
Abstract
Reactive oxygen species (ROS) may cause cellular damage and oxidative stress-induced cell death. Autophagy, an evolutionarily conserved intracellular catabolic process, is executed by autophagy (ATG) proteins, including the autophagy initiation kinase Unc-51-like kinase (ULK1)/ATG1. Although autophagy has been implicated to have both cytoprotective and cytotoxic roles in the response to ROS, the role of individual ATG proteins, including ULK1, remains poorly characterized. In this study, we demonstrate that ULK1 sensitizes cells to necrotic cell death induced by hydrogen peroxide (H2O2). Moreover, we demonstrate that ULK1 localizes to the nucleus and regulates the activity of the DNA damage repair protein poly (ADP-ribose) polymerase 1 (PARP1) in a kinase-dependent manner. By enhancing PARP1 activity, ULK1 contributes to ATP depletion and death of H2O2-treated cells. Our study provides the first evidence of an autophagy-independent prodeath role for nuclear ULK1 in response to ROS-induced damage. On the basis of our data, we propose that the subcellular distribution of ULK1 has an important role in deciding whether a cell lives or dies on exposure to adverse environmental or intracellular conditions.
Similar content being viewed by others


Main
Reactive oxygen species (ROS), such as superoxide and hydrogen peroxide (H2O2), are formed by the incomplete reduction of oxygen during oxidative phosphorylation and other enzymatic processes. ROS are signaling molecules that regulate cell proliferation, differentiation, and survival.1, 2, 3 Accumulation of ROS (i.e., oxidative stress) on exposure to xenobiotic agents or environmental toxins can cause cellular damage and death via apoptotic or nonapoptotic pathways.4, 5, 6 Oxidative stress-induced cellular damage and death have been implicated in aging, ischemia-reperfusion injury, inflammation, and the pathogenesis of diseases (e.g., neurodegeneration and cancer).7 Oxidative stress also contributes to the antitumor effects of many chemotherapeutic drugs, including camptothecin8, 9 and selenite.10, 11
Autophagy, an evolutionarily conserved intracellular catabolic process, involves lysosome-dependent degradation of superfluous and damaged cytosolic organelles and proteins.12 It is typically upregulated under conditions of perceived stress and in response to cellular damage. The consequence of autophagy activation – whether cytoprotective or cytotoxic – appears to depend on the cell type and the nature and extent of stress. Although most studies indicate a cytoprotective role for autophagy, some evidence suggests that it contributes to cell death in response to oxidative stress.13, 14, 15, 16, 17 Studies have also indicated that autophagy may be suppressed in response to oxidative stress, thereby sensitizing certain cells to apoptosis.18, 19 Unc-51-like kinase/autophagy 1 (ULK1/ATG1) is a mammalian serine–threonine kinase that regulates flux through the autophagy pathway by activating the VPS34 PI(3) kinase complex and facilitating ATG9-dependent membrane recycling.20 Results from two studies suggest that ULK1 expression is altered in response to oxidative stress, and that the corresponding effects on autophagy contribute to cell death.18, 21For example, p53-mediated upregulation of ULK1 and increase in autophagy promote cell death in osteosarcoma cells exposed to sublethal doses of camptothecin,21 yet mutant p53-mediated suppression of ULK1 impairs autophagic flux and promotes apoptosis in selenite-treated NB4 cells.18 Here we investigated the role of ULK1 in cells exposed to H2O2.
Results
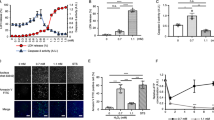
ULK1 facilitates nonapoptotic cell death after H2O2 treatment
To study the role of ULK1 following ROS-induced cellular damage, we treated wild-type (WT) and Ulk1-knockout (KO), SV40-transformed murine embryonic fibroblasts (MEFs) with H2O2, a diffusible nonradical oxidant that has been extensively used to study ROS damage in vitro.22 Trypan blue-exclusion assays revealed a significant (P<0.01) delay in cell death in Ulk1-KO MEFs (Figure 1a). Overexpression of full-length WT ULK1 in Ulk1-KO MEFs, at levels comparable to those used to rescue their defect in autophagy,23 restored sensitivity to H2O2-induced cell death (Figure 1b). Similarly, overexpression of ULK1 in 293 T cells, which have low endogenous levels of ULK1, increased sensitivity to H2O2-induced cell death (Figure 1c). These results suggest a prodeath role for ULK1 in response to oxidative stress. Reverse-transcription (RT)-qPCR analyses indicated a transient decline in steady-state levels of Ulk1 mRNA in H2O2-treated MEFs (Figure 1d); however, ULK1 protein levels were unchanged (Figures 1e and f).

ULK1 sensitizes cells to H2O2-induced cell death. (a) WT (Ulk1+/+) and Ulk1-deficient (Ulk1−/−) MEFs were treated for up to 12 h with 500 μM H2O2. Trypan blue-exclusion assays demonstrate a delay in H2O2-induced cell death in Ulk1−/− MEFs compared with WT MEFs. The graph shows the mean (±S.D.) percentage of live cells. (b) Trypan blue-exclusion assays demonstrate a reduction in survival of Ulk1−/− MEFs stably reconstituted with the ULK1 (WT) expression vector compared with those reconstituted with the empty vector after treatment for 6 h with 500 μM H2O2. The graph shows the mean (±S.D.) percentage of live cells. Representative immunoblots show the levels of ULK1 and ACTB expression. (c) Trypan blue-exclusion assays demonstrate that transient overexpression of ULK1 in 293T cells accelerates cell death induced by H2O2 (500 μM). The graph shows the mean (±S.D.) percentage of dead cells. Representative immunoblots show the levels of ULK1 and ACTB expression. (d) Total RNA collected from cells at various time points after H2O2 treatment was analyzed by RT-qPCR analyses. Graphs show relative Ulk1 mRNA expression (calibrated to 18 S and normalized to levels in untreated MEFs) (mean±S.E.M.). (e–f) Whole-cell extracts prepared from Ulk1+/+ and Ulk1−/− MEFs treated with 500 μM H2O2 for the indicated time periods (in hours) or with 1 μM staurosporine (St) for 6 h were subjected to immunoblot analyses. Representative immunoblots and graph (mean±S.D.) demonstrate that H2O2 treatment does not significantly change ULK1 protein levels or promote cleavage of caspase 3 and PARP1. (g) Caspase 3/7 activity was evaluated at the indicated time points (in hours) after treating Ulk1+/+ and/or Ulk1−/− MEFs with 500 μM H2O2 or 1 μM staurosporine (St). Graphs show the mean (±S.D.). (h) Live-cell imaging of Ulk1+/+ and Ulk1−/− MEFs stained with Hoechst, SYTOX green, and Annexin-V was performed using a × 40 objective after treating the cells with 500 μM H2O2 or 1 μM staurosporine (St). The graph shows the percentage of SYTOX+ and Annexin V+ cells at each time point (n>50 cells per condition) after H2O2 treatment. A corresponding graph of WT MEFs treated with St is shown in Supplementary Figure S1b and representative images are shown in Supplementary Figure S1c. (i) Flow cytometry was performed on MEFs treated with 500 μM H2O2 for 6 h or 1 μM St for 12 h before staining with Annexin V-FITC/PI. Early-stage apoptotic cells: lower-right quadrant (Annexin V+/PI−); necrotic cells: upper-left quadrant (Annexin V−/PI+). Representative FACS plots generated from analysis of H2O2-treated cells are shown in i; corresponding FACS plots generated from analysis of untreated cells and those treated with St are shown in Supplementary Figure S1d and e, respectively. (j) Trypan blue-exclusion assays performed on Ulk1+/+ and Ulk1−/− MEFs treated with 20 μM ZVAD in the presence or the absence of H2O2 demonstrate that caspase inhibition does not protect against H2O2-induced cell death in MEFs. Graph shows the mean (±S.D.) percentage of live cells. *P≤0.01. n=3 biologic replicates per condition, unless otherwise indicated
H2O2 treatment did not induce cleavage of apoptotic caspases or poly (ADP-ribose) polymerase 1 (PARP1) in MEFs (Figures 1e and g).Conversely, these hallmarks of apoptosis were detected in WT and Ulk1-KO MEFs treated with staurosporine, a proapoptotic protein kinase inhibitor (Figures 1e and g, and Supplementary Figure S1a). Staurosporine also reduced steady-state levels of ULK1 in WT MEFs (Figure 1e and Supplementary Figure S1a). Imaging of live cells incubated in the presence of Annexin V (AlexaFluor-555 conjugate) and SYTOX green, and flow cytometric analyses of cells stained with propidium iodide (PI) and Annexin V were performed to further distinguish between apoptotic and nonapoptotic cell death induced by H2O2. Annexin V provides a quick, reliable method for detecting the externalization of phosphatidylserine that occurs during apoptosis; SYTOX green and PI are nucleic acid stains that are impermeant to live cells but penetrate the compromised membranes of dead cells. Consistent with the results of the trypan blue assays, live-cell imaging and flow cytometry of Ulk1-KO MEFs showed a delay in the accumulation of SYTOX green+ (Figure 1h and Supplementary Figure S1c) and PI+ dead cells (Figure 1i and Supplementary Figure S1d), respectively. Contrary to the staurosporine treatment, the H2O2 treatment resulted in no intact (i.e., SYTOX green– or PI–) Annexin V+ cells (Figures 1h–i and Supplementary Figure S1b–e). Finally, treatment of WT or Ulk1-KO MEFs with the pan-caspase inhibitor z-VAD did not improve viability on exposure to H2O2, instead it potentiated cell death (Figure 1j). These findings indicate that ULK1 promotes nonapoptotic cell death in H2O2-treated MEFs.
