 70: 440–446.
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282: 1497–1501.
Innocente SA, Abrahamson JL, Cogswell JP, Lee JM . p53 regulates a G2 checkpoint through cyclin B1. Proc Natl Acad Sci USA 1999; 96: 2147–2152.
Barckhausen C, Roos WP, Naumann SC, Kaina B . Malignant melanoma cells acquire resistance to DNA interstrand cross-linking chemotherapeutics by p53-triggered upregulation of DDB2/XPC-mediated DNA repair. Oncogene 2014; 33: 1964–1974.
Russell P, Nurse P . Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 1987; 49: 559–567.
Carr AM, Moudjou M, Bentley NJ, Hagan IM . The chk1 pathway is required to prevent mitosis following cell-cycle arrest at 'start'. Curr Biol 1995; 5: 1179–1190.
Clay-Farrace L, Pelizon C, Santamaria D, Pines J, Laskey RA . Human replication protein Cdc6 prevents mitosis through a checkpoint mechanism that implicates Chk1. EMBO J 2003; 22: 704–712.
Vogel C, Hager C, Bastians H . Mechanisms of mitotic cell death induced by chemotherapy-mediated G2 checkpoint abrogation. Cancer Res 2007; 67: 339–345.
Nakai A, Ishikawa T . Cell cycle transition under stress conditions controlled by vertebrate heat shock factors. EMBO J 2001; 20: 2885–2895.
Garcia-Morales P, Carrasco-Garcia E, Ruiz-Rico P, Martinez-Mira R, Menendez-Gutierrez MP, Ferragut JA et al. Inhibition of Hsp90 function by ansamycins causes downregulation of cdc2 and cdc25c and G(2)/M arrest in glioblastoma cell lines. Oncogene 2007; 26: 7185–7193.
Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 2003; 35: 165–170.
Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S . A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998; 391: 43–50.
Karanja KK, Lee EH, Hendrickson EA, Campbell JL . Preventing over-resection by DNA2 helicase/nuclease suppresses repair defects in Fanconi anemia cells. Cell Cycle 2014; 13: 1540–1550.
Stingl L, Stuhmer T, Chatterjee M, Jensen MR, Flentje M, Djuzenova CS . Novel HSP90 inhibitors, NVP-AUY922 and NVP-BEP800, radiosensitise tumour cells through cell-cycle impairment, increased DNA damage and repair protraction. Br J Cancer 2010; 102: 1578–1591.
Stecklein SR, Kumaraswamy E, Behbod F, Wang W, Chaguturu V, Harlan-Williams LM et al. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proc Natl Acad Sci USA 2012; 109: 13650–13655.
Noguchi M, Yu D, Hirayama R, Ninomiya Y, Sekine E, Kubota N et al. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem Biophys Res Commun 2006; 351: 658–663.
Dote H, Burgan WE, Camphausen K, Tofilon PJ . Inhibition of hsp90 compromises the DNA damage response to radiation. Cancer Res 2006; 66: 9211–9220.
Yao Q, Weigel B, Kersey J . Synergism between etoposide and 17-AAG in leukemia cells: critical roles for Hsp90, FLT3, topoisomerase II, Chk1, and Rad51. Clin Cancer Res 2007; 13: 1591–1600.
Koll TT, Feis SS, Wright MH, Teniola MM, Richardson MM, Robles AI et al. HSP90 inhibitor, DMAG, synergizes with radiation of lung cancer cells by interfering with base excision and ATM-mediated DNA repair. Mol Cancer Ther 2008; 7: 1985–1992.
Ha K, Fiskus W, Rao R, Balusu R, Venkannagari S, Nalabothula NR et al. Hsp90 inhibitor-mediated disruption of chaperone association of ATR with hsp90 sensitizes cancer cells to DNA damage. Mol Cancer Ther 2011; 10: 1194–1206.
Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov 2012; 2: 524–539.
Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther 2010; 9: 514–522.
