
Abstract
The acetyltransferase Tip60/Kat5 acetylates both histone and non-histone proteins, and is involved in a variety of biological processes. By acetylating p53, Tip60 controls p53-dependent transcriptional activity and so is implicated as a tumor suppressor. However, many breast cancers with low Tip60 also show p53 mutation, implying that Tip60 has a tumor suppressor function independent of its acetylation of p53. Here, we show in a p53-null mouse model of sporadic invasive breast adenocarcinoma that heterozygosity for Tip60 deletion promotes mammary tumorigenesis. Low Tip60 reduces DNA repair in normal and tumor mammary epithelial cells, both under resting conditions and following genotoxic stress. We demonstrate that Tip60 controls homologous recombination (HR)-directed DNA repair, and that Tip60 levels correlate inversely with a gene expression signature associated with defective HR-directed DNA repair. In human breast cancer data sets, Tip60 mRNA is downregulated, with low Tip60 levels correlating with p53 mutations in basal-like breast cancers. Our findings indicate that Tip60 is a novel breast tumor suppressor gene whose loss results in genomic instability leading to cancer formation.
Similar content being viewed by others
Main
The acetyltransferase Tip60 (Kat5) acetylates histone and non-histone proteins, and is involved in diverse biological processes, including apoptosis, cell cycle, and DNA damage responses (DDR).1 Tip60 acetylates histones H4 and H2A,1 as well as the non-histone proteins p53,2, 3 c-Myc,4 and androgen receptor.5 Because of its pleiotropic functions, Tip60’s role in cancer suppression is complex. Tip60 is a haplo-insufficient tumor suppressor in a mouse lymphoma model,6 and Tip60 heterezygosity promotes tumorigenesis in a chemically-induced mouse colon cancer model.7 In melanoma patients, low Tip60 levels correlate with poor survival and increased metastasis.8 In human breast cancers, decreased Tip60 correlates with accelerated tumor cell proliferation and increased tumor grade.6 Interestingly, abnormal Tip60 protein levels coincide with p53 mutations in these malignancies. Because Tip60 is required for p53 activation,3, 9, 10 one might expect that Tip60 downregulation alone would impair the p53 pathway in tumor cells. However, the inactivation of both Tip60 and p53 is required for breast cancer progression, implying that Tip60 has another function in tumor suppression that is p53 independent.
Tip60 is involved in DDR at various levels, including in signal transduction following DDR induction and during DNA repair.1 Tip60 acetylates and activates the ataxia telangiectasia mutant (ATM) kinase that senses DNA damage,11, 12 and also acetylates the γ-phosphorylated form of the H2AX histone (γH2AX) that accumulates in response to DNA damage.13 Tip60-mediated γH2AX acetylation is also required for γH2AX ubiquitination that releases γH2AX from DNA breakage sites.13 In addition, Tip60 influences the balance between BRCA1 and 53BP1 at sites of DNA damage and regulates the choice of DNA repair mechanism.14 As DDR constitutes a barrier to tumorigenesis,15, 16, 17 any alterations to Tip60’s effects on DDR may affect cancer development. Here, we use a mouse model of human breast cancer driven by mammary-specific p53 inactivation to investigate Tip60-mediated tumor suppression.
Results
Heterozygous Tip60 deletion cooperates with p53 deletion to promote mouse mammary tumorigenesis
Human and mouse studies have demonstrated functional cooperation between Tip60 and p53. By acetylating human p53 at lysine-120 (K120) within the DNA-binding domain, Tip60 redirects p53 transcriptional activity toward apoptotic genes.3 In mutant mice in which p53 K117 (equivalent to human K120) is replaced by arginine, p53-mediated apoptosis is abrogated but p53-dependent cell-cycle arrest and senescence remain intact.18
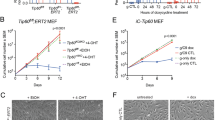
To investigate combinatorial effects of Tip60 and p53 mutations on tumor suppression in breast cancer, we crossed mice heterozygous for Tip60 deletion (Tip60+/−) with mice bearing a mammary gland-specific conditional p53 knockout mutation (K14cre;p53f/f; KP) to generate K14cre;p53f/f;Tip60+/− (KPT) progeny. K14cre-controlled recombination drives p53 deletion in the mammary epithelium and in other K14-expressing tissues such as skin and salivary glands.19 Female KP mice develop intermediate- to high-grade invasive ductal mammary carcinomas that resemble human estrogen receptor (ER)-negative and basal-like breast cancers. We monitored virgin nulliparous KP and KPT females for mammary tumor formation over 400 days. Although both groups developed single mammary tumors, KPT mice did so at higher frequency and with shorter latency than KP mice (Figure 1a). Histopathology showed that tumors arising in KP and KPT mice were high-grade mammary adenocarcinomas as determined by the Nottingham histologic scoring system (Figure 1b, Supplementary Figure S1a). Most of these malignancies were poorly differentiated and ER negative and progesterone receptor (PR) negative, contained spindle-shaped neoplastic cells, and exhibited cribriform and solid morphology (Supplementary Figure S1b and c). However, only cells in KPT tumors had abnormal nuclear morphology (Figure 1b, Supplementary Figure S1a), which is associated with higher tumor grade, aggression, and genetic instability.20, 21

Heterozygosity for Tip60 deletion accelerates mammary tumorigenesis in a p53-null background. (a) Kaplan–Meier survival curves of KP and KPT mice (n=15/group). *P<0.05. Mean tumor latency is 35.5 weeks for KP mice and 28 weeks for KPT mice. (b) Hematoxylin-eosin (H&E) stained sections of high-grade mammary carcinomas from a KP and a KPT mouse from (a). Scale bars, 50 μm. Results are representative of 10 tumors examined/group. (c) PCR analysis to detect the WT and mutated (mut) Tip60 alleles in genomic DNA extracted from KP (n=2) and KPT (n=8) tumors. Results are representative of 3 trials involving a total of 10 KP and 10 KPT tumors. (d) Quantitation of RT-PCR analysis of relative Tip60 mRNA levels in KP (n=10) and KPT (n=10) tumors. Data are the mean±S.E.M. normalized to mouse rsp9 RNA and are representative of three trials. **P<0.01. (e) Immunoblot to detect the α and β Tip60 isoforms in KPT (n=7) and KP (n=3) tumors. Actin, loading control. Numbers between the blots are image densitometric values of the Tip60 isoform bands above. Results are representative of three trials. (f) Quantitation of densitometric values as reported in (e). Data are the mean±S.E.M. (g) Immunostaining to detect Tip60 protein in normal WT virgin female mouse mammary gland and mammary tumors from KP and KPT mice, as indicated. Scale bars, 50 μm. Images are representative of five glands and five tumors per group
We next investigated the Tip60 gene in KP and KPT tumors using qualitative PCR. Both the WT and mutant Tip60 alleles were present in KPT tumor DNA (Figure 1c), indicating that there was no loss-of-heterozygosity of Tip60. Subsequent mRNA analysis confirmed that Tip60 mRNA was reduced by 50% in KPT versus KP tumors (Figure 1d). Immunoblotting revealed variable levels of the 60 kDa Tip60 β isoform and the 50-kDa α splice variant22 in KP and KPT tumors (Figure 1e). These Tip60 isoforms are differentially expressed in skin cancers and cardiomyocytes, but their specific functions remain unknown.23, 24 Quantitative image densitometry confirmed that both Tip60 isoforms were decreased in KPT tumors (Figure 1f). Histological analysis demonstrated that Tip60 was expressed in the nucleus and cytoplasm of myoepithelial and luminal cells within normal WT mammary gland, a pattern holding for KP tumor cells (Figure 1g). However, the majority of KPT tumor cells showed negligible Tip60 staining in the nucleus and slightly positive staining in the cytoplasm (Figure 1g). Both KP and KPT tumors showed extremely low p53 mRNA (Supplementary Figure S1d). These data indicate that Tip60 is a haplo-insufficient tumor suppressor whose loss cooperates with p53 inactivation to promote murine mammary tumorigenesis.
