
Abstract
Inducing senescence in cancer cells is an effective approach to suppress cancer growth, and it contributes significantly to the efficacy of therapeutic drugs. Previous studies indicated that transcription factors NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) and C/EBPβ (CCAAT/enhancer-binding protein-β) play a critical role in the establishment of senescence by upregulating proinflammatory cytokines, notably interleukin-6 (IL-6) and interleukin-8 (IL-8). However, it is not clear how these two factors are activated in response to senescence-inducing stimuli and subsequently regulate gene transcription. Here, we reveal Bcl-2-associated transcription factor 1 (Bclaf1) as a novel player in the therapeutic drug doxorubicin-induced senescence (TIS) in multiple cancer cells. Bclaf1 is upregulated through the ATM/Nemo/NF-κB pathway during TIS and is a direct target of p65 and c-Rel. The induction of Bclaf1 by NF-κB is essential for C/EBPβ upregulation and IL-6/IL-8 transcription during TIS. Bclaf1 can interact with the leucine zipper region of C/EBPβ and cooperate with C/EBPβ to upregulate IL-8. Furthermore, we show that Bclaf1 is required for the effectiveness of doxorubicin (Dox) treatment-induced tumor suppression in a xenograft tumor model. These finding suggest that Bclaf1 plays a crucial role in transducing the senescence-inducing signal from NF-κB to C/EBPβ during TIS, thus amplifying the signals for the establishment of senescence. Given the recent revelation that Bclaf1 is involved in tumorigenesis, our data indicate that the responsiveness of Bclaf1 to NF-κB may determine the effectiveness of therapeutic drugs.
Similar content being viewed by others


Main
Cellular senescence is a protective cellular response to a variety of external and internal stresses and serves to prevent damaged cells from proliferating.1 The types of stresses include but are not limited to telomere attrition,2 excessive mitogenic stimulation3 and DNA damage.4 Accumulative evidence supports the notion that cellular senescence physiologically functions as an essential tumor suppression mechanism especially at the premalignant stage.5, 6 Numerous chemotherapeutic agents, including the commonly used chemotherapeutic agent doxorubicin (Dox), induce senescence in cancer cells,7 significantly contributing to their effectiveness. We refer to therapeutic drug doxorubicin-induced senescence as TIS.
A key feature of cellular senescence is stable cell cycle arrest that is often executed by the p53 and Rb tumor suppressor pathways.3, 8 Senescent cells also display other features such as an increased senescence-associated galactosidase (SA-β-gal) activity9 and the secretion of various factors including chemokines and cytokines.10, 11 The latter phenomenon is referred to as senescence-associated secretory phenotype (SASP), and this process may contribute to the elimination of senescent cells by immune cells in vivo.6 Although SASP was initially considered a hallmark of senescence, it has become clear that numerous SASP components, such as interleukin-6 (IL-6) and interleukin-8 (IL-8), are produced at the early stage of the senescent process and actively promote the initiation and reinforcement of senescence.12, 13, 14 In addition, during the course of cellular senescence, cells undergo massive changes in gene expression profiles, collectively leading to the establishment of the senescence program that includes SASP.11, 15
Recent studies suggest that the transcriptional factors NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) and C/EBPβ (CCAAT/enhancer-binding protein-β), which are critically involved in cytokine production in immune response, play a prominent role in gene regulation associated with the induction of SASP and senescence.12, 15, 16 However, the mechanism by which these two factors are activated in response to senescence-inducing stimuli and subsequently induce transcriptional changes in senescence remains unclear.
A persistent DNA damage response (DDR) appears to be a pivotal trigger for cellular senescence.4 It is also the key mechanism of many cancer therapeutic drugs. DNA damage foci and activation of the key components of DDR are often observed in cells undergoing senescence induced by various stresses.4, 10 DDR induces cell cycle arrest through the p53/p21 and Rb/p16 pathways17, 18 and is essential for SASP induction typically with the involvement of DDR components, such as ataxia telangiectasia mutated protein (ATM), checkpoint kinase 2 (CHK2) and NBS.4, 16 However, the critical players and exact pathways linking DDR to NF-κB and C/EBPβ as well as SASP have not been well defined.