ULK1-mediated cell death in response to H2O2 occurs in an autophagy-independent manner
Next, we sought to determine the contribution of autophagy to H2O2-induced cell death in MEFs. We examined levels of lipidated LC3 (i.e., LC3II) and P62 by immunoblot analyses in the presence or the absence of the lysosomal inhibitor bafilomycin A1. LC3 is a ubiquitin-like molecule that is conjugated to phosphatidylethanolamine (i.e., lipidated) on induction of autophagy and undergoes lysosomal degradation as autophagosomes mature.24 P62 is a ubiquitin-binding scaffold protein that is degraded by autophagy and accumulates when autophagy is inhibited.24 H2O2 treatment increased LC3II, but adding bafilomycin A did not enhance that effect; P62 levels were also unchanged (Figures 2a–c). These findings suggest that H2O2 mildly impairs autophagic flux in MEFs. We used a quantitative, reproducible firefly luciferase-based assay, which specifically exploits the autophagy-dependent turnover of LC3b, to measure flux through the autophagy pathway.23 Unlike the ULK1-dependent LC3 degradation induced by starvation (Figure 2e),23 H2O2 did not stimulate autophagy-mediated LC3 degradation in either WT or Ulk1-KO MEFs (Figure 2d).

ULK1-mediated cell death occurs in an autophagy-independent manner. (a–c) WT MEFs were incubated in complete media (CM), CM with 500 μM H2O2, or EBSS for 2 h in the absence or the presence of Bafilomycin A1 (BafA1). Extracts prepared from Atg7-deficient (Atg7−/−) cells were included as a control in the representative immunoblots. The graphs show relative LC3II (b) and p62 (c) levels (normalized to ACTB) (mean±S.D.). (d and e) Flux through the autophagy pathway was monitored using a luciferase-LC3-based reporter that quantifies the rate of autophagy-mediated LC3 turnover, as described previously.23 The assays were performed with WT (Ulk1+/+) and Ulk1-deficient (Ulk1−/−) MEFs stably expressing Luc-LC3 (LC3) or the control construct luciferase-LC3-G120A (G120A), which cannot be modified by PE and, therefore, cannot be degraded by the autophagy pathway. Paired WT and Ulk1-deficient MEFs stably expressing Luc-LC3 or Luc-G120A were treated with vehicle or 500 μM H2O2 in CM for the indicated times (d). Paired WT and Ulk1-deficient MEFs stably expressing Luc-LC3 or Luc-G120A were transfected with nontargeting (Ctrl) or Atg7 siRNA for 48 h, washed twice in either prewarmed CM (used for normalization of data) or EBSS, and then incubated in the respective media for the indicated times (e). Data are represented as the ratio of Luc-LC3 to Luc-G120A (mean±S.D.). Autophagy-mediated degradation of Luc-LC3 is observed in amino acid-deprived cells but not in cells treated with H2O2. (f) WT (Ulk1+/+) and Ulk1-deficient (Ulk1−/−) MEFs were transfected with nontargeting (Ctrl) or Atg7 siRNA for 48 h before incubation with or without 500 μM H2O2 in CM for 6 h. Trypan blue-exclusion assays show a mild increase in death of H2O2-treated cells after Atg7 silencing. The levels of Atg7 in Ctrl- and Atg7-siRNA-transfected MEFs are shown in Supplementary Figure S1. Graph shows the mean (±S.D.) percentage of live cells. (g) WT and Ulk1-deficient MEFs were transfected with nontargeting (Ctrl) or Atg7-siRNA for 48 h before incubation with CM or EBSS for 24 h. Trypan blue-exclusion assays show comparable levels of starvation-induced cell death in Ulk1-deficient and/or Atg7-silenced MEFs. Graph shows the mean (±S.D.) percentage of dead cells. (h) Trypan blue-exclusion assays show increased cell death in Atg7-deficient (Atg7−/−) MEFs. Graph shows the mean (±S.D.) percentage of dead cells. (i) Representative transmission electron micrographs of WT and Ulk1-deficient MEFs treated with H2O2 for 5 h. Morphologic changes in WT cells include segregation of chromatin at the periphery of the nucleus (N), swelling of the mitochondria (M) and endoplasmic reticulum (ER), the presence of inclusions within the mitochondria, and disruption of the the plasma membrane. Although Ulk1-deficient MEFs show mitochondrial changes similar to those in WT MEFs, their nuclei remain intact with prominent nucleoli and a normal chromatin distribution pattern. Autophagosomes were rare in WT and Ulk1 KO samples at 2 h (not shown) and 5 h. n=3 biologic replicates per condition, unless otherwise indicated. *P<0.01
We then examined the contribution of autophagy to cell viability in H2O2-treated WT and Ulk1-deficient MEFs. Atg7 is an E1-like ligase that promotes elongation of autophagosome membranes by facilitating the conjugation of ATG5 to ATG12 and that of LC3 to phosphatidylethanolamine.25 The level of RNAi-mediated knockdown of Atg7, confirmed by RT-qPCR (Supplementary Figure S2), impaired starvation-induced LC3 degradation in WT MEFs, although not to the same level as in Ulk1-deficient MEFs (Figure 2e), and increased susceptibility to cell death after starvation comparable with that observed in Ulk1-deficient MEFs (Figure 2g).26, 27 Atg7 knockdown did not protect against H2O2-induced cell death in WT or Ulk1-KO MEFs (Figure 2f). Instead, MEFs with Atg7 knockdown or KO were more sensitive to H2O2 treatment than were WT MEFs (Figures 1f and h). Thus, the minimal levels of H2O2-induced autophagy may have a cytoprotective role, and the cytotoxic effects of ULK1 are unlikely to be due to altered flux through the canonical (ATG7-dependent) autophagy pathway.
As ULK1 has been implicated in an ATG7-independent autophagy,28 we examined H2O2-treated WT and Ulk1-deficient MEFs by transmission electron microscopy. WT MEFs treated with H2O2 for 5 h had abnormal nuclear morphology, with chromatin segregated to the periphery and mitochondria that were swollen and contained small, electron-dense inclusions (Figure 2i). Necrotic cells with disrupted plasma membranes were also frequent in the WT sample (Figure 2i) but not in Ulk1-KO samples (Figure 2i). Although H2O2-treated Ulk1-KO MEFs showed mitochondrial abnormalities similar to those in intact WT MEFs, their nuclei were more similar to those of untreated MEFs (Figure 2i). Autophagosomes and autophagolysosomes were rare in WT and Ulk1-KO samples at 2 and 5 h after H2O2 treatment. These findings indicate that the H2O2-induced ULK1-dependent cell death is not caused by excessive induction of autophagy or impaired autophagosome maturation; it occurs via an autophagy-independent mechanism.
ULK1 interacts with PARP1
To elucidate the mechanism of ULK1-dependent cell death, we immunoprecipitated flag-tagged ULK1 from whole-cell extracts derived from 293T cells and identified interacting partners by liquid chromatography–tandem mass spectrometry (MS/MS). PARP1, a nuclear enzyme involved in DNA repair and cell death,29 was among the ULK1-interacting partners (Figure 3a and Supplementary Tables S1 and S2). The ULK1–PARP1 interaction was confirmed by immunoblot detection of endogenous PARP1 in flag-ULK1 (Figure 3b) and GFP-ULK1 immunoprecipitates (Supplementary Figure S3), and that of overexpressed ULK1 from GFP-PARP immunoprecipitates in 293T cells (Figure 3c). ULK1 shares a significant homology with ULK2, a related ATG1 family member that has also been implicated in autophagy.30 Although PARP1 interacted with ULK2 (data not shown), we could not confirm this interaction using green fluorescent protein (GFP)-tagged ULK2 (Supplementary Figure S3); therefore, subsequent studies focused on the function of the PARP1–ULK1 interaction.