Paulson JR, Taylor SS . Phosphorylation of histones 1 and 3 and nonhistone high mobility group 14 by an endogenous kinase in HeLa metaphase chromosomes. J Biol Chem 1982; 257: 6064–6072.
Eastman A . The mechanism of action of cisplatin: from adducts to apoptosis. In: Lippert (ed). Cisplatin. Chemistry and Biochemistry of a Leading Anticancer Drug. Wiley-VCH: Zurich, Switzerland, 1999, pp 111–134.
Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H et al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med 2003; 9: 568–574.
Jacquemont C, Simon JA, D'Andrea AD, Taniguchi T . Non-specific chemical inhibition of the Fanconi anemia pathway sensitizes cancer cells to cisplatin. Mol Cancer 2012; 11: 26.
Bast RC Jr., Hennessy B, Mills GB . The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer 2009; 9: 415–428.
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474: 609–615.
Kumar S, Burgers PM . Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes Dev 2013; 27: 313–321.
Hu J, Sun L, Shen F, Chen Y, Hua Y, Liu Y et al. The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 2012; 149: 1221–1232.
Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M et al. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 2015; 208: 545–562.
Higgs MR, Reynolds JJ, Winczura A, Blackford AN, Borel V, Miller ES et al. BOD1L is required to suppress deleterious resection of stressed replication forks. Mol Cell 2015; 59: 462–477.
Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL et al. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 2010; 467: 112–116.
Zhou C, Pourmal S, Pavletich NP . Dna2 nuclease-helicase structure, mechanism and regulation by Rpa. Elife 2015; 4: e09832.
Dobbelstein M, Sorensen CS . Exploiting replicative stress to treat cancer. Nat Rev Drug Discov 2015; 14: 405–423.
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA . DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008; 8: 193–204.
Kopper F, Bierwirth C, Schon M, Kunze M, Elvers I, Kranz D et al. Damage-induced DNA replication stalling relies on MAPK-activated protein kinase 2 activity. Proc Natl Acad Sci USA 2013; 110: 16856–16861.
Acknowledgements
We thank Hyungjin Kim (Stony Brook University) for the pcDNA_FancA construct, Gary Kupfer (Yale University) for the pMMP_FancA construct and Aziz Sancar for the pcDNA4-Chk1-flag plasmid. We thank Hannes Landmann and Cathrin Bierwirth for their assistance in performing cell culture experiments. Our work was supported by the EU 7th Framework project Gannet53, by NCI 1R01CA176647 to UMM, and by the Wilhelm Sander-Stiftung 2011.029.1 to UMM.
Author contributions
DK, UMM and MD conceived the project and designed the experiments. MD and DK drafted the manuscript. DK, UMM and NC revised the manuscript. DK, NS and LR conducted the in vitro experiments, NS, SE and RS-H conducted the xenograft experiments. NE performed the live-cell imaging analysis. HB, NC, UMM and MD supervised the experiments. All the authors read and approved the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by S Fulda
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
About this article

Cite this article
Kramer, D., Stark, N., Schulz-Heddergott, R. et al. Strong antitumor synergy between DNA crosslinking and HSP90 inhibition causes massive premitotic DNA fragmentation in ovarian cancer cells. Cell Death Differ 24, 300–316 (2017). https://doi.org/10.1038/cdd.2016.124
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.124
This article is cited by
-
Ganetespib selectively sensitizes cancer cells for proximal and distal spread-out Bragg peak proton irradiation
Radiation Oncology (2022)
-
The HSP90 inhibitor Onalespib exerts synergistic anti-cancer effects when combined with radiotherapy: an in vitro and in vivo approach
Scientific Reports (2020)
-
USP22-dependent HSP90AB1 expression promotes resistance to HSP90 inhibition in mammary and colorectal cancer
Cell Death & Disease (2019)
-
Cisplatin based therapy: the role of the mitogen activated protein kinase signaling pathway
Journal of Translational Medicine (2018)

{kind=link}
{kind=link}