Bclaf1 (Bcl-2-associated transcription factor1) was first identified in a yeast two-hybrid screen as a binding protein for the adenoviral Bcl2 homolog E1B19K.19 Bclaf1 knockout mice are embryonic lethal because of defects in lung development.20 Ectopical Bclaf1 expression induces apoptosis in various cell types or autophagic cell death in myeloma cells.19, 21 However, thymocytes and splenocytes isolated from Bclaf1 knockout mice exhibit normal responsiveness to various apoptotic stimulations, thus arguing its role in apoptosis at physiological levels.20 The Bclaf1 protein contains homologies to the basic zipper (bZIP) and Myb DNA-binding domain and can bind to DNA.19 Several studies suggested a role of Bclaf1 in DDR. Bclaf1 promotes TP53 gene transcription through interaction with PKCδ in response to DNA damage22, 23 and interacts with γ-H2AX in response to ionizing radiation.24 Although most of studies suggest that Bclaf1 is a tumor suppressor, a recent report indicated that Bclaf1 was oncogenic in colorectal cancers.25 Thus, the function of Bclaf1 in carcinogenesis is controversial.
Here, we report that Bclaf1 is an important player in TIS in multiple cancer cells. We provide evidence to suggest that NF-κB is activated by the ATM/NF-κB essential modulator (Nemo) axis and that Bclaf1 is a downstream gene of NF-κB that is required for C/EBPβ upregulation and cooperates with C/EBPβ to regulate transcription of SASP components.
Results
Bclaf1 is involved in TIS in multiple cancer cells
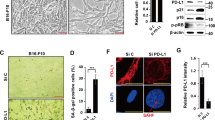
Senescence is an important mechanism contributing to the effectiveness of DNA damage-based chemotherapies.26, 27 In an effort to understand the underlying molecular mechanism, we chronically treated MCF-7 breast cancer cells as well as HCT116 and HT-29 colon carcinoma cells with low concentrations of Dox that induces DNA double-strand breaks.28 As reported previously,7 extensive cellular senescence was observed in three types of cells on day 7. Western blot analysis and immunostaining demonstrated that Bclaf1, a protein recently implicated in DDR, was upregulated during TIS (Figures 1a and b and Supplementary Figures S1A–C). This upregulation was not evident during the first 2 days of Dox treatment (Supplementary Figure S1B). Bclaf1 knockdown with two individual small interfering RNAs (siRNAs; Figures 1g–i) significantly reduced senescence in three types of cells (Figures 1d–f), indicating that Bclaf1 is involved in the regulation of senescence. In addition, both Ki67 staining (Supplementary Figure S1D) and cell proliferation assay by MTT (Supplementary Figures S1E and F) showed that Bclaf1 knockdown slightly retarded cell growth in unstressed conditions, but markedly decreased Dox-induced cell growth inhibition in HCT116 and HT-29 cells (Figures 1j and k and Supplementary Figure S1D), supporting the role of Bclaf1 in TIS. However, these data also suggest that Bclaf1 may play a different role in unstressed cells.