ULK1 interacts with PARP1. (a) PARP1 is identified as a putative ULK1-interacting partner by using an unbiased proteomics-based approach. Proteins significantly (P<0.05) enriched in the flag-ULK1 immunoprecipitates compared with control immunoprecipitates (Table 1) were submitted to ConsensusPathDB for induced network-module gene-set analysis to visualize previously described relationships among the proteins. Interactions between ULK1 and the HSP90 chaperone complex, AMPK subunits, 14-3-3, and RB1CC1 have been previously validated.23, 27(b) Whole-cell extracts (WCEs) and anti-flag immunoprecipitates from 293T cells transfected with Flag-GFP or Flag-ULK1 were subjected to immunoblot analyses by using antibodies against the indicated proteins (PARP1, ULK1, and GFP). PARP1 was detected in the Flag-ULK1 immunoprecipitates but not in Flag-GFP immunoprecipitates. (c) WCEs and anti-GFP immunoprecipitates from 293 T cells cotransfected with Flag-ULK1 and either GFP or PARP1-GFP were subjected to immunoblot analyses by using antibodies against the indicated proteins. ULK1 was more abundant in the PARP1-GFP immunoprecipitates than in the GFP immunoprecipitates. (d) Nuclear (N) and cytosolic (C) fractions were prepared from Ulk1+/+ and Ulk1−/− MEFs and subjected to immunoblot analyses using antibodies against PARP1, GAPDH, and ULK1. ULK1 immunoreactivity in the nuclear-enriched fraction shows the presence of ULK1 in the nucleus. (e) Nuclear fractions prepared from Ulk1+/+ and Ulk1−/− MEFs were subjected to IP with an anti-ULK1 antibody (N17, Santa Cruz) in the presence or the absence of an antibody-specific blocking peptide. Endogenous PARP1 immunoprecipitated with endogenous ULK1 from nuclear extracts prepared from WT MEFs
ULK1 presumably functions primarily in the cytosol. However, PARP1 is almost exclusively located in the nucleus.31 Therefore, we examined the subcellular distribution of endogenous ULK1 in MEFs. Immunoblot analyses showed that although ULK1 is more abundant in the cytoplasm, it is also present in the nucleus (Figure 3d). We then immunoprecipitated ULK1 from nuclear extracts and assessed the presence of endogenous PARP1. Immunoblot analyses confirmed the ULK1–PARP1 interaction in nuclear extracts (Figure 3e).
PARP1 is the primary source of PAR-modified proteins and cell death in H2O2-treated MEFs
On activation by DNA-strand breaks, PARP1 transfers 50–200 branched chains of ADP-ribose to various nuclear proteins, including PARP1, which transiently increases PAR-modified (PARylated) proteins.32 Although PARP1 participates in the DNA repair process, excessive DNA damage results in PARP1 hyperactivation, ATP depletion, and cell death from bioenergetic collapse.33 Consistent with PARP1 being the major source of PAR polymers in H2O2-treated MEFs, the increase in PARylated proteins after H2O2 treatment was abolished with PARP1 knockdown (Figure 4a). PARP1 knockdown also inhibited cell death in WT MEFs (Figures 4b and c). PARP1 hyperactivation associated with H2O2 treatment decreases intracellular ATP levels in WT MEFs.33 The decline in cellular ATP after H2O2 treatment was attenuated in WT MEFs pretreated with the PARP inhibitor 3,4-dihydro-5[4-(1-piperindinyl)butoxy]-1(2H)-isoquinoline (DPQ) and in Ulk1-KO MEFs (Figure 4d). Thus, PARP1 is central to the bioenergetics crisis and cell death resulting from H2O2 treatment.

PARP1 is a major source of PAR formation and cell death. (a) WT MEFs transfected with nontargeting (Ctrl) or PARP1 siRNA for 48 h were treated with 500 μM H2O2 for 10 min. PARP activation was measured by immunoblot analyses using an anti-PAR antibody. PAR-polymer formation was decreased with PARP1 knockdown. (b) WT MEFs transfected with nontargeting (Ctrl) or PARP1 siRNA and Ulk1−/− MEFs transfected with Ctrl siRNA were treated with 500 μM H2O2 for 6 h. The percentage of live cells was determined by performing the trypan blue-exclusion assay. The protection conferred by the lack of ULK1 in MEFs was comparable to that after PARP1 silencing. (c) Immunoblot analyses demonstrate the efficiency of PARP1 knockdown in WT MEFs. GAPDH was used as a loading control. (d) ATP levels were measured in WT or Ulk1−/− MEFs treated with H2O2 in the presence or the absence of 40 nM 3,4-dihydro-5[4-(1-piperindinyl)butoxy]-1(2H)-isoquinoline (DPQ), as indicated. The experiments were performed in triplicate. *P<0.001
ULK1 regulates PARP1 activity
PARP1 localizes to the nucleoli in live or mildly fixed cells and redistributes to the nucleoplasm on excessive DNA damage.34, 35 To gain insight into the functional consequence of the ULK1–PARP1 interaction, we monitored GFP-labeled PARP1 (PARP1-GFP) localization in WT and Ulk1-KO MEFs subjected to H2O2 or ultraviolet (UV) radiation, both of which damage DNA.36, 37 As expected, transiently transfected PARP1-GFP was concentrated in the nucleoli of WT MEFs under normal conditions and migrated to the nucleoplasm after exposure to H2O2 (Figures 5a and b) or UV radiation (Figure 5b). Although the DNA damage was comparable in UV-irradiated WT and Ulk1-KO MEFs, as suggested by the similar levels of γH2AX (a biomarker of DNA damage)38 up to 4 h after treatment (Supplementary Figure S4), PARP1-GFP was retained in the nucleoli of Ulk1-KO MEFs exposed to UV radiation or H2O2 (Figures 5a and b).

ULK1 facilitates PARP1 redistribution and activation. (a) Localization of PARP1 was monitored in Ulk1+/+ and Ulk1−/− MEFs transfected with PARP1-GFP with or without exposure to 500 μM H2O2 for 1 h. After H2O2 exposure, PARP1 was depleted from the nucleoli in ULK1 WT MEFs but not in the Ulk1-KO MEFs. Representative images are shown. Scale bars=10 μM. (b) The percentage of cells showing nucleolar PARP1 was quantified in Ulk1+/+ and Ulk1−/− MEFs transfected with PARP1-GFP for 1 h after exposure to UV radiation or treatment with 500 μM H2O2 (n>100 cells per condition from three independent experiments). (c and d) Representative immunoblots and graphs (mean±S.D.) show the levels of PARylated proteins in Ulk1+/+ and Ulk1−/− MEFs treated with 500 μM H2O2 for the indicated time periods. (e and f) Ulk1+/+ and Ulk1−/−MEFs, and Ulk1−/−MEFs stably expressing ULK1 were incubated with or without 500 μM H2O2 for 10 min before preparation of extracts for immunoblot detection of PARylated proteins. Representative immunoblots and graphs (mean±S.D.) show the levels of PARylated proteins. Each condition was replicated in triplicate, unless otherwise indicated. *P<0.001
Next, we performed fluorescence recovery after photobleaching experiments to determine whether PARP1 mobility was altered by ULK1. Neither the rate of fluorescence recovery nor the percent recovery after photobleaching nucleoli (Supplementary Figures S5a–c) or nucleoplasm (data not shown) differed between WT and Ulk1-deficient MEFs, suggesting that the impaired PARP1 redistribution in H2O2-treated Ulk1-KO MEFs was not caused by altered mobility. On laser-induced DNA damage in Ulk1-KO MEFs, PARP1 mobilized (Supplementary Figure S5d).
A recent study demonstrated that PARP1 activation is required for DNA damage-induced suppression of rRNA synthesis within the nucleoli and relocation of PARP1 into the nucleoplasm.39 Therefore, we assessed PARP1 activation by measuring the levels of PARylated proteins in H2O2-treated WT and Ulk1-KO MEFs. The level of PARylated proteins in Ulk1-KO MEFs was lower than that in WT MEFs (Figures 5c and d), consistent with reduced PARP1 activation in the absence of ULK1. Reconstitution of ULK1 in Ulk1-deficient MEFs resulted in higher levels of PARylated proteins than those in WT MEFs at baseline and after H2O2 treatment (Figures 5e and f), and restored H2O2 treatment-induced ATP depletion in Ulk1-deficient MEFs (Figure 5g). Thus, the retention of PARP1 within the nucleoli of Ulk1-KO MEFs subjected to H2O2 treatment or UV irradiation is probably caused by reduced PARP1 activation resulting from the absence of ULK1.
ULK1 activity is required for nuclear localization and interaction with PARP1
To characterize the determinants of ULK1 nuclear localization, we examined the subcellular localization of overexpressed ULK1 by immunofluorescence confocal microscopy. Although GFP-ULK1 (Supplementary Figure S6a) and untagged ULK1 (Figure 6a) were more abundant in the cytosol than in the nuclei in most MEFs and 293T cells, occasional cells showed clear nuclear localization (Figure 6a). The proportion of cells with prominent nuclear localization increased from 1% to nearly 30% within 30 min after H2O2 treatment in MEFs (Supplementary Figure S6a) and 293T cells (Figure 6b). Fractionation studies confirmed the increased nuclear localization of ULK1 after H2O2 treatment (Figure 6d), which was accompanied by increased ULK1 (and ATG13) level in PARP1 immunoprecipitates by 30 min (Supplementary Figure S6b). ULK1 facilitated the translocation of ATG13 into the nucleus in the presence or absence of H2O2 (Supplementary Figure S6c). These data suggest that low levels of ULK1 are present in the nuclei of cells under normal growth conditions, and that H2O2 increases the levels of nuclear ULK1 in a subset of cells. Moreover, the ULK1-dependent increase in nuclear localization of ATG13 after H2O2 treatment may explain the presence of endogenous ATG13 detected in PARP1-GFP immunoprecipitates derived from H2O2-treated cells (Supplementary Figure S6b).