Tip60 regulates DNA repair in normal and tumor mammary epithelial cells
Tip60 has been implicated in DNA repair12, 14 and oncogene-induced DDR.6 Because DDR deregulation contributes to genomic instability leading to tumor formation,15, 16, 17 we examined the role of Tip60 in DDR in normal and tumor mammary epithelial cells (MEC).
First, we transfected immortalized mouse MEC (COMMA-1D cells, termed COMMA) with scrambled (scr; control) siRNA oligomers or siRNA oligomers directed against mouse Tip60 (siTip60). siTip60-transfected cells showed a 50% reduction in Tip60 mRNA and protein (Figure 2a and b), reflecting the maximum Tip60 decrease tolerated by a cell since complete Tip60 downregulation is lethal in vivo and in vitro.6 We subjected COMMA cells to γ-irradiation (IR, 5 Gy) and assessed responses to IR-induced DNA damage at 1, 8, or 24 h post IR by immunofluorescence staining to detect γH2AX. Although IR exposure induced γH2AX+ nuclear focus formation in both scr- and siTip60-expressing COMMA cultures within 1 h post IR, quantitation confirmed a greater number of γH2AX+ cells in Tip60-silenced cultures than in controls at 8 and 24 h (Figure 2c, Supplementary Figure S2). Because excessive γH2AX+ foci indicate unrepaired DNA,25 we immunostained scr- and siTip60-expressing COMMA cells to detect the DNA repair factors BRCA1 and Rad51. Before IR, only very low levels of BRCA1 and Rad51 were present in scr- and siTip60-expressing cells (Figure 2d and e, Supplementary Figure S2). After IR, scr-expressing cells accumulated many BRCA1+Rad51+ nuclear foci, whereas Tip60-silenced cultures had fewer cells with such foci (Figure 2d and e,Supplementary Figure S2). Thus, loss of Tip60 decreases DNA repair capacity following genotoxic stress.

Tip60 expression regulates HR-directed DNA repair in normal and tumor MEC. (a) Quantitation of RT-PCR analysis of relative Tip60 mRNA levels in COMMA cells transfected with scrambled (scr; control) or Tip60-specific siRNA (siTip60). The analysis was performed at 48 h post transfection. Data were analyzed as in Figure 1d. (b) Immunoblot to detect the α and β isoforms of Tip60 protein in cells treated as in (a). Results are representative of three trials. (c) Quantitation of cells with γH2AX+ nuclear foci as determined by immunofluorescence staining of COMMA cells that were treated as in (a), then left untreated or subjected to 5 Gy IR and stained at 1, 8, or 24 h post IR. Data are the mean±S.E.M. (n=4). ***P<0.001. (d and e) Quantitation of cells with BRCA1+ or Rad51+ nuclear foci as determined by immunofluorescence staining of COMMA cells that were left untreated (ctr) or subjected to IR and stained as in (c), and evaluated at 8 h post IR. Data are the mean±S.E.M. (BRCA1, n=4; RAD51, n=5). (f) Assay of HR efficiency (see Materials and methods) in COMMA cells treated as in (c). siBRCA1, positive control. Data are the mean±S.E.M. (n=5) relative to the scr value. (g) Quantitation of RT-PCR analysis of relative Tip60 mRNA levels in WAPcre;PTENf/f tumor cells that were infected with lentiviral-based shRNAs against GFP (shGFP; control) or Tip60 (shTip60). Data are the mean±S.E.M. (n=3) relative to the shGFP value and are representative of three trials. (h) Immunoblot to detect γH2AX in WAPcre;PTENf/f tumor cells treated as in (g). Results are representative of three trials. (i) Assay of HR efficiency in WAPcre;PTENf/f tumor cells cells treated as in (g). Data were analyzed as in (f)
IR causes DNA double-strand breaks (DSBs) that are resolved mainly by homologous recombination (HR)-directed DNA repair.26 The failure of IR-exposed Tip60-silenced cells to form numerous BRCA1+Rad51+ foci indicated a defect in this mechanism. We manipulated COMMA cells to stably express the DR-GFP reporter plasmid containing two differently mutated green fluorescent protein (GFP) genes oriented as direct repeats (DR).27 We exposed scr- and siTip60-expressing COMMA cells also expressing DR-GFP to IR to examine the efficiency of HR-directed DNA repair. Like BRCA1 depletion, Tip60 silencing decreased HR-directed DNA repair (Figure 2f). siTip60-expressing COMMA cells also showed heightened Rad51 accumulation at 24 h and increased γH2AX indicative of unrepaired DSBs (Supplementary Figure S2).