Bclaf1 is involved in cellular senescence. (a and b) Bclaf1 is upregulated upon Dox treatment. MCF-7 (a) or HCT116 (b) cells either untreated or treated with Dox (50 ng/ml) for the indicated days were subject to western blot analysis using the indicated antibodies. (c–f) Knockdown of Bclaf1 reduces the Dox-induced senescence. MCF7 (c and d), HCT116 (e) or HT-29 (f) cells transfected with the control (siCtrl) or two independent siRNAs against Bclaf1 were treated with Dox (50 ng/ml) for 7 days, and then stained for SA-β-gal. Representative images of stained MCF-7 cells of each treatment are shown in (c). The percentages of cells positive for SA-β-gal were calculated and graphed as shown in (e) and (f). At least 200 cells from four random chosen areas were counted for each group. Error bars represent mean+S.D. (n=4). The knockdown efficiency is presented in (g–i). (j and k) Bclaf1 knockdown decreased Dox-induced inhibition of cancer cell growth. 7.5 × 103 of HCT116 (j) or HT-29 (k) cells expressing the indicated shRNAs were seeded in 96-well plates and treated with 50 ng/ml doxorubicin. Cells were then analyzed using the MTT assay. Values of each point are means of triplicate. ***P<0.001
DNA damage-mediated Bclaf1 upregulation is dependent on ATM/ATR and Nemo
To delineate the pathways involved in Bclaf1 upregulation, we first individually knocked down ATM and ATR (ataxia telangiectasia and rad3 related) using their respective siRNAs. ATM and ATR are central kinases in the signaling cascade of DDR.29 As expected, on day 3 of treatment, DNA damage-induced phosphorylation of the effector kinases checkpoint kinase 1 (Chk1) and Chk2 was diminished upon knockdown of either ATM or ATR (Figure 2a). Not surprisingly, Bclaf1 upregulation was also abolished at both protein and mRNA levels (Figure 2a and Supplementary Figure S2A), suggesting that Bclaf1 is upregulated in response to DDR. Accordingly, the reduction in senescence resulting from ATM or ATR knockdown was not further enhanced upon simultaneous knockdown of Bclaf1 (Figure 2b), confirming that the action of Bclaf1 is downstream of ATM/ATR. Although p53 is a downstream effector in DDR and critically involved in senescence, Bclaf1 was upregulated by Dox in p53-deficient HCT116 cells but to a lesser extent (Supplementary Figures S2B and C). Although this result cannot exclude the involvement of p53, it suggests that other transcription factors play a role in this process.

Bclaf1 upregulation by DNA damage is dependent on ATM/ATR and Nemo. HCT116 cells were transfected with siCtrl or siRNAs against ATM or ATR (a and b) or Nemo (c and d). At 24 h after transfection, the cells were untreated or treated with Dox (50 ng/ml) for 3 days followed by western blot analysis (a and c), or for 7 days followed by SA-β-gal staining and cell counting as described above (b and d). (e) Cells treated as described in (c) were subjected to nuclear–cytoplasm fractionation. β-Tubulin and lamin B were used as cytoplasmic and nuclear loading controls, respectively. **P<0.01 and ***P<0.001
Given that NF-κB is a master regulator of gene transcription associated with SASP and senescence, we hypothesize that it may upregulate Bclaf1. To examine this hypothesis, we knocked down Nemo using a specific siRNA given that Nemo is the critical link between ATM and NF-κB activation in other scenarios of DDR.30 Nemo depletion inhibited Dox-induced nuclear translocation of p65 and c-Rel (Figure 2e), a critical event in NF-κB activation. Bclaf1 upregulation was also abrogated (Figure 2c and Supplementary Figure S2D). Moreover, compared with the control siRNA-transfected cells, fewer senescent and more proliferative cells, which were measured by Ki67 staining, were observed in Nemo or Bclaf1 siRNA-transfected cells after Dox treatment. However no additive effects were observed when Bclaf1 and Nemo were simultaneously knocked down (Figure 2d and Supplementary Figure S3). These results indicate that Bclaf1 upregulation occurs downstream of the ATM/Nemo axis.
Bclaf1 is a direct target of NF-κB
To directly investigate whether Bclaf1 is regulated by NF-κB, we transfected each individual member of NF-κB into 293T cells and found that p65 and c-Rel increased the endogenous expression of Bclaf1 (Figure 3a). To confirm this finding, we transfected siRNAs against p65 and c-Rel either alone or together into HCT116 cells before Dox treatment. As expected, induction of Bclaf1 expression by DNA damage was partially inhibited by downregulation of either p65 or c-Rel, and a more complete inhibition was observed when both proteins were downregulated (Figure 3b and Supplementary Figure S2E). In addition, similar phenotype was also observed in HCT116 p53−/− cells, despite that depletion of both p65 and c-Rel did not further enhance this impact (Supplementary Figure S2F) To investigate whether Bclaf1 was directly regulated by NF-κB, we analyzed the promoter region of human Bclaf1 and identified a potential NF-κB-binding consensus sequence as indicated (Figure 3c). We fused this region upstream of a luciferase reporter gene. Co-transfection of p65 with the reporter stimulated the luciferase activity (Figure 3d), indicating that Bclaf1 is a target of NF-κB. To further confirm this finding, we performed chromatin immunoprecipitation (ChIP) assays using p65- or c-Rel-specific antibodies in both untreated and Dox-treated cells. The results suggested that the binding of p65 to the potential NF-κB-binding region within the Bclaf1 promoter is induced by Dox treatment, whereas the binding of c-Rel is constitutive (Figure 3e). Taken together, these data demonstrated that p65 and c-Rel are bound to the Bclaf1 promoter and transcriptionally activate Bclaf1 during TIS.