ULK1 kinase activity is required for nuclear localization of ULK1 and its interaction with PARP1. (a and b) The 293T cells cotransfected with PARP1-GFP and untagged WT ULK1 were incubated in complete media (CM) in the absence (a) or the presence (b) of H2O2 for 30 min. The percentage of cells showing clear nuclear localization of ULK1 increased from 1% to nearly 30% after H2O2 treatment. Representative images and line scans indicate the degree of colocalization between PARP1-GFP and ULK1 in cells treated with or without H2O2. (c) The 293T cells cotransfected with PARP1-GFP and the nuclear localization-defective R266A-ULK1 mutant were treated for 30 min with 500 μM H2O2 before imaging. Representative images and a line scan show the degree of colocalization of R266A-ULK1 and PARP1-GFP, highlighting the nuclear localization defect of the R266A mutant. Scale bars=10 μM. (d) Representative immunoblots of nuclear and cytoplasmic fractions prepared from 293T cells transfected with WT or mutant ULK1 (R266A, KI, or 4SA) and incubated in CM with or without 500 μM H2O2 for 30 min. The levels of WT ULK1 and the 4SA mutant in nuclear fractions increase after H2O2 treatment, but those of the KI and R66A mutants do not. (e) Whole-cell extracts prepared from 293 T cells cotransfected with PARP1-GFP and untagged WT or mutant (KI, R266A, or 4SA) ULK1 were subjected to IP using anti-GFP-conjugated beads before SDS-PAGE and immunoblot analyses. Immunoblot analyses demonstrate a decrease in PARylated proteins in cells transfected with the R266A mutant. (f) The 293T cells were transfected with HA-tagged ATG13 and the indicated ULK1 expression constructs. Whole-cell extracts were analyzed by SDS-PAGE followed by immunoblot analyses. ATG13 phosphorylated at S318 was detected only in lanes containing WT ULK1 or the AMPK-resistant mutant (4SA)
Next, we introduced point mutations into a predicted importin α-dependent nuclear-localization signal (NLS) in murine ULK1 (aa261–289) that was identified using the cNLS Mapper (Supplementary Figure S7a).40 Individual arginine (R) and lysine (K) residues within the 29-residue NLS were replaced with alanines (A) and the resulting mutants were screened for their ability to disrupt ULK1’s nuclear localization. Immunofluorescence microscopy of transiently transfected 293T cells revealed that unlike WT ULK1, the R266A-ULK1 mutant was excluded from the nuclei, even after H2O2 treatment, and did not alter nuclear localization of PARP1-GFP (Figure 6c). Confocal microscopic assessment of the nuclear localization of kinase-inactive (KI, K46A)23 and AMPK-resistant (4SA)27 ULK1 mutants revealed that the KI-ULK1 mutant was excluded from the nuclei, similar to the R266A mutant; however, the 4SA mutant behaved in a manner similar to WT ULK1 (Supplementary Figure S7b).
Fractionation studies showed a marked reduction in the amount of R266A and KI-ULK1 mutants in the nuclear fractions compared with that of the 4SA mutant or WT ULK1; the levels remained unchanged after H2O2 treatment (Figure 6d). Consistent with having defective nuclear localization, the KI mutant, which still interacts with ATG13,23 failed to promote ATG13’s translocation into the nucleus to the same extent as WT ULK1 did (Supplementary Figure S6c). In addition to defective nuclear localization, or perhaps as a consequence of it, the R266A and KI mutants were not efficiently incorporated into PARP1-GFP immunoprecipitates (Figure 6e).
Immunoblot analyses (Figures 6d and e) also revealed that the electrophoretic mobility of the R266A-ULK1 mutant was comparable to that of the KI mutant; both migrated faster than WT ULK1 and the 4SA mutant. The structure of ULK1 crystals demonstrates that the R266 residue interacts with the glutamic acid (E) at 191 via a buried salt bridge (Supplementary Figures S8a–c).41 Both residues are conserved among closely related kinases (Supplementary Figure S8b) and the R266A mutation may alter folding of the kinase domain and impair ULK1 catalytic activity.42 ATG13 is phosphorylated at serine (S) 318 in a ULK1-dependent manner. Therefore, to test the effect of the R266A mutation on ULK1’s catalytic activity, we cotransfected 293T cells with individual ULK1 constructs (WT, R266A, KI, and 4SA) and ATG13, and then examined phosphorylated S318 levels by immunoblot analyses. Although phosphorylated ATG13 was detected in cells expressing WT ULK1 or the 4SA mutant, it was not detected in cells expressing the KI or the R266A-ULK1 mutants (Figure 6f). Thus, ULK1’s catalytic activity is required for its localization to the nucleus and interaction with PARP1.
ULK1 activity is required for ULK1-mediated PARP1 activation
To assess the importance of ULK1’s activity and nuclear localization on the regulation of PARP1 activity, we examined PARP1’s localization and activity in Ulk1-deficient MEFs stably expressing either WT ULK1 or the KI mutant. PARP1 localization experiments were performed using Ulk1-KO MEFs with stable Ulk2 knockdown;27 these cells were prepared without using GFP, thereby enabling us to detect transiently transfected PARP1-GFP by confocal microscopy. PARP1-GFP was rapidly depleted from the nucleoli of Ulk1-KO/Ulk2 shRNA MEFs stably expressing WT ULK1 on treatment with H2O2; only 15% of cells retained nucleolar localization of PARP1 after 20 min. Conversely, PARP1-GFP persisted in the nucleoli of more than 90% of Ulk1-KO/Ulk2 shRNA MEFs and those expressing KI ULK1 (Figure 7a) for more than 1 h posttreatment with H2O2. Ulk1-KO/Ulk2 shRNA and Ulk1-deficient MEFs expressing the KI-ULK1 mutant had lower levels of PARylated proteins after H2O2 treatment than did those expressing WT ULK1 (Figures 7b and c). WT ULK1 in Ulk1-KO/Ulk2 shRNA and Ulk1-deficient MEFs restored sensitivity to H2O2-induced cell death, whereas expression of the KI mutant did not (Figures 7d and e). Similar results were obtained in 293T cells transfected with either WT ULK1 or the R266A mutant (Figures 7f and h). Immunoblot analyses demonstrated lower levels of PARylated proteins in 293T cells expressing the ULK1 R266A mutant than in those expressing comparable levels of WT ULK1 (Figure 7f); ectopic expression of WT ULK1 but not that of the R266A mutant sensitized 293T cells to H2O2-induced ATP depletion (Figure 7g) and cell death (Figure 7h). Therefore, kinase-dependent localization of ULK1 to the nucleus and ULK1 interaction with PARP1 are required for PARP1 hyperactivation, ATP depletion, and cell death in H2O2-treated cells.

ULK1 kinase activity promotes PARP1 activation and sensitizes cells to oxidative stress-induced death. (a) Ulk1 KO/Ulk2 shRNA MEFs27 stably expressing vector control, WT ULK1, or KI ULK1 were cotransfected with a vector containing PARP1-GFP and incubated in complete media (CM) with or without H2O2 for 20 min. PARP1 was depleted from the nucleoli of cells expressing WT ULK1 but not from those of cells expressing the KI mutant. Scale bars=10 μM. Representative immunoblots (b) and a graph (c) show the levels of PARylated proteins in the indicated MEF lines incubated in the presence or the absence of 500 μM H2O2 for 10 min. (d and e) Trypan blue-exclusion assays demonstrate that the KI-ULK1 mutant fails to promote cell death to the same extent as WT ULK1 does when stably expressed in Ulk1-deficient MEFs. (f) The 293T cells cotransfected with PARP1-GFP and either WT ULK1 or the R266A-ULK1 mutant were treated for 10 min with 100 μM H2O2. Immunoblot analyses demonstrate a decrease in PARylated proteins in cells transfected with the R266A mutant. (g) The 293T cells transfected with the indicated expression constructs were treated with 100 μM H2O2. ATP levels were analyzed in cells collected 10, 30, or 60 min after treatment with 100 μM H2O2 and normalized to levels in untreated cells. (h) Trypan blue-exclusion assays performed 6 h after treatment with the indicated dose of H2O2 demonstrate increased viability of 293T transfected with the R266A-ULK1 mutant expression construct compared with that of those transfected with the WT ULK1 construct. (i) The 293T cells were transfected with empty vector (−), WT ULK1 (WT), or the following mutant forms of ULK1: K46A (kinase-defective mutant), 4SA (AMPK-resistant, autophagy-defective mutant), or R266A (NLS mutant). Transfected cells were treated with 4.5 μM β-lapachone for 72 h and then cell viability was assessed using CellTiter-Glo assays. Signals were normalized to those in DMSO-treated controls (n=4 biologic replicates per condition), P<0.001. (j) The 293T cells transfected with the indicated expression vectors were arrayed in a 96-well format and treated with either camptothecin for 72 h (starting at a concentration equal to 7 mM followed by threefold dilutions) or DMSO (negative control). Signals (in relative light units, RLUs) from the CellTiter-Glo assays were normalized by subtracting the median RLU of the DMSO control wells and then fit to the four-parameter log-logistic function (‘Hill equation’) to yield ‘activity.’ EC50: log concentration at half-maximal observed response. The results demonstrate an eightfold difference between the EC50 of WT ULK1 and that of the R266A-ULK1 mutant. (k) Representative immunoblot showing the levels of ULK1 in cells transfected with the indicated constructs and used for the experiments described in i and j (n=3 biologic replicates per condition, unless otherwise indicated)
Finally, to determine whether the effect of ULK1 was specific to H2O2-induced cell death, we assessed the viability of 293T cells transiently overexpressing WT ULK1 or one of several ULK1 mutants before treatment with β-lapachone or camptothecin. The orthonapthoquinone β-lapachone inhibits the catalytic activity of DNA topoisomerase 1, thereby causing PARP1 hyperactivation.43, 44, 45 The efficacy of β-lapachone as an anticancer agent is being evaluated in multiple Phase II trials (http://www.cancer.gov/clinicaltrials). The AMPK-resistant 4SA mutation sensitized cells to a sublethal dose of β-lapachone to the same extent as WT ULK1 did. Conversely, the KI and R266A mutants did not sensitize the cells (Figures 7i and k). Thus, we observed good correlation between nuclear localization and ULK1 sensitization to β-lapachone-induced cell death. Similar responses were observed in cells treated with camptothecin (Figures 7j and k, and Supplementary Figure S9).