Because DDR and DNA repair must be coordinated to suppress tumorigenesis, we investigated links between Tip60 and DNA damage in mouse tumor MEC. We employed two cell lines previously established from mouse primary mammary tumors isolated from MMTV-PyMT or WAPcre;PTENf/f mice.28, 29 We infected both tumor cell lines with lentiviral-based shRNAs against mouse Tip60 (shTip60) or GFP (shGFP). Compared with shGFP-infected controls, both lines of shTip60-infected tumor cells exhibited reduced Tip60 mRNA (Figure 2g, Supplementary Figure S3a) and elevated γH2AX (Figure 2h, Supplementary Figure S3b). Like COMMA cells, shTip60-infected tumor cells showed defective HR following IR (Figure 2i).
Because Tip60 is required for ATM activation controlling DDR signaling,11, 30 we examined ATM status in siTip60-expressing COMMA cells and shTip60-expressing WAPcre;PTENf/f tumor cells. The low Tip60 levels in these cells did not affect IR-induced ATM activation or p53 phosphorylation (Supplementary Figure S3c and d), or mdm2 mRNA and protein levels (Supplementary Figure S3e and f), suggesting that Tip60 does not influence ATM or p53 stability. Thus, in both normal and transformed MEC, Tip60 regulates HR-directed DNA repair rather than DNA damage-induced ATM activation.
Tip60 influences tumor cell sensitivity to cisplatin-induced DNA damage
Malignant cells with defective DDR are particularly sensitive to chemotherapy-induced genotoxic stress. Human breast cancers are often treated with cisplatin, whose platinum atom bonds covalently to purine residues in DNA and creates cisplatin-DNA adducts.31 The resolution and excision of these adducts causes DSB that kill the tumor cell unless they are fixed by HR-directed repair.32
Because Tip60 was important for HR-directed repair in MEC, we evaluated the sensitivity of Tip60-silenced cells to cisplatin-induced DNA damage. We transfected COMMA cells with scr or siTip60 siRNAs and treated them 48 h later with 5 μM cisplatin. Like IR, cisplatin caused these Tip60-silenced normal MEC to accumulate γH2AX (Figure 3a) but did not increase their apoptosis (Figure 3b). Indeed, siTip60 provided cells with modest protection against cisplatin-induced death, as confirmed by their lack of cleaved PARP (Figure 3c). Thus, Tip60 depletion allows normal MEC to resist cisplatin-induced death, regardless of DNA damage.

Tip60 depletion influences MEC sensitivity to cisplatin. (a) Immunoblot to detect γH2AX in COMMA cells expressing scr or siTip60 siRNAs and left untreated (ctr) or treated for 24 h with IR (10 Gy) or cisplatin (10 μM). Actin, loading control. (b) Quantitation of AnnexinV/7-AAD+ apoptotic cells in cultures of scr- or siTip60-expressing COMMA cells that were left untreated or treated for 24 h with the indicated doses of cisplatin. Ctr, vehicle control. Data are the mean±S.E.M. (n=3) and are representative of two trials. (c) Immunoblot to detect cleaved PARP in scr- or siTip60-expressing COMMA cells that were left untreated or treated for 24 h with 10 μM cisplatin as in (a). Results are representative of three trials. Vinculin, loading control. (d) Quantitation of apoptosis as in (b) of WAPcre;PTENf/f cells that were infected with shGFP- or shTip60-expressing lentivirus and treated for 24 h with the indicated doses of cisplatin. Data are the mean±S.E.M. (n=3) and are representative of two trials. (e) Quantitation of RT-PCR analysis of relative p21/Cdkn1a and Puma/Bbc3 mRNA levels in WAPcre;PTENf/f cells infected as in (c) and treated with 2 μM cisplatin for 24 h. Data were analyzed as in Figure 1d and are representative of two trials. *P<0.05 and ***P<0.001
We then investigated the role of Tip60 in responses to cisplatin by tumor MEC. We treated shGFP- or shTip60-expressing WAPcre;PTENf/f tumor cells with cisplatin and analyzed apoptosis 24 h later. Once again, shTip60-expressing cultures contained surprisingly few apoptotic cells compared with shGFP cultures (Figure 3d) and showed an abnormal cell-cycle profile (Supplementary Figure S4). Flow cytometry revealed that shTip60-expressing tumor cells did not accumulate in G1/S-like control cells and instead entered S phase. Thus, Tip60-silenced tumor cells are resistant to cisplatin despite their increase in unrepaired DNA.
Because cisplatin-induced DNA damage triggers p53-dependent apoptosis,33 and Tip60 regulates p53 activation, we investigated the p53 targets p21/Cdkn1a and puma/bbc3 in cisplatin-treated shGFP- or shTip60-expressing WAPcre;PTENf/f tumor cells. The latter contained marginally less p21 mRNA but significantly less puma mRNA than cisplatin-treated shGFP cells (Figure 3d). Thus, Tip60 is required for p53-dependent expression of PUMA, which regulates p53-dependent apoptosis following genotoxic insults.3 These data indicate that Tip60 is involved in both DNA damage repair and DNA damage-induced cell death.
Tip60 expression correlates with the ‘HR defect’ gene signature
To further link downregulated Tip60 to genomic instability in MEC, we examined a gene expression profile associated with defective HR-directed repair.34 Peng et al.34 infected MCF10A human MEC with lentiviral shRNAs against BRCA1, RAD51 or BRIT1, which regulate HR-directed repair at different steps. Analysis of microarray profiles of these cells generated an ‘HR defect’ (HRD) gene signature that was validated in human cancer cell lines carrying known mutations in HR genes. We performed a gene set expression analysis (GSEA) of the Peng data34 to determine whether Tip60 mRNA levels correlated with the HDR signature in human breast cancers. We found that Tip60 expression correlated negatively with the HRD signature (Figure 4a), suggesting that normal Tip60 expression is required for functional HR-directed repair.