Bclaf1 upregulation by DNA damage is regulated by NF-κB. (a) NF-κB overexpression upregulates Bclaf1. 293T cells were transfected with either empty vector (−) or various doses (0.25, 0.5 or 1 μg) of p65-pCDNA3 (upper) or Flag-c-Rel-pRK5 (lower). After 24 h, the cells were analyzed by western blot (b). NF-κB knockdown reduces DNA damage-induced Bclaf1 upregulation. HCT116 cells were transfected with siCtrl or siRNAs against p65 or c-Rel individually or together. After 24 h, the cells were untreated or treated with Dox (50 ng/ml) for 3 days, lysed and analyzed by western blot. (c) Schematic diagram of the putative promoter region of Bclaf1. The relative positions of PCR primer sets used for ChIP assay below are presented (P1, P2). RE refers to the potential NF-κB responding element in the nucleotide sequence; NS refers to the nonspecific distal Bclaf1 promoter region. (d) Bclaf1 RE was fused with a luciferase reporter and co-transfected with or without the p65 expression vector into 293T cells. The experiments were performed in triplicate with a Renilla reporter in the transfection mixture for normalization. Error bars represent mean+S.D. (n=3). (e) p65 and c-Rel bind to the promoter region of Bclaf1. HCT116 cells untreated or treated with Dox for 3 days were subject to ChIP analysis using anti-p65, anti-c-Rel and control IgG antibodies for immunoprecipitation followed by PCR analysis of promoter regions. ***P<0.001
Bclaf1 is required for C/EBPβ upregulation during TIS
C/EBPβ is another important transcription factor for the establishment of the SASP. C/EBPβ works synergistically with NF-κB to induce transcription of various cytokines including IL-6 and IL-8.12, 13 C/EBPβ is upregulated in a NF-κB-dependent manner during TIS given that knockdown of Nemo or p65 and c-Rel abrogated C/EBPβ upregulation (Figure 4a and Supplementary Figure S5A). Strikingly, siRNA- or short hairpin RNA (shRNA)-mediated depletion of Bclaf1 also profoundly impaired the induction of C/EBPβ by Dox in both HCT116 and HT-29 cells (Figure 4b and Supplementary Figure S5B), but p65 nuclear translocation was not affected in HCT116 cells (Supplementary Figures S4A and B). The induction of p15, a downstream target of C/EBPβ and inhibitor of cell cycle,13 by Dox treatment was also reduced upon Bclaf1 knockdown, whereas inductions of other cell cycle inhibitors p21 and p27 as well as p53 and p53 downstream genes were not affected (Figure 4b and Supplementary Figure S5B). However, p16 was not detected in HCT116 and HT-29 cells (Figure 4b and Supplementary Figure S5B), probably because of epigenetic silencing by methylation that occurs in the promoter region of p16 as previously reported.31 These results suggest that Bclaf1 may mediate C/EBPβ upregulation by NF-κB.