Discussion
Here we demonstrated that ULK1 potentiates cell death after oxidative stress and PARP1 activation. Although cytoprotective and cytotoxic roles have been ascribed to ULK1’s autophagic function,21, 23, 46 our results provide evidence that ULK1 acts as a prodeath molecule in an Atg7-independent manner. In Drosophila, overexpression of Atg1 promotes cell death through an apoptotic pathway,47 whereas in human osteosarcoma cells treated with camptothecin the p53-dependent upregulation of ULK1 potentiates cell death.21 Our results indicate that ULK1 is one of several ATG proteins that induces cell death through autophagy-independent pathways.48 They do not exclude the possibility that ULK1 overexpression promotes apoptosis or autophagy-mediated cell death under certain circumstances but rather provide an alternative mechanism for ULK1-dependent cell death that is associated with nuclear localization of ULK1.
Previous studies showed that ULK1 expression can be modulated in a p53-dependent manner in response to oxidative stress; however, we bypassed the p53-dependent changes in ULK1 expression and detected a novel function of ULK1 that is unrelated to its autophagy-inducing function.
We identified a pool of ULK1 within the nucleus that interacts with PARP1 and enhances its activation after exposure to ROS-inducing and/or DNA-damaging agents. Decreased PARP1 activation in Ulk1-deficient cells was manifested by reduced redistribution of PARP1, decreased accumulation of PARylated proteins, reduced ATP depletion, and reduced cell death after exposure to lethal doses of H2O2. These findings reveal an uncharacterized role for nuclear ULK1 in regulating PARP1 activity and highlight the importance of ULK1’s catalytic activity in facilitating nuclear translocation of ULK1. ATG13 may accompany ULK1 into the nucleus, and given the role of ATG13 in stabilizing ULK1 protein it is likely to be that ATG13 regulates PARP1 activity; however, additional studies are required to determine whether ATG13 is a component of the ULK1–PARP1 complex or simply maintains ULK1 in a stable conformation until it transfers to the PARP1-containing complex. Increased localization of ULK1 in nuclei after H2O2 treatment may limit the autophagic response, whereas promoting PARP1-dependent necrosis under conditions resulting in excessive ROS (Figure 8), especially in cells lacking p53. Therefore, we propose that the contribution of ULK1 in health and disease may be its ability to regulate not only autophagy but also PARP1 activity.

Model showing the pro-survival role of cytoplasmic ULK1and the pro-death role of nuclear ULK1. Cytoplasmic ULK1 promotes cell survival by promoting autophagy; by contrast, nuclear ULK1 interacts with PARP1 and promotes its activation in response to oxidative stress, thereby facilitating ATP depletion and necrosis
Balancing the regulation of autophagy and PARP1-mediated cell death by ULK1 is most likely relevant to the development and/or treatment of certain cancers. ULK1 overexpression in patients with esophageal squamous cell carcinoma,49 hepatocellular carcinoma,50 or breast cancer46 is associated with poor prognosis. Conventional wisdom posits that autophagy confers a survival advantage to tumor cells within the nutrient- and oxygen-deprived tumor microenvironment. Our results show that it may also facilitate PARP1 hyperactivation and necrotic cell death, which can induce a protumor inflammatory response.51 The heterogeneity of ULK1 localization within a given tumor population may be beneficial for overall tumor growth. Although ULK1 overexpression is associated with poor prognosis with current treatment regimens, the observation that ULK1 expression sensitizes cells to ROS-induced cell death could be exploited to select appropriate ROS-inducing drugs to advance treatment of patients with ULK1-overexpressing tumors.
Materials and Methods
Plasmid constructs
Flag-tagged ULK1 (gift from Dr. Toshifumi Tomoda), pMXs-IP-EGFP-ULK1 (gift from Noboru Mizushima, Addgene plasmid 38193),52 pMXs-IP-EGFP-ULK2 (gift from Noboru Mizushima, Addgene plasmid 38201),52 HA-tagged ATG13 (gift from Dr. Do-Hyung Kim),53 and PARP1-GFP (gift from Dr. Guy G Poirier)54 were described previously. ULK1 kinase-inactive (KI, K46A), AMPK phosphorylation site (4SA), and nuclear localization-defective (R266A) mutants were created via site-directed mutagenesis by using the Quikchange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). The pBlueScript SK+ plasmid containing mULK1 cDNA was used as the starting template with the following mutagenic oligonucleotides (altered codons are underlined and are in bold):
Ulk1-K46A: 5′-CGACCTGGAGGTGGCCGTCGCATGCATTAACAAGAAGA-3′ and 5′-CTTCTTGTTAATGCATGCGACGGCCACCTCCAGGTCG-3′; Ulk1-S467A: 5′-ATCCGAAGGTCAGGGGCCACCAGCCCCCTG-3′ and 5′-CAGGGGGCTGGTGGCCCCACTCCTTCGGAT-3′; ULK1-S555A: 5′-TGCCGCCTGCACGCTGCCCCTAACCTGTCC-3′ and 5′-GGACAGGTTAGGGGCAGCGTGCAGGCGGCA-3′; Ulk1-T574A: 5′-CTGCCTAAGCCCCCAGCAGACCCACTGGGA-3′ and 5′-TCCCAGTGGGTCTGCTGGGGGCTTAGGCAG-3′; Ulk1-S637A: 5′-TTCCCCAAAACCCCCGCCTCTCAGAATTTG-3′ and 5′-CAAATTCTGAGAGGCGGGGGTTTTGGGGAA-3′; ULK1-R266A: 5′-CGGAACCACAAGGACGCCATGGACTTTGAT-3′ and 5′-ATCAAAGTCCATGGCGTCCTTGTGGTTCCG-3′.
Each ULK1 mutant was subcloned into the 5′- and 3′-EcoRI restriction enzyme sites of the MSCV-IRES-GFP-MII (gift from Dr. Terrence Geiger)55 and pcDNA3.1 (Invitrogen, Grand Island, NY, USA) vectors for retrovirus generation and transient transfection, respectively.
Cell culture and drug treatment
MEFs and 293T cells (ATCC, Manassas, VA, USA) were grown in Dulbecco’s modified Eagle medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), penicillin/streptomycin (Invitrogen), and Glutamax (Invitrogen) at 37 °C (5% CO2). For amino acid starvation, cells were washed twice with either prewarmed complete DMEM (control) or Earle’s balanced salt solution (EBSS; experimental) and incubated in either DMEM or EBSS supplemented with 10% dialyzed FBS for the indicated times. H2O2 (Sigma-Aldrich, St. Louis, MO, USA) was diluted to the appropriate concentration in media; the PARP inhibitor III DPQ (EMD Millipore, Billerica, MA, USA) was reconstituted in methanol and was used at a final concentration of 40 nM. Cells were prepared via standard cell culture techniques. The β-lapachone (CAS 4707-32-8, Tocris 1293, Bristol, UK) was dissolved in DMSO and used at a final concentration of 4.5 μM.