Tip60 mRNA expression correlates inversely with defective HR-directed DNA repair. (a) GSEA of the association of Tip60 (Kat5) with the HRD gene signature of Peng et al.34 was performed using default values for all parameters and a specific gene set. (b) Quantitation of RT-PCR analysis of relative Tip60 mRNA levels in one group of mammary tumors with high Tip60 expression and another group of mammary tumors with low Tip60 expression. Tumors were from MMTV-PyMT and MMTV-Neu mice (n=5/group). Data were analyzed as in Figure 1d and are representative of two trials. (c) Quantitation of RT-PCR analysis of relative mRNA levels of 8 genes associated with the HRD gene signature in mammary tumors with high or low Tip60 expression. Data are expressed as absolute values for individual tumors. Horizontal lines are mean values±S.E.M. *P<0.005, **P<0.01, and ***P<0.001
Our GSEA also identified eight genes that are downregulated in the HRD signature and whose abnormal expression patterns correlate strongly with Tip60 downregulation (Figure 4a). These genes are involved in: the mitotic spindle checkpoint (ASPM); nucleotide metabolism,35 mismatch repair and recombination (EXO1); assembly of small nuclear ribonucleoproteins (Gemin6); cell survival (HSPE1); cytokinesis (KIF14); cell-cycle progression (MTBP); coenzyme Q biosynthesis (PDSS1); DNA replication (POLQ); and telomere integrity (TERF1). We then validated these correlations in 10 mammary tumors from mouse tumor models available in the laboratory, including MMTV-PyMT mice or MMTV-Neu mice. Tumors were ranked according to their Tip60 mRNA levels, and expression levels of HDR-associated genes were compared between the top and bottom tertiles (Figure 4b, Supplementary Figure S5). This analysis confirmed that the mRNA levels of the eight HDR genes correlated positively with Tip60 mRNA levels, reaching statistical significance in most cases (Figure 4c). Thus, elevated Tip60 mRNA is an indicator of competent HR-directed DNA repair in MEC. Conversely, tumor cells with low Tip60 expression may be more prone to accumulating unrepaired DNA damage and developing genomic instability.
Tip60 gene expression is downregulated in human breast cancers
To investigate the status of Tip60 in human breast cancers, we examined the publicly available Perou and Richardson databases.36, 37 Tip60 was indeed downregulated in most breast cancer samples regardless of subtype (Table 1). The use of a publicly available Kaplan–Meier plotting tool38 showed that patients with basal-like, luminal A or luminal B breast cancers exhibiting with low Tip60 levels tended to have shorter disease-free survival (Figure 5a and d). Breast tumors of the Her2+ subtype showed a similar trend but failed to reach statistical significance due to their relatively small number.

Tip60 expression is downregulated in human breast cancers and correlates with poor prognosis and p53 mutation. (a–d) Kaplan–Meier analyses of risk-free survival of human breast cancer patients grouped by the indicated breast cancer subtypes and by high or low Tip60 expression. (e) Correlation between Tip60 (Kat5) gene copy number and relative Tip60 mRNA expression in breast cancers (n=1098) of the indicated subtypes from the TCGA data set. (f) Cluster analysis of relative Tip60 mRNA levels in the human breast tumors in (e). Tumors were clustered based on their expression of the PAM50 signature,58 which assigns tumors to the indicated breast cancer subtypes. (g) Non-recurrence plot of basal-like breast tumors in patients listed in the TCGA data set and grouped by high or low relative Tip60 mRNA expression. (h) Cluster analysis of relative Tip60 mRNA levels and p53 mutations in breast tumors listed in the TCGA data set. Tumors were clustered by subtype as in (f) and subdivided by the presence of the WT or mutated13 p53 allele. Data were analyzed as in (f). (i) Cluster analysis of relative Tip60 mRNA expression in all breast tumors, and in ER− breast tumors only, from a study by Miller et al. (GSE3494, PMID: 16141321), where 58 malignancies had mutated p53 and 193 malignancies had WT p53. Data were analyzed by unpaired, one-tailed t-test
A similar analysis of data in The Cancer Genome Atlas (TCGA) confirmed that Tip60 expression was low in a significant proportion of human breast cancers (Figure 5e). Tumors expressing the lowest Tip60 levels were mainly of the basal-like subtype (Figure 5f), were associated with poor recurrence-free survival (Figure 5g), and were likely to have p53 mutations (Figure 5h). This latter correlation was particularly significant in basal-like breast cancers, with a similar trend for Her2+ malignancies. Analysis of data from a different breast cancer study39 further corroborated the association between low Tip60 and p53 mutation (Figure 5i).
These human data bolster our mouse findings and support our contention that heterozygous Tip60 deletion contributes to mammary tumorigenesis in KPT mice.
Tip60 expression correlates with genomic rearrangements in mouse and human breast cancers
Because defects in DNA repair are associated with genomic instability,40 we investigated the correlation between Tip60 level and genomic integrity in mouse and human breast tumors. First, we performed comparative genomic hybridization (CGH) of KP and KPT mammary tumors. As expected, KP tumors showed increased genomic rearrangements compared with diploid reference control tissue (Figure 6a). Interestingly, KPT tumors did not show any increase in instability compared with KP tumors (Figure 6a). However, a deep analysis of CGH profiles revealed a pattern of genomic rearrangements that was specific to KPT tumors (Supplementary Figure S6). Thus, the effects of Tip60 heterozygosity on DNA repair favor the occurrence of particular genomic changes.

Tip60 expression is important for maintenance of genomic integrity in mouse mammary tumors and human breast cancers. (a) Quantitation of CNA (i.e., percent genome altered) in genomic DNA of KP (n=3) and KPT (n=4) tumors determined as described in Materials and methods. Data are values for individual tumors. Horizontal lines are mean±S.E.M. n.s., not significant. (b) CNA in TCGA tumors with WT p53 (i.e., not mutated and not homozygously deleted) versus TCGA tumors with mutated p53. Data were analyzed using the Mann–Whitney test. (c) CNA in TCGA tumors ranked in the top or bottom quartiles of Tip60 expression. Data were expressed and analyzed as in (b). (d) CNA in TCGA tumors with WT or mutated p53 as in (b) and classified by level of Tip60 expression as in (c). Data were expressed and analyzed in (c). ***P<0.001
We next analyzed copy number alterations (CNA) in the TCGA data set of human breast cancers. We confirmed a strong correlation between genomic instability and not only p53 status (Figure 6b) but also Tip60 expression (Figure 6c). When we examined only breast tumors carrying WT p53, a strong correlation persisted between Tip60 levels and CNA. This difference between mouse and human tumors may exist because, in the mouse model, the complete ablation of p53 may induce such severe genomic instability that any additional effect due to low Tip60 expression is masked. Overall, our mouse and human data show that Tip60 downregulation accelerates tumorigenesis by favoring genomic rearrangements.