Bclaf1 upregulates C/EBPβ. (a) Western blot analysis of HCT116 cells transfected with siCtrl or siNemo and treated with Dox for 3 days. (b) HT-29 cells expressing shCtrl or two shRNAs against Bclaf1 were treated with Dox for 7 days followed by western blot analysis. (c) HCT116 transfected with siCtrl or siNemo together with or without the expression plasmids for Bclaf1 or its mutants were treated with Dox for 3 days followed by western blot analysis. FL, full length; ΔRS, RS deleted mutant; ΔM, Myb domain deleted mutant. (d and e) Bclaf1 overexpression upregulates C/EBPβ. HCT116 cells transfected with either empty vector (−) or Flag-Bclaf1 were harvested at the indicated times and analyzed by western blot (d) or quantitative real-time PCR (e). (f) The schematic representation of the C/EBPβ-LAP promoter with the indicated regions (p1 to p3) for PCR analysis. The position is relative to the start of the first exon. ChIP analysis of the C/EBPβ promoter in HCT116 cells transfected with siCtrl or siC/EBPβ followed by 3 days of treatment with Dox. Immunoprecipitation was performed using anti-Bclaf1 and control IgG antibodies followed by PCR analysis. (g) SA-β-gal analysis of HCT116 cells transfected with the indicated siRNAs and treated with Dox for 7 days. The cells positive for SA-β-gal were counted and graphed. **P<0.01 and ***P<0.001
To prove this hypothesis, we co-transfected the Bclaf1 expression plasmid with Nemo siRNA into HCT116 cells and treated these cells with Dox. Nemo knockdown suppressed C/EBPβ and Bclaf1 upregulation (Figure 4a). However, forced expression of Bclaf1 restored C/EBPβ upregulation, whereas the Myb deletion mutant did not (Figure 4c). These results indicate that Bclaf1 is required for C/EBPβ upregulation during TIS and its Myb domain may play a role in this process.
We next examined the effect of Bclaf1 overexpression on C/EBPβ. Despite causing mild cell death, Bclaf1 overexpression in HCT116 increased C/EBPβ mRNA and protein expression at 72 h, confirming that Bclaf1 upregulation induces C/EBPβ expression (Figures 4d and e). To explore the mechanisms by which Bclaf1 upregulates C/EBPβ, we performed a ChIP assay to examine whether Bclaf1 binds to the promoter region of the transcriptionally active isoform of C/EBPβ-LAP (liver-enriched transcriptional activator protein). We designed three sets of primers that spanned –15 to –543 bp (Figure 4f, +1 indicates the first bp of exon 1) of the C/EBPβ promoter and demonstrated that Bclaf1 was specifically recruited to the p3 region (Figure 4f) and the amount was increased after Dox treatment (Supplementary Figure S5D).
To further examine the functional relationship between Bclaf1 and C/EBPβ in TIS, we knocked down these two proteins individually or in combination and demonstrated that senescence was inhibited by depleting either protein. In addition, no additive effect was observed when both proteins were simultaneously depleted, indicating these two proteins function in the same pathway (Figure 4g). Taken together, our results suggest that Bclaf1-induced C/EBPβ upregulation is a key event in TIS.
Bclaf1 interacts with C/EBPβ
C/EBPβ is subject to autoregulation driven by homo- or hetero-dimerization with other members of the C/EBPβ family.32, 33 We therefore examined whether Bclaf1 interacts with C/EBPβ. We untreated or treated HCT116 cells with Dox for 3 days and extracted chromatin-binding proteins for immunoprecipitation. We found C/EBPβ was present in the Bclaf1 immunocomplex in both treated and untreated cells (Figure 5a). We further determined the regions in both proteins that mediated their interaction via immunoprecipitation of Flag-tagged either Bclaf1 or C/EBPβ and their deletion mutants followed by western blot analysis of the presence of HA-C/EBPβ or endogenous Bclaf1. The results indicated that the leucine zipper region of C/EBPβ (Figure 5c) and the Myb domain of Bclaf1 (Figure 5b) are involved in the interaction. In addition, other regions of Bclaf1 also made contributions.

Bclaf1 interacts with C/EBPβ. (a) Interaction between C/EBPβ and Bclaf1 was analyzed by co-immunoprecipitation assay with the chromatin exacts of HCT116 cells untreated or treated with Dox for 3 days. (b and c) The regions in Bclaf1 (b) and C/EBPβ (c) mediating their interaction were determined. 293T (b) or HCT116 (c) cells transfected with the indicated plasmids were immunoprecipitated with an anti-Flag antibody followed by western blot analysis. A diagram of full-length Bclaf1 (b) or C/EBPβ (c) and their deletion mutants is shown
Bclaf1 cooperates with C/EBPβ to upregulate IL-8
Similar to its effect on C/EBPβ, shRNA-mediated Bclaf1 depletion dramatically reduced IL-6 and IL-8 induction by DNA damage at both protein and mRNA levels (Figures 6a–c). Moreover, Bclaf1 overexpression increased IL-8 transcription in 293T cells (Figure 6d). We failed to detect IL-6 mRNA probably because IL-6 is not transcribed in 293T cells. To examine whether C/EBPβ plays a role in Bclaf1-mediated IL-8 upregulation, we depleted C/EBPβ before Bclaf1 transfection and demonstrated that IL-8 upregulation is abolished upon C/EBPβ knockdown (Figure 6d). These results suggest that Bclaf1-induced transcription of some of the NF-κB downstream genes, such as IL-8, is mediated by C/EBPβ. To confirm this finding, we cloned the −764 to +1 region of the IL-8 promoter (Figure 6e) and fused it with a luciferase reporter. Consistent with the mRNA data, Bclaf1 overexpression increased IL-8 reporter activity in a C/EBPβ-dependent manner (Figure 6g).