For camptothecin treatment, cells were plated in 96-well plates (Corning catalogue number 3917, Corning, NY, USA) at the following densities: untransfected WT and Ulk1-deficient MEFs (2.0 × 104 cells/well) and transfected 293T cells (2.5 × 104 cells/well). Approximately 24 h after plating, each cell line was treated by using a Biomek FX (Beckman Coulter, Fullerton, CA, USA) liquid handler equipped with a pin tool, with a compound plate containing the following solutions: camptothecin (CAS 7689-03-4, Toronto Research C175150, Toronto, ON, Canada) arrayed in dose–response in rows A–D, with columns 1–10 having a starting concentration equal to 7 mM and threefold dilution, and DMSO (negative control) in rows A–D, columns 11–12. The pin tool transferred 150 nl compound stock, resulting in 740-fold compound dilution. At 72 h posttreatment, cells were lysed with the addition of 100 μl/well CellTiter-Glo reagent (‘CTG’, Promega, Fitchburg, WI, USA; catalogue number G7573) and the fluorescence was read by using a Perkin Elmer (Waltham, MA, USA) EnVision plate reader to measure the relative light units (RLUs). Concentration–response data were analyzed by using a custom script written in the R language (www.R-project.org). RLU values were first normalized by subtracting the median RLU of the DMSO control wells, then fit to the four-parameter log-logistic function (‘Hill equation’): y=y0+(yFin−y0)/(1+(EC50/x)−Hill), where y is the activity, x is the log concentration, EC50 is the log concentration at the half-maximal observed response, and y0 and yFin are the responses at zero and infinite concentrations, respectively.
Generation of MEFs
MEFs were prepared from the E12.5 embryos of Atg7+/− × Atg7+/− or Ulk1+/− × Ulk1+/− crosses. Embryos were collected, washed twice with PBS, incubated in trypsin, and then separated into single-cell suspensions. Cells were expanded in DMEM supplemented with 10% FBS, MEM nonessential amino acids (Life Technologies, Grand Island, NY, USA), L-glutamine (Life Technologies), β-mercaptoethanol (Life Technologies), and gentamycin (Life Technologies). These primary MEFs were then immortalized by using FuGENE 6 (Promega, Madison, WI, USA) to transfect the cells with 1 μg of an expression vector expressing SV40, as described previously.56 After transfection, cells were maintained in normal growth media. To generate stable MEF lines, retrovirus was produced as described previously.57 MSCV-IGFP-MII containing helper vector and either no cDNA, WT, or mutant ULK1 were cotransfected into 293T cells by using FuGENE 6; cells were then incubated in DMEM with 10% FBS for 48 h. Supernatant was collected twice daily and used to infect GP+E86 retroviral producer cells in the presence of Polybrene (8 μg/ml). Transduced GP+E86 cells were FACS sorted by the presence of GFP and cell-free supernatant was used to transduce Ulk1−/− MEFs. MEFs stably expressing the Luc+LC3 and Luc+LC3-G120A constructs (gift from Dr. John Cleveland)23 and Ulk1−/− Ulk2shRNA MEFs stably expressing WT or KI ULK1 (from of Dr. Reuben Shaw)27 were described previously.
Gene silencing and quantitative real-time PCR
WT and Ulk1-deficient MEFs were transfected with pooled nontargeting siRNA (Dharmacon, Lafayette, CO, USA), Atg7 siRNA (Dharmacon), or PARP1 siRNA (Dharmacon) by using Lipofectamine RNAi Max (Life Technologies) according to the manufacturer’s protocol. Knockdown was confirmed by RT-qPCR (TaqMan, Life Technologies), immunoblot analyses, or both. Total RNA was isolated from cells by using TRIzol Reagent (Life Technologies). The RT reaction was carried out using the SuperScript III first-strand synthesis kit (Life Technologies) according to the manufacturer’s instructions. TaqMan Gene Expression Assays containing FAM-labeled primer/probe sets specific for Atg7, PARP1, or 18S were obtained from Applied Biosystems (Grand Island, NY, USA). The real-time PCR reactions were performed in a total reaction volume of 25 μl by using FastStart TaqMan Probe Master (Roche, Mannheim, Germany) reagent and results were analyzed by using the iCycler IQ real-time PCR detection system (Bio-Rad, Hercules, CA, USA). Relative expression (log2) was calculated using the Pfaff quantification method58 after normalization of cycle thresholds to 18S RNA and calibration to respective controls.
Cell viability, ATP, and caspase activity assays
MEFs grown in complete media were washed with DMEM or EBSS and incubated either in DMEM or EBSS supplemented with 10% dialyzed FBS or in DMEM with 500 μM H2O2 for the indicated times. At the desired intervals, cells were trypsinized and counted by using trypan blue-exclusion assays to determine the percentages of viable and nonviable cells. Cells were also subjected to Annexin V-FITC/PI double staining before flow cytometry. The staining was performed according to the protocol of the manufacturer (BD Pharmingen, San Jose, CA, USA). Flow cytometry was performed using a FACSCalibur (BD Biosciences, San Jose, CA, USA) and results were analyzed using FlowJo V10 (Tree Star, Ashland, OR, USA). Cell viability was also measured using the CellTiter-Glo reagent (‘CTG’, Promega catalogue number G7573) as per the manufacturer’s protocol. Intracellular ATP was measured using the EnzyLight ATP assay kit (BioAssay Systems, Atlanta, GA, USA) or Celltiter Glo (Promega) as per the manufacturer’s protocols. The Caspase-Glo 3/7 Luminescent Assay (Promega) was used according to the manufacturer’s instructions. In brief, WT and KO MEFs were seeded into 96-well plates (2 × 104 cells/well) in triplicate. Then, 1 μM staurosporine and 500 μM H2O2 were added. Caspase activity was detected on a Synergy HT luminometer (BioTek, Winooski, VT).
Luc-LC3 luciferase-degradation assay
Luc-LC3-degradation assays were performed as described previously.23 Briefly, 1 × 104 cells expressing Luc-LC3 or the control Luc-LC3G120A mutant were plated in a 96-well white plate (Costar 3917, Fisher Scientific, Pittsburgh, PA, USA) with a final volume of 100 μl per well. To analyze luciferase activity, an equal volume of BriteLite Plus reagent (Perkin Elmer) was added to all wells and incubated at room temperature for 2 min; the resulting luminescence was measured using an Envision reader (Perkin Elmer) with a dwell time of 0.1 s/well. Resulting data were normalized to the control wells (normal media) by using the formula (100 × experimental/average (control)), to determine the percentage–control values. To normalize data to the Luc-LC3G120A-negative control, we calculated the ratio of the Luc-LC3 signal (% control) to the Luc-LC3G120A signal (% control).
Western blot analysis and antibodies
Unless otherwise indicated, cells were lysed in modified RIPA buffer (150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 50 mM Tris-HCl pH 8.0) supplemented with complete EDTA-free protease-inhibitor cocktails (Sigma-Aldrich) and phosphatase-inhibitor cocktails (Sigma-Aldrich). Cleared lysates were size separated on 4–12% Bis-Tris gels (Invitrogen). Proteins were transferred to either a nitrocellulose or PVDF membrane. After incubation with 5% milk or 5% BSA block, blots were probed with the following antibodies: Flag-M2 (F3165, Sigma-Aldrich), ULK1 (A7481, Sigma-Aldrich), PARP1 (9542, Cell Signaling Technology, Danvers, MA, USA), PAR (ab14459, Abcam, Cambridge, MA, USA), Caspase-3 (8G10, Cell Signaling Technology), and GAPDH (G9545, Sigma-Aldrich). Membranes were then incubated with either of the following: (1) horseradish peroxidase-conjugated secondary antibody, with bands detected by chemiluminescence detection kits (Amersham, Pittsburgh, PA, USA), or (2) secondary antibodies labeled with a 700- or 800-nm channel dye (LI-COR Biosciences, Lincoln, NE, USA), with bands detected by the Odyssey Infrared Imaging System (LI-COR Biosciences).
Subcellular fractionation
Subcellular fractionation was conducted as described previously, with slight modifications.59 Briefly, cells were washed in ice-cold PBS and lysed by incubation for 10 min in ice-cold hypotonic buffer (10 mM HEPES/KOH pH 7.9, 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 1 mM dithiothreitol (DTT), 0.5 mM PMSF, protease inhibitors, and NP-40). The lysates were centrifuged at 13 000 × g for 2 min. The supernatant was stored as the cytoplasmic fraction. The nuclear pellet was washed and nuclear proteins were extracted by incubation on ice for 20 min in cold buffer (20 mM HEPES/KOH pH 7.9, 1.5 mM MgCl2, 0.2 mM EDTA, 650 mM NaCl, 25% v/v glycerol, 1 mM DTT, 0.5 mM PMSF, and protease inhibitors). After centrifugation at 13 000 × g for 20 min at 4 °C, supernatants containing nuclear proteins were stored.
Immunoprecipitation
For immunoprecipitation (IP) of GFP- or Flag-tagged proteins, 293T cells were transfected with the relevant constructs. After 48 h, cells were lysed with hypotonic lysis buffer (10 mM Tris-HCl pH 7.5, 10 mM NaCl, 2 mM EDTA, 0.5% Triton X-100) supplemented with phosphatase inhibitor (Sigma-Aldrich) and complete EDTA-free protease-inhibitor cocktails (Sigma-Aldrich). Then, 500 μg of lysate was incubated overnight at 4 °C with anti-Flag M2 antibody-conjugated beads (Sigma-Aldrich) or anti-GFP antibody-conjugated sepharose beads (ab69314, Abcam). The beads were washed with Net-2 buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.05% Triton X-100) and bound proteins were eluted with the Flag peptide (Sigma-Aldrich) or incubated at 95 °C for 5 min in SDS sample buffer (Sigma Aldrich).