Discussion
In healthy tissues, cellular responses to DNA damage are tightly controlled to avoid the accumulation of unrepaired lesions that can lead to gene mutations. The acetyltransferase Tip60 has been implicated in DDR at various levels: (i) Tip60 acetylates and activates ATM, which is a key transducer of DNA damage signaling;11 (ii) Tip60-mediated acetylation of γH2AX promotes dynamic changes to chromatin that are important for the correct assembly of DNA repair factor complexes;13 and (iii) Tip60 controls the localization on chromatin of factors involved in HR-directed DNA repair, such as BRCA1 and 53BP1.14 On the basis of these observations, we postulated that Tip60 downregulation might contribute to tumorigenesis by decreasing the fidelity of DNA repair, thereby promoting genomic instability which often drives cancerous transformation. If such genomic rearrangements in pre-cancerous cells activate certain crucial genes, then these cells may acquire a selective growth advantage and become aggressive cancers.
Our study showed that Tip60 regulates the capacity of both normal and tumor MEC to mount efficient DDR under both basal conditions and following genotoxic stress induced by IR or cisplatin. Even under basal conditions, persistent shRNA-mediated Tip60 downregulation induced an increase in γH2AX, an indicator of unrepaired DNA damage. Our DR-GFP reporter assay showed that Tip60-silenced cells have a defect in HR-directed DNA repair. When we exposed cells expressing low Tip60 to IR, they did not form DNA damage-associated foci containing BRCA1 and Rad51 and instead accumulated γH2AX. Tip60 levels also correlated inversely with the HRD gene signature associated with defective HR-directed repair. Interestingly, low Tip60 did not affect DNA damage-induced ATM activation. We conclude that Tip60 is an orchestrator of HR-directed DNA repair that coordinates DNA repair factor foci formation. This function is crucial for the maintenance of genomic integrity, and its malfunction can contribute to cancer-associated genomic instability. Our analysis of mouse tumor tissues and cells showed that low Tip60 causes γH2AX accumulation and morphological changes to the nucleus, both of which are linked to tumor aggressiveness and genomic instability. Indeed, we found that Tip60 downregulation favors genomic rearrangements both in human and in mouse tumors.
Our work also addresses the relationship between Tip60 and p53. Examination of the TCGA breast cancer data set showed that tumors with low Tip60 mRNA have p53 mutations. Numerous studies have shown that Tip60 is an important co-factor for p53 transcriptional activation. It would be logical to assume that Tip60 downregulation would be sufficient to reduce p53 activity and therefore relieve the cell from the pressure to mutate p53 itself. However, we have previously shown that Tip60 heterozygosity neither altered the p53 transcriptional signature in Myc-induced lymphomas nor prevented the p53-dependent embryonic lethality of mdm2 knockout mice.6 The present study has demonstrated in vivo cooperation between Tip60 heterozygosity and p53 deletion in mice, and suggests that some of Tip60’s tumor-suppressive functions are p53 independent. Indeed, the correlation we established between Tip60 levels and genomic alterations is independent of p53 status. That being said, how Tip60 expression is modulated during tumorigenesis is still unclear. Multiple cancer-associated pathways have been shown to affect Tip60 expression, stability and function, including acetylation/deacetylation by Sirt1 and HDAC3,41 USP7-mediated deubiquitination,42 and phosphorylation by Abl kinase.43 These issues remain to be clarified.
Our work has important clinical implications. Three studies have recently explored Tip60 as a therapeutic target and proposed the use of Tip60 inhibitors to treat various cancers.44, 45, 46 However, our in vivo mouse results suggest that caution is warranted because any inhibition of Tip60 strong enough to be effective might impair both DNA damage-induced apoptosis and DNA repair, inadvertently promoting cancer cell survival and increasing genomic instability.
In conclusion, our study supports a scenario where the tumor-associated downregulation of Tip60 expression or activity reduces the capacity of the cell to repair DNA damage, leading to an unstable genome and favoring the acquisition of specific mutations that drive tumor progression. This effect is at least partially independent of p53 mutation, making Tip60 an important novel tumor suppressor in its own right.
Materials and Methods
Mice
Heterozygous Tip60-deficient (Tip60+/−) mice were kindly provided by B Amati (Italian Institute of Technology, Milan, Italy) and J Lough (Medical College of Wisconsin, Milwaukee, WI, USA) and were of a mixed 129 × B6 genetic background.6 For mammary tumorigenesis studies, Tip60+/− mice were backcrossed for 10 generations onto the FVB background. We used Tip60+/− mice for this cross because total ablation of the mouse Tip60 gene is embryonic lethal,6, 47 and a recently reported Tip60 conditional knockout mouse48 was not available at the time of this study. Cytokeratin 14-cre transgenic (K14cre) and p53 conditional knockout mice (p53f/f) were kindly provided by A Berns (Netherlands Cancer Institute, Amsterdam, The Netherlands). Primary tumor samples from MMTV-Neu and MMTV-PyMT mice were kindly provided by V Stambolic and P Dutt (Ontario Cancer Institute, Toronto, Canada). Tumor-bearing mice were monitored twice a week and killed when they reached the humane end point (tumor diameter of 1cm). All mice were maintained and handled according to protocols approved by the Animal Care and Use Committee of the University Health Network, Toronto, Canada.