Bclaf1 upregulates IL-8 through C/EBPβ. (a and b) ELISA analysis of the secreted IL-6 (a) and IL-8 (b) in the supernatants of HCT116 cells expressing control shRNA or shBclaf1 treated with Dox for 7 days. (c) Real-time RT-PCR analysis of IL-6 and IL-8 mRNA expression in HCT116 cells expressing control shRNA or shBclaf1 treated with Dox for 3 days relative to untreated controls. Error bars represent mean+S.D. (n=3). (d) 293T cells treated with siCtrl or siC/EBPβ were transfected with empty vector or Bclaf1-expressing plasmids and then treated with Dox for 3 days. IL-8 mRNAs were analyzed by real-time RT-PCR. Error bars represent mean+S.D. (n=3). (e) Diagram of the IL-8 promoter. Binding sites for AP-1, C/EBPβ and NF-κB are presented. The position is relative to the start of the first exon. (f) ChIP analysis of IL-6 and IL-8 promoters in HCT116 cells treated with Dox for 3 days. Immunoprecipitation was performed using anti-Bclaf1 and control IgG antibodies followed by PCR analysis of the C/EBPβ-binding region within the IL-6 or IL-8 promoters. (g) Relative luciferase activities of the IL-8 promoter were analyzed in 293T cells transfected with empty vector or 100 ng Bclaf1-expressing plasmid and treated with siCtrl or siRNAs against p65, c-Rel or C/EBPβ. Error bars represent mean+S.D. (n=3). (h) ChIP analysis of the C/EBPβ, IL-6 and IL-8 promoters in HCT116 cells transfected with siCtrl or siC/EBPβ and treated with Dox for 3 days. Immunoprecipitation and PCR are performed as described in (f). *P<0.05, **P<0.01 and ***P<0.001
We next examined whether C/EBPβ recruited Bclaf1 to the IL-6 and IL-8 promoters. ChIP assays indicated Dox treatment increased amounts of Bclaf1 bound to the promoter regions of IL-6 and IL-8 that harbor C/EBPβ-binding motifs (Figure 6f and Supplementary Figure S5D). This binding is indeed mediated by C/EBPβ given that C/EBPβ depletion greatly reduced promoter occupancy by Bclaf1 (Figure 6h). Examination of whether the recruitment of Bclaf1 to the p3 region of C/EBPβ is dependent on C/EBPβ (Figure 6h) showed that in contrast to the IL-6 and IL-8 promoters, Bclaf1 binding to the C/EBPβ promoter was not affected by C/EBPβ knockdown.
Bclaf1 knockdown reduces the efficiency of Dox-induced inhibition of tumor growth in a HT-29 colon cancer xenograft model in vivo
To determine whether Bclaf1 contributes to drug efficiency in vivo, we used an HT-29 colon cancer xenograft model. HT-29 cells expressing the control or shBclaf1 were subcutaneously inoculated into athymic nude mice. Mice were randomly divided into two groups 7 days after inoculation. One group was treated with Dox (3 mg/kg) intraperitoneally every 2 days for 2 weeks and the other group with the carrier PBS. After 4 weeks, the mice were killed and the tumors were harvested and analyzed.