Endogenous ULK1 was immunoprecipitated from nuclear extracts as described previously.23 Briefly, after nuclear–protein extraction, lysates were incubated overnight at 4 °C with either anti-ULK1 antibody (Santa Cruz, Dallas, TX, USA) or anti-ULK1 antibody preblocked with the ULK1 peptide (Santa Cruz). ULK1 and its interacting partners were precipitated using Protein-G agarose beads (Pierce, Grand Island, NY, USA). The beads were washed five times with NET-2 buffer and incubated at 95 °C for 5 min in the sample buffer (Invitrogen).
Proteomics
The MS analysis was performed according to an optimized platform as previously reported.60 Ulk1-interacting proteins were visualized by performing SYPRO Ruby Protein Gel Staining (Sigma-Aldrich) according to the manufacturer’s protocol. Proteins in gel bands were reduced by adding DTT and then alkylated by adding iodoacetamide. The gel bands were then washed, dried in a speed vacuum, and rehydrated with a trypsin-containing buffer for overnight proteolysis. The digested peptides were extracted, dried, reconstituted, and loaded onto a capillary reverse-phase C18 column by an HPLC system (Waters ACQUITY UPLC, Milford, MA, USA). Peptides were eluted in a gradient, ionized by electrospray ionization, and detected using an inline mass spectrometer (Thermo Scientific LTQ Orbitrap Elite, Grand Island, NY, USA). MS spectra were collected and the top 20 abundant ions were sequentially isolated for MS/MS analysis. This process was cycled over the entire liquid chromatography gradient. The acquired MS/MS spectra were used to search protein databases to obtain possible peptide matches. The quality of the IP-MS data were examined and we found that the total spectral counts of three controls and two ULK1-IP were 234, 959, 408, 1064, and 909, respectively. The first control appeared to be an outlier and was therefore removed. Obvious contaminants such as keratin were also deleted from the protein table.
We analyzed the statistically significant protein changes among these IP samples by using a previously published protocol based on G-test.50 Briefly, the G-value of a given protein was calculated according to the detected spectral counts in any of two samples for comparison. Assuming that the G-values approximately fit the χ2 distribution (1 degree of freedom), we derived the P-values. We next established a reliable P-value threshold to identify protein changes with statistical significance. Ideally, the threshold should accept a very small number of proteins (i.e., false discoveries) in null experiments (e.g., comparison of technical replicates, control A versus control B, or ULK1A versus ULK1B). When the same threshold is used for control-ULK1 comparisons, the list is deemed acceptable with a low false-discovery rate (FDR). To find the appropriate threshold, we dynamically adjusted the P-value from 0.4 to 0.001 and identified that a P-value of 0.02 was a reasonable threshold with balanced sensitivity and specificity (FDR of ~10%, Supplementary Table S1). After manual interpretation, we accepted 14 proteins in the ULK1 IP samples (Supplementary Table S2).
Microscopy
For live-cell imaging, cells were incubated with SYTOX green, Hoechst, and Annexin V (Invitrogen) in the presence or the absence of H2O2 as per the manufacturer’s protocol. Images were acquired every 15 min for up to 24 h on a Marianas spinning-disk confocal (SDC) imaging system (Intelligent Imaging Innovations/3i, Denver, CO) consisting of a Carl Zeiss Axio Observer motorized inverted microscope (Carl Zeiss MicroImaging, Peabody, MA, USA) equipped with a CSUX SDC head (Yokogowa Electric Corporation, Tokyo, Japan) and a LaserStack (3i), including the lasers 405, 488, and 561 nm. Images were acquired using Zeiss Plan-Apochromat × 63 1.4 NA DIC objectives, an Evolve 512 EMCCD camera (Photometrics, Tucson, AZ, USA), and SlideBook 5.0 software (3i). For nucleolar localization imaging, PARP1-GFP-transfected cells were fixed in 0.25% paraformaldehyde for 15 min without permeabilization. For localization of untagged ULK1, cells were fixed in 4% formaldehyde and permeabilized with digitonin. After being blocked with 1% BSA, cells were stained with an anti-ULK1 antibody and a secondary antibody conjugated to Alexa Fluor 488 or 555 (Invitrogen).
Samples for electron microscopy were fixed in 2.5% glutaraldehyde and 2.0% paraformaldehyde in 0.1 M sodium cacodylate buffer pH 7.4, overnight at 4 °C, and processed as described previously.61 Sections were examined by using a JEOL 1010 electron microscope (JEOL USA, Peabody, MA, USA) fitted with a Hamamatsu digital camera system (Hamamatsu Photonics, Hamamatsu City, Japan).
Fluorescence recovery after photobleaching
Fluorescence recovery after photobleaching (FRAP) experiments were conducted as described previously.54 FRAP analyses were performed on live WT or Ulk1-deficient MEFs transfected with PARP1-GFP fusion proteins. All photobleaching experiments were performed on a Marianas SDC imaging system (Intelligent Imaging Innovations/3i). A 488-nm laser and × 63 objective with 1.4 NA were used to acquire FRAP data. Fluorescence recovery was monitored over a 50-s period. Data obtained for recovery were corrected for the background intensity and loss of total fluorescence.
Two-photon microirradiation
Laser microirradiation was carried out as previously described.54 Briefly, cells were grown on coverslips and labeled with 1 μm 5′-bromo-2′-deoxyuridine overnight. Cells were exposed to 1 μg/ml Hoechst 33258 for 1 h and then placed on the stage of a Marianas SDC microscope. Double-strand breaks (DSBs) were generated along a 1-μm-wide region across the nucleus of a single living cell by using a near-infrared, 780-nm titanium-sapphire laser line to excite the Hoechst 33258 dye. The laser output was set to 10% (unless stated otherwise) and we used 10 iterations to generate localized DSBs that were clearly traceable with a × 40 objective. Protein accumulation within the laser path was compared with that in an undamaged region within the same microirradiated cell. Therefore, each cell provided a reference unirradiated region that provided high sensitivity to detect and measure small changes in the concentration of fluorescently tagged proteins. For microirradiation, we selected cells with low expression levels and normalized the fluorescence intensity in the microirradiated area to the initial fluorescence in the whole nucleus, to compensate for photobleaching during acquisition.
Statistical analyses
All statistical analyses were performed on data from at least three independent experiments. For nucleolar localization, more than 100 cells from 3 independent experiments were counted per condition. Error bars represent S.D. Statistical analysis was performed by using SigmaPlot (Systat Software, Inc., San Jose, CA, USA). Significance was assessed by either one-way ANOVA testing followed by Student–Newman–Keuls post-hoc analysis or by Student’s t-testing.
Abbreviations
- ATG:
-
autophagy protein
- DPQ:
-
3,4-dihydro-5[4-(1-piperindinyl)butoxy]-1(2H)-isoquinoline
- GFP:
-
green fluorescent protein
- H2O2:
-
hydrogen peroxide
- KI:
-
kinase inactive
- KO:
-
knockout
- MEF:
-
murine embryonic fibroblast
- NLS:
-
nuclear-localization signal
- PARP1:
-
poly (ADP-ribose) polymerase 1
- PI:
-
propidium iodide
- ROS:
-
reactive oxygen species
- ULK1:
-
Unc-51-like kinase
- UV:
-
ultraviolet
- WT:
-
wild type
References
Orient A, Donko A, Szabo A, Leto TL, Geiszt M . Novel sources of reactive oxygen species in the human body. Nephrol Dial Transplant 2007; 22: 1281–1288.
Ray PD, Huang BW, Tsuji Y . Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012; 24: 981–990.
D'Autreaux B, Toledano MB . ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 2007; 8: 813–824.
Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal 2007; 9: 49–89.
Kwon YW, Masutani H, Nakamura H, Ishii Y, Yodoi J . Redox regulation of cell growth and cell death. Biol Chem 2003; 384: 991–996.
Gardner AM, Xu FH, Fady C, Jacoby FJ, Duffey DC, Tu Y et al. Apoptotic vs. nonapoptotic cytotoxicity induced by hydrogen peroxide. Free Radic Biol Med 1997; 22: 73–83.
Navarro-Yepes J, Burns M, Anandhan A, Khalimonchuk O, Del Razo LM, Quintanilla-Vega B et al. Oxidative stress, redox signaling and autophagy: cell death vs survival. Antioxid Redox Signal 2014; 21: 66–85.
Wenzel U, Nickel A, Kuntz S, Daniel H . Ascorbic acid suppresses drug-induced apoptosis in human colon cancer cells by scavenging mitochondrial superoxide anions. Carcinogenesis 2004; 25: 703–712.