Cell lines
COMMA-1D (COMMA) cells were obtained from S Muthuswamy (Ontario Cancer Institute, Toronto, Canada). Cells from primary mammary tumors originating in MMTV-PyMT and WAPcre;PTENf/f mice were isolated as described.28, 29 All mouse mammary cell lines were maintained in DMEM/F12 medium containing 10% FBS, l-glutamine, 1 μg/ml hydrocortisone (Sigma), 5 μg/ml insulin (Sigma, Oakville, ON, Canada), and 5 ng/ml epidermal growth factor (Sigma). For cisplatin treatments, COMMA and WAPcre;PTENf/f cells were seeded at 70% confluence and treated with 2, 5, or 10 μM cisplatin for 24 h. For DDR analysis, COMMA cells were irradiated at 5 Gy and stained 1, 8, or 24 h later to detect BRCA1, Rad51, or γH2AX as described below.
Immunoblotting
Immunoblotting to detect specific proteins in cell extracts prepared in RIPA buffer was performed according to standard procedures. The following antibodies (Abs) were used: anti-Tip60 (CLHF), the kind gift of B Amati (Italian Institute of Technology, Milan, Italy); anti-γH2AX (Millipore, Burlington, ON, Canada); anti-cleaved PARP (Cell Signaling, Danvers, MA, USA); anti-vinculin (Abcam, Cambridge, United Kingdom), and anti-β-actin (Sigma). Anti-mouse and anti-rabbit HRP-conjugated secondary Abs were obtained from Amersham (Mississauga, ON, Canada). Immunoblots were developed both on X-ray films and with a Microchemi chemoluminescence analyser (DNR Bio-Imaging Systems Ltd, Jerusalem, Israel). Immunoblot quantification was performed using the ImageJ software (NIH Image, Bethesda, MD, USA).
Tip60 silencing
Non-targeting scrambled siRNA (scr; Cat. No. D-001810-10) and Tip60-directed siRNA (Cat. No. L-057795-02) oligomers were from Dharmacon (Lafayette, CO, USA). Lentiviral-based pLKO shRNAs against the non-target or mouse Tip60 genes were from Sigma. For siRNA and shRNA studies, cells were processed as previously described.49
Immunofluorescence
Cells cultured on glass coverslips were rinsed in PBS, fixed with 3.7% formaldehyde in PBS for 10 min at room temperature (RT), permeabilized with PBS plus 0.5% Triton X-100 for 5 min, blocked overnight at 4°C with PBS containing 1% bovine serum albumin,47 and then incubated with primary Abs recognizing RAD51 (Santa Cruz, Dallas, TX, USA, 8349, 1 : 500), γH2AX (Millipore 05-636, 1 : 1000) or BRCA1 (Santa Cruz 646, 1 : 100). After three washes with PBS for 5 min at RT, samples were incubated for 30 min with a 1 : 400 dilution (in PBS) of goat anti-rabbit IgG conjugated to the fluorescent Alexa 488 dye or with goat anti-mouse IgG conjugated to the fluorescent Alexa 546 dye (Invitrogen Molecular Probes, Grand Island, NY, USA). Stained slides were washed three times, counterstained with DAPI, and mounted in Mowiol. Stained cells were imaged using an Axio Observer inverted microscope (Carl Zeiss Canada Ltd, Toronto, ON, Canada). Stained cells were considered positive for DNA repair factor staining if they contained at least 3 foci/cell.
HR assay
To assess HR efficiency, COMMA cells were stably transduced with the DR-GFP construct (Addgene #26475, Cambridge, MA, USA) and with siRNA oligomers against Tip60 or BRCA1. Cells were first stably transfected with pCMV3xNLS-I SceI (a gift of R Bristow, Ontario Cancer Institute, Toronto, Canada) and then transfected with siRNA oligomers using Lipofectamine (Invitrogen, Grand Island, NY, USA). At 2 days post transfection, GFP signals were quantified in 10 000 cells/group using an FACSCalibur flow cytometer (BD Biosciences, Mississauga, ON, Canada). Recombination efficiency was calculated as the number of GFP+ cells in a sample divided by the number of GFP+ cells among control COMMA-D cells. The same experiment was conducted in WAPcre;PTENf/f cells stably expressing shGFP or shTip60.
Tissue immunohistochemistry
Tissue sections (4 μm) that were formalin-fixed and paraffin-embedded were dewaxed in xylene, stained lightly with Mayer’s hematoxylin, dehydrated in an alcohol series, cleared in xylene, and mounted in Permount (Fisher Scientific Company, Ottawa, Canada; Cat. No. SP15-500). Staining to detect ER, PR, and Ki67 was performed as previously described,50 as was Tip60 staining.6 Specimens were viewed with a brightfield microscope (Leica DM2500, Leica Microsystems, Concord, ON, Canada) equipped with Micropublisher 3.3-QI imaging camera), analyzed using the Q-Capture Pro software (QImaging, Surrey, BC, Canada), and processed with Adobe Photoshop CS5.
Genomic PCR
Genomic PCR was conducted as previously described.49 Primer sequences used are listed in Supplementary Table 1.
RT-PCR
RNA was isolated using the Nucleospin RNA II kit (Macherey-Nagel, Bethehem, PA, USA) and reverse-transcribed using the iScript cDNA synthesis kit (Bio-Rad, Mississauga, ON, Canada) according to the manufacturers’ instructions. Quantitative RT-PCR was performed using SYBR Green primers (Applied Biosystems, Foster City, CA, USA). The mouse ribosomal protein S9 (rps9) was used as the housekeeping gene for normalization and determination of relative mRNA expression. Primer sequences used are listed in Supplementary Table 2.
Cell-cycle analysis
Cell-cycle analysis was performed by propidium iodide (PI) staining as described.49
Apoptosis assay
Apoptosis was evaluated by AnnexinV/7-AAD staining and flow cytometry. Briefly, 4 × 105 cells were stained for 15 min at RT with FITC-conjugated AnnexinV plus 7-AAD in 1 × AnnexinV binding buffer (all from BD Biosciences). Stained cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences).