Evaluation of tumor sizes and weights indicated that Bclaf1 knockdown did not affect tumor growth, but dramatically reduced the responsiveness of the tumors to Dox treatment (Figures 7a and b). Similar results were obtained using Ki67 staining, showing that shBclaf1 xenografts contained more proliferative cells than the controls in the Dox-treated group, but had similar number of Ki67-positive cells to the controls in the PBS-treated group (Figure 7c). Western blot analysis of the excised tumor samples demonstrated that Dox treatment dramatically induced upregulations of Bclaf1, C/EBPβ, p15 and IL-6 in the control tumors, but this effect was diminished in the Bclaf1 knockdown tumors (Figure 7d). All these in vivo results are consistent with our in vitro data, confirming the important role of Bclaf1 in mediating Dox treatment-induced upregulation of C/EBPβ, p15 and IL-6 and tumor growth inhibition.

Bclaf1 deficiency leads to chemoresistance of xenografts. HT-29 cells expressing control shRNA or shBclaf1 were injected subcutaneously at the back of athymic BALB/c nu/nu mice. The mice were then grouped and treated with Dox or PBS. (a) Images of tumors that were excised at the end of the treatment are shown. (b) The tumor weights of each group are depicted graphically. (c) Western blot analysis of the tumor samples using the indicated antibodies. (d) Tumors from each group were stained for Ki67. Representative images are shown. Arrowheads indicate Ki67-positive cells. (f) A schematic model showing the pathways that Bclaf1 is involved in TIS. **P<0.01
Discussion
Persistent DDR is a hallmark of senescence that continuously fires the upstream signals characterized by activation of ATM/ATR and other proteins and eventually induces cellular senescence;4 however, the signaling pathways and critical players connecting DDR signaling to senescence are not well characterized. In this study, we identified Bclaf1 as a critical player in DNA damage-induced senescence by stimulating C/EBPβ upregulation and activities downstream of NF-κB. Bclaf1 depletion impaired the upregulation of IL-6 and IL-8 as well as p15 and senescence likely through C/EBPβ (Figure 7e). Thus, Bclaf1 is an important regulator for the development of the SASP. It may also contribute to Dox-induced cell cycle arrest via p15.
NF-κB and C/EBPβ are important transcription factors for the upregulation of various nonsecreted and secreted factors required for the development of SASP and senescence.11, 12 These two proteins are often upregulated and activated and cooperatively induce the transcriptions of the SASP components during senescence.15, 34 C/EBPβ upregulation is observed following NF-κB activation (Supplementary Figure S5A). However, the mechanism by which NF-κB activation leads to C/EBPβ upregulation is unclear. We demonstrated that Bclaf1 is the critical mediator between NF-κB and C/EBPβ upregulation. Bclaf1 knockdown severely impaired C/EBPβ upregulation without affecting NF-κB activation (Figure 4b and Supplementary Figures S4 and S5B), and forced expression of exogenous Bclaf1 restored C/EBPβ upregulation in Nemo knockdown cells (Figure 4c). Our data indicate that Bclaf1 is required and NF-κB activation alone is not sufficient for C/EBPβ upregulation. ChIP assays revealed that Bclaf1 bound to a region of the C/EBPβ promoter (Figure 4f and Supplementary Figure S5D) that does not contain any defined transcription factor-binding elements. A previous study demonstrated that Bclaf1 interacts with the p53 promoter region and promotes its transcription.22 Thus, Bclaf1 may function as a transcriptional activator to promote C/EBPβ upregulation.
IL-6 and IL-8 play pivotal roles in the development of SASP and senescence that are co-regulated by NF-κB and C/EBPβ.12, 13 We demonstrated that Bclaf1 was recruited to the IL-6 and IL-8 promoters (Figure 6f and Supplementary Figure S5D) in a C/EBPβ-dependent manner (Figure 6h) and cooperates with C/EBPβ to enhance IL-8 transcription (Figures 6d and g). Thus, Bclaf1 may not only mediate C/EBPβ upregulation but also influence its ability to transactivate IL-6 and IL-8.
Bclaf1 may also play a role in Dox-induced cell cycle arrest through upregulation of p15. Bclaf1 depletion dramatically decreased Dox-induced inhibition of tumor cell growth both in vitro (Figures 1j and k) and in vivo (Figures 7a and b). Among the cell cycle inhibitors examined, only the upregulation of p15 is suppressed by Bclaf1 knockdown (Figures 4b and 7d and Supplementary Figure S5B). It has been demonstrated that C/EBPβ is a key transcription factor regulating p15.13 Thus, Bclaf1 likely regulates p15 through C/EBPβ.