Xia S, Rosen EM, Laterra J . Sensitization of glioma cells to Fas-dependent apoptosis by chemotherapy-induced oxidative stress. Cancer Res 2005; 65: 5248–5255.
Li J, Zuo L, Shen T, Xu CM, Zhang ZN . Induction of apoptosis by sodium selenite in human acute promyelocytic leukemia NB4 cells: involvement of oxidative stress and mitochondria. J Trace Elem Med Biol 2003; 17: 19–26 Epub 2003/05/21.
Wang HT, Yang XL, Zhang ZH, Lu JL, Xu HB . Reactive oxygen species from mitochondria mediate SW480 cells apoptosis induced by Na2SeO3. Biol Trace Elem Res 2002; 85: 241–254.
Kroemer G, Marino G, Levine B . Autophagy and the integrated stress response. Mol Cell 2010; 40: 280–293.
Wang Y, Singh R, Massey AC, Kane SS, Kaushik S, Grant T et al. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem 2008; 283: 4766–4777.
Qiang L, Wu C, Ming M, Viollet B, He YY . Autophagy controls p38 activation to promote cell survival under genotoxic stress. J Biol Chem 2013; 288: 1603–1611.
Samara C, Syntichaki P, Tavernarakis N . Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ 2008; 15: 105–112.
Shen S, Kepp O, Kroemer G . The end of autophagic cell death? Autophagy 2012; 8: 1–3.
Denton D, Nicolson S, Kumar S . Cell death by autophagy: facts and apparent artefacts. Cell Death Differ 2012; 19: 87–95.
Ci Y, Shi K, An J, Yang Y, Hui K, Wu P et al. ROS inhibit autophagy by downregulating ULK1 mediated by the phosphorylation of p53 in selenite-treated NB4 cells. Cell Death Dis 2014; 5: e1542.
Jiang P, Huang Z, Zhao H, Wei T . Hydrogen peroxide impairs autophagic flux in a cell model of nonalcoholic fatty liver disease. Biochem Biophys Res Commun 2013; 433: 408–414.
Wirth M, Joachim J, Tooze SA . Autophagosome formation—the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol 2013; 23: 301–309.
Gao W, Shen Z, Shang L, Wang X . Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ 2011; 18: 1598–1607.
Gough DR, Cotter TG . Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis 2011; 2: e213.
Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell 2011; 43: 572–585.
Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8: 445–544.
Hale AN, Ledbetter DJ, Gawriluk TR, Rucker EB 3rd . Autophagy: regulation and role in development. Autophagy 2013; 9: 951–972.
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169: 425–434.
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331: 456–461.
Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461: 654–658.
Thomas C, Tulin AV . Poly-ADP-ribose polymerase: machinery for nuclear processes. Mol Aspects Med 2013; 34: 1124–1137.
Chan EY, Tooze SA . Evolution of Atg1 function and regulation. Autophagy 2009; 5: 758–765.
Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG . PARP inhibition: PARP1 and beyond. Nature Rev Cancer 2010; 10: 293–301.
Schreiber V, Dantzer F, Ame JC, de Murcia G . Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 2006; 7: 517–528.
Ha HC, Snyder SH . Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA 1999; 96: 13978–13982.
Meder VS, Boeglin M, de Murcia G, Schreiber V . PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J Cell Sci 2005; 118: 211–222.
Rancourt A, Satoh MS . Delocalization of nucleolar poly(ADP-ribose) polymerase-1 to the nucleoplasm and its novel link to cellular sensitivity to DNA damage. DNA Rep 2009; 8: 286–297.
Henle ES, Linn S . Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J Biol Chem 1997; 272: 19095–19098.
Ravanat JL, Douki T, Cadet J . Direct and indirect effects of UV radiation on DNA and its components. J Photochem Photobiol B Biol 2001; 63: 88–102.
Cleaver JE . gammaH2Ax: biomarker of damage or functional participant in DNA repair "all that glitters is not gold!". Photochem Photobiol 2011; 87: 1230–1239.
Calkins AS, Iglehart JD, Lazaro JB . DNA damage-induced inhibition of rRNA synthesis by DNA-PK and PARP-1. Nucleic Acids Res 2013; 41: 7378–7386.
Kosugi S, Hasebe M, Tomita M, Yanagawa H . Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci USA 2009; 106: 10171–10176.
Lazarus MB, Novotny CJ, Shokat KM . Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem Biol 2015; 10: 257–261.
Reva B, Antipin Y, Sander C . Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 2011; 39: e118.
Park EJ, Min KJ, Lee TJ, Yoo YH, Kim YS, Kwon TK . beta-Lapachone induces programmed necrosis through the RIP1-PARP-AIF-dependent pathway in human hepatocellular carcinoma SK-Hep1 cells. Cell Death Dis 2014; 5: e1230.
Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA . NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J Biol Chem 2000; 275: 5416–5424.
Docampo R, Cruz FS, Boveris A, Muniz RP, Esquivel DM . beta-Lapachone enhancement of lipid peroxidation and superoxide anion and hydrogen peroxide formation by sarcoma 180 ascites tumor cells. Biochem Pharmacol 1979; 28: 723–728.
Pike LR, Singleton DC, Buffa F, Abramczyk O, Phadwal K, Li JL et al. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem J 2013; 449: 389–400.
Scott RC, Juhasz G, Neufeld TP . Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol 2007; 17: 1–11.
Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8: 1124–1132.
Jiang S, Li Y, Zhu YH, Wu XQ, Tang J, Li Z et al. Intensive expression of UNC-51-like kinase 1 is a novel biomarker of poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Sci 2011; 102: 1568–1575.
Bai B, Hales CM, Chen PC, Gozal Y, Dammer EB, Fritz JJ et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer's disease. Proc Natl Acad Sci USA 2013; 110: 16562–16567.
Giansanti V, Dona F, Tillhon M, Scovassi AI . PARP inhibitors: new tools to protect from inflammation. Biochem Pharmacol 2010; 80: 1869–1877.
Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181: 497–510.
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20: 1992–2003.
Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ et al. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem 2008; 283: 1197–1208.
Alli R, Nguyen P, Geiger TL . Retrogenic modeling of experimental allergic encephalomyelitis associates T cell frequency but not TCR functional affinity with pathogenicity. J Immunol 2008; 181: 136–145.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–730.
Alli R, Nguyen P, Boyd K, Sundberg JP, Geiger TL . A mouse model of clonal CD8+ T lymphocyte-mediated alopecia areata progressing to alopecia universalis. J Immunol 2012; 188: 477–486.
Pfaffl MW . A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29: e45.
Eftekharzadeh B, Maghsoudi N, Khodagholi F . Stabilization of transcription factor Nrf2 by tBHQ prevents oxidative stress-induced amyloid beta formation in NT2N neurons. Biochimie 2010; 92: 245–253.
Xu P, Duong DM, Peng J . Systematical optimization of reverse-phase chromatography for shotgun proteomics. J Proteome Res 2009; 8: 3944–3950.
Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 2007; 67: 6745–6752.
Acknowledgements
We thank Dr. Toshifumi Tomoda (Beckman Research Institute of City of Hope, Duarte, CA), Dr. Noboru Mizushima (University of Tokyo, Tokyo, Japan), Dr. Do-Hyung Kim (University of Minnesota, Twin Cities, MN), Dr. Guy G Poirier (Centre Hospitalier Universitaire de Québec, QC, Canada), Dr. John Cleveland (The Moffitt Cancer Center and Research Institute, Tampa, FL), Dr. Reuben Shaw (Salk Institute for Biological Studies, La Jolla, CA), and Dr. Terrence Geiger (St. Jude Children’s Research Hospital, Memphis, TN) for reagents. We thank Dr. Cherise Guess and Dr. Angela McArthur for editing the manuscript, and Feenalie Patel for technical assistance. Imaging data were acquired at the Cell and Tissue Imaging Center, which is supported by St. Jude Children’s Research Hospital and NCI P30 CA021765. Proteomics studies were performed in the Proteomics and Mass Spectrometry Core Facility, which is supported by St. Jude Children’s Research Hospital and NCI P30 CA021765. The research was supported by ALSACand by grants to MK from the National Institutes of Health (R01HL114697), Burroughs Wellcome Fund (1006062.01), and the American Society of Hematology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Baehrecke
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Joshi, A., Iyengar, R., Joo, J. et al. Nuclear ULK1 promotes cell death in response to oxidative stress through PARP1. Cell Death Differ 23, 216–230 (2016). https://doi.org/10.1038/cdd.2015.88
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.88
This article is cited by
-
Autophagy and autophagy-related pathways in cancer
Nature Reviews Molecular Cell Biology (2023)
-
Discovery of a signaling feedback circuit that defines interferon responses in myeloproliferative neoplasms
Nature Communications (2022)
-
Autophagy-related DjAtg1-1 plays critical role in planarian regeneration by regulating proliferation and cell death
Cell and Tissue Research (2022)
-
GSK3B induces autophagy by phosphorylating ULK1
Experimental & Molecular Medicine (2021)
-
Cell death in head and neck cancer pathogenesis and treatment
Cell Death & Disease (2021)