Analyses of human breast cancer data sets
The Richardson37 and Perou36 data sets were independently analyzed to determine HTATIP (Tip60) mRNA expression. The Kaplan–Meier plotting tool (http://kmplot.com/analysis/)51 was used to evaluate the risk-free survival of breast cancer patients expressing low or high levels of Tip60. Tip60 expression was scored using the Jetset best probe set.52 Pre-processed and normalized gene expression and copy number data, as well as recurrence information, were downloaded from the corresponding TCGA breast cancer repository (version 3 July 2012; http://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp).53 Tumor profiles were clustered based on expression of the PAM50 signature, which assigns tumors to the breast cancer subtypes basal-like, HER2-enriched, luminal A, or luminal B. Clustering and correlation analyses were performed using R software (R Foundation for Statistical Computing, Vienna, Austria). The GSEA54 tool was employed using default values for all parameters. Information on specific gene sets was downloaded from the published report on the HRD signature34 or from the Biocarta repository. Normalized gene expression and p53 mutation data were obtained for a cohort of 251 breast cancer patients from the Gene Expression Omnibus (GSE3494). Tip60 expression levels (Affymetrix U133A Probe 206689_x_at) were compared between p53 mutant (n=58) and p53 WT (n=193) tumors using a one-tailed Student's t-test.
Array aCGH
aCGH (4x180k Agilent array) was performed by the Princess Margaret Genomics Centre according to the manufacturer’s instructions. Experimental and reference diploid DNA samples were labeled with Cy5 (red) and Cy3 (green) dyes, respectively. Agilent feature extraction files were analyzed using DNA Nexus 7.5 (BioDiscovery, Hawthorne, CA, USA). Briefly, log2 ratios (Cy5/Cy3) were normalized using a linear systematic correction. Genomic segmentation to identify CNA was performed using the Rank Segmentation algorithm applying the default settings with a minimum number of probes per segment equal to 30. Genomic coordinates were mapped to mouse build mm9/NCBI37.
The percentage of the genome altered and the total number of CNA were compared between KP and KPT tumors using an unpaired t-test, with a two-tailed P-value of <0.05 considered as significant. Regions of CNA were compared between KP and KPT tumors using the Nexus Comparison function and the default settings (minimum frequency difference between groups=25% and two-tailed Fisher’s exact test P-value <0.05).
TCGA BRCA data set
The fraction of genome altered (FGA) by CNA for BRCA patients was downloaded from the clinical summary tab in cBioportal.55, 56 Gene expression levels, copy number calls, and mutation status for Tip60 (KAT5) and TP53 were obtained from the Cancer Genomics Browser.57 Correlations between gene expression and FGA were calculated by comparing FGA in tumors with Tip60 expression in the top and bottom quartiles of all p53 WT BRCA tumors (defined as neither mutated nor homozygously deleted). A P-value of <0.05 by the Mann–Whitney test was considered as significant.
Statistical analyses
For mouse Kaplan–Meier survival curves, P-values were determined using the log-rank test. Most results were reported as the mean±standard error of the mean (S.E.M.). The two-sided independent Student's t-test without equal variance assumption was used to determine P-values of differences between groups. *P<0.05, **P<0.01, ***P<0.001.
Abbreviations
- Tip60/KAT5:
-
60 kDa Tat-interactive protein/Lysine acetyltransferase 5
- HR:
-
homologous recombination
- DDR:
-
DNA damage response
- ATM:
-
ataxia telangiectasia mutant
- BRCA1:
-
breast cancer type 1 susceptibility protein
- 53BP1:
-
p53-binding protein 1
- KP:
-
K14cre;p53f/f
- KPT:
-
K14cre;p53f/f;Tip60+/−
- ER:
-
estrogen receptor
- LOH:
-
loss-of-heterozygosity
- WT:
-
wild type
- MEC:
-
mammary epithelial cells
- IR:
-
ionizing radiation
- DR-GFP:
-
direct repeat green fluorescent protein
- MMTV-PyMT:
-
mouse mammary tumor virus/polyoma middle T-antigen
- MMTV-Neu:
-
mouse mammary tumor virus/proto-oncogene Neu
- WAPcre:
-
whey acidic protein activated CRE
- GSEA:
-
Gene Set Expression Analysis
- HRD:
-
homologous recombination defect
- CNA:
-
copy number alterations
- CGH:
-
comparative genomic hybridization
References
Squatrito M, Gorrini C, Amati B . Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol 2006; 16: 433–442.
Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 2006; 24: 841–851.
Tang Y, Luo J, Zhang W, Gu W . Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 2006; 24: 827–839.
Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen CJ et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol Cell Biol 2004; 24: 10826–10834.
Gaughan L, Logan IR, Cook S, Neal DE, Robson CN . Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J Biol Chem 2002; 277: 25904–25913.
Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007; 448: 1063–1067.
Chevillard-Briet M, Quaranta M, Grezy A, Mattera L, Courilleau C, Philippe M et al. Interplay between chromatin-modifying enzymes controls colon cancer progression through Wnt signaling. Hum Mol Genet 2014; 23: 2120–2131.
Chen G, Cheng Y, Tang Y, Martinka M, Li G . Role of Tip60 in human melanoma cell migration, metastasis, and patient survival. J Invest Dermatol 2012; 132: 2632–2641.
Legube G, Linares LK, Tyteca S, Caron C, Scheffner M, Chevillard-Briet M et al. Role of the histone acetyl transferase Tip60 in the p53 pathway. J Biol Chem 2004; 279: 44825–44833.
Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004; 428: 431–437.
Sun Y, Jiang X, Chen S, Fernandes N, Price BD . A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA 2005; 102: 13182–13187.
Kaidi A, Jackson SP . KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature 2013; 498: 70–74.
Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol 2007; 27: 7028–7040.
Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 2013; 20: 317–325.
Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434: 864–870.
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444: 638–642.
Gorgoulis VG, Zoumpourlis V, Rassidakis GZ, Karameris A, Rassidakis AN, Spandidos DA et al. A molecular and immunohistochemical study of the MDM2 protein isoforms and p53 gene product in bronchogenic carcinoma. J Pathol 1996; 180: 129–137.
Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012; 149: 1269–1283.
Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci USA 2007; 104: 12111–12116.
Elston CW, Ellis IO . Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology 1991; 19: 403–410.
Gisselsson D, Bjork J, Hoglund M, Mertens F, Dal Cin P, Akerman M et al. Abnormal nuclear shape in solid tumors reflects mitotic instability. Am J Pathol 2001; 158: 199–206.
McAllister D, Merlo X, Lough J . Characterization and expression of the mouse tat interactive protein 60 kD (TIP60) gene. Gene 2002; 289: 169–176.