The role of Bclaf1 in transcriptional regulation is poorly understood. Bclaf1 may participate in the epigenetic regulation of chromatin structure as it was found to be associated with L3MBTL3, a member of the malignant brain tumor (MBT) family of chromatin-interacting transcriptional repressors.35 Recently, Bclaf1 was identified as a component of the RNA splicing complex and regulates the stability of cyclin D and BRCA1 mRNA.36, 37 Whether this function of Bclaf1 is related to C/EBPβ upregulation requires further investigation.
Bclaf1 responds to NF-κB activation by upregulation. Bclaf1 contains a putative NF-κB-binding element within its promoter. Both ChIP and reporter assays indicate that NF-κB binds to this region and activates Bclaf1 transcription (Figures 3d and e). Kong et al.38 have also demonstrated that RelA (p65) binds to the Bclaf1 promoter. However, Bclaf1 upregulation by NF-κB may require its activity to reach a certain threshold. Compared with severe and acute DNA damage treatment, mild DNA damage induces senescence. NF-κB activation is low on day 3 of drug treatment, and a relatively full NF-κB activation was achieved on day 7 as indicated by efficient p65 nuclear translocation and dramatic p65 upregulation (Supplementary Figure S4A). Thus, NF-κB signaling during DNA damage-induced senescence appears to be a gradual process and subject to further amplification through upregulation. As reported, Nemo can transmit DDR to NF-κB activation;30 however, it did not mediate NF-κB upregulation (Figure 2e).
Recent studies have linked Bclaf1 to DDR. Lee et al.24 reported that Bclaf1 interacted with γH2AX and may regulate the Ku70/DNA-PKcs complex in response to DNA damage. Savage et al.37 demonstrated that Bclaf1 complexes with BRCA1 and influences the radiosensitivity of cells. We have demonstrated that Bclaf1 is involved in persistent and mild DNA damage-induced senescence downstream of NF-κB activation. Depending on the extent of DNA damage, Bclaf1 probably reacts differently. In the acute DNA damage response, Bclaf1 may be recruited to DNA damage foci and facilitates DNA repair. However, under chronic DNA damage conditions, Bclaf1 expression is upregulated in response to NF-κB activation and subsequently amplifies NF-κB downstream signaling to induce cellular senescence.
Materials and Methods
Plasmids, antibodies and reagents
Full-length Bclaf1 was kindly provided by Dr Tokuko Haraguchi39 and subcloned into the pRK5 vector with an N-terminal FLAG tag. Bclaf1 and control shRNAs were inserted into the pLKO.1-puro vector. To assess putative NF-κB-binding sites, the following oligo pairs containing four copies of binding sites were used: 5′-CGGGGCTTGCCGGGGCTTGCCGGGGCTTGCCGGGGCTTGCCG-3′ and 5′-AATTCGGCAAGCCCCGGCAAGCCCCGGCAAGCCCCGGCAAGCCCCGGTAC-3′. The annealed oligonucleotide pairs were cloned into the KpnI and EcoRI sites of pBVLuc.40 The IL-8 promoter sequence flanking the 5′ region from −764 to +26 was cloned into the pGL3-Basic vector (Promega, Madison, WI, USA).
The following primary antibodies were used for analysis: anti-Bclaf1, anti-p53, anti-p65, anti-cRel, anti-C/EBPβ and anti-p15INK4B (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-phospho-CHK1, anti-phospho-CHK2, anti-Nemo and anti-Ki67 (Cell Signaling Technology, Danvers, MA, USA); and anti-p21 (BD Biosciences, San Jose, CA, USA). Doxorubicin (Sigma, St. Louis, MO, USA) was dissolved in sterile water.
Cells and transfections
Human colon cancer cells HCT116 and human breast adenocarcinoma cells MCF7 were cultured in medium supplemented with 10% (v/v) FBS. HCT116 cells stably expressing Bclaf1 shRNA were generated by lentiviral infection followed by 2 μg/ml of puromycin selection for 4 days. HCT116 cells