Fisher JB, Kim MS, Blinka S, Ge ZD, Wan T, Duris C et al. Stress-induced cell-cycle activation in Tip60 haploinsufficient adult cardiomyocytes. PLoS One 2012; 7: e31569.
Hobbs CA, Wei G, DeFeo K, Paul B, Hayes CS, Gilmour SK . Tip60 protein isoforms and altered function in skin and tumors that overexpress ornithine decarboxylase. Cancer Res 2006; 66: 8116–8122.
Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S et al. GammaH2AX and cancer. Nat Rev Cancer 2008; 8: 957–967.
van Gent DC, Hoeijmakers JH, Kanaar R . Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2001; 2: 196–206.
Pierce AJ, Johnson RD, Thompson LH, Jasin M . XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999; 13: 2633–2638.
Cappello P, Blaser H, Gorrini C, Lin DC, Elia AJ, Wakeham et al. Role of Nek2 on centrosome duplication and aneuploidy in breast cancer cells. Oncogene 2014; 33: 2375–2384.
Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ et al. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 2013; 341: 395–399.
Sun Y, Xu Y, Roy K, Price BD . DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol 2007; 27: 8502–8509.
Jamieson ER, Lippard SJ . Structure, recognition, and processing of cisplatin-DNA Adducts. Chem Rev 1999; 99: 2467–2498.
Borst P, Rottenberg S, Jonkers J . How do real tumors become resistant to cisplatin? Cell Cycle 2008; 7: 1353–1359.
Basu A, Krishnamurthy S . Cellular responses to cisplatin-induced DNA damage. J Nucleic Acids 2010; 2010: 2010–2026.
Peng G, Chun-Jen Lin C, Mo W, Dai H, Park YY, Kim SM et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat Commun 2014; 5: 3361.
Dutka J, Lowe SS, Michaud M, Watanabe S . Long-term use of methylnaltrexone for the management of constipation in advanced cancer. J Support Oncol 2009; 7: 177–179.
Hoadley KA, Weigman VJ, Fan C, Sawyer LR, He X, Troester MA et al. EGFR associated expression profiles vary with breast tumor subtype. BMC Genomics 2007; 8: 258.
Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 2006; 9: 121–132.
Gyorffy B, Lanczky A, Szallasi Z . Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer 2012; 19: 197–208.
Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA 2005; 102: 13550–13555.
Negrini S, Gorgoulis VG, Halazonetis TD . Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010; 11: 220–228.
Yi J, Huang X, Yang Y, Zhu WG, Gu W, Luo J . Regulation of histone acetyltransferase TIP60 function by histone deacetylase 3. J Biol Chem 2014; 289: 33878–33886.
Gao Y, Koppen A, Rakhshandehroo M, Tasdelen I, van de Graaf SF, van Loosdregt J et al. Early adipogenesis is regulated through USP7-mediated deubiquitination of the histone acetyltransferase TIP60. Nat Commun 2013; 4: 2656.
Shin SH, Kang SS . Phosphorylation of Tip60 Tyrosine 327 by Abl Kinase Inhibits HAT Activity through association with FE65. Open Biochem J 2013; 7: 66–72.
Gao C, Bourke E, Scobie M, Famme MA, Koolmeister T, Helleday T et al. Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Sci Rep 2014; 4: 5372.
Yang C, Ngo L, Zheng YG . Rational design of substrate-based multivalent inhibitors of the histone acetyltransferase Tip60. ChemMedChem 2014; 9: 537–541.
Coffey K, Blackburn TJ, Cook S, Golding BT, Griffin RJ, Hardcastle IR et al. Characterisation of a Tip60 specific inhibitor, NU9056, in prostate cancer. PLoS One 2012; 7: e45539.
Hu Y, Fisher JB, Koprowski S, McAllister D, Kim MS, Lough J . Homozygous disruption of the Tip60 gene causes early embryonic lethality. Dev Dynamics 2009; 238: 2912–2921.
Xiao Y, Nagai Y, Deng G, Ohtani T, Zhu Z, Zhou Z et al. Dynamic interactions between TIP60 and p300 regulate FOXP3 function through a structural switch defined by a single lysine on TIP60. Cell Rep 2014; 7: 1471–1480.
Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J Exp Med 2013; 210: 1529–1544.
Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL et al. Progesterone induces adult mammary stem cell expansion. Nature 2010; 465: 803–807.
Gyorffy B, Surowiak P, Budczies J, Lanczky A . Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 2013; 8: e82241.
Li Q, Birkbak NJ, Gyorffy B, Szallasi Z, Eklund AC . Jetset: selecting the optimal microarray probe set to represent a gene. BMC Bioinformatics 2011; 12: 474.
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490: 61–70.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1.
Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D et al. Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Sci Rep 2013; 3: 2652.
Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009; 27: 1160–1167.
Acknowledgements
We thank Bruno Amati and Jos Jonkers for insightful discussions, Jillian Haight and Drew Wakeham for assistance with mouse studies, Meegan Larsen for mouse tumor histopathology, and Mary Saunders for scientific editing of this manuscript. This work was supported by an FIRC Fellowship for Abroad (CG), an EMBO Long Term Fellowship (CG), and grants from the AGAUR Generalitat de Catalunya (2014-SGR364) (MAP), the Spanish Ministry of Health ISCIII FIS (PI12/01528) (MAP), and RTICC (RD12/0036/0008) (MAP).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Bassi, C., Li, YT., Khu, K. et al. The acetyltransferase Tip60 contributes to mammary tumorigenesis by modulating DNA repair. Cell Death Differ 23, 1198–1208 (2016). https://doi.org/10.1038/cdd.2015.173
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.173
This article is cited by
-
NAA60 (HAT4): the newly discovered bi-functional Golgi member of the acetyltransferase family
Clinical Epigenetics (2022)
-
Loss of TIP60 (KAT5) abolishes H2AZ lysine 7 acetylation and causes p53, INK4A, and ARF-independent cell cycle arrest
Cell Death & Disease (2022)
-
Potential role of human papillomavirus proteins associated with the development of cancer
VirusDisease (2022)
-
MYC: a multipurpose oncogene with prognostic and therapeutic implications in blood malignancies
Journal of Hematology & Oncology (2021)
-
SRPK1 acetylation modulates alternative splicing to regulate cisplatin resistance in breast cancer cells
Communications Biology (2020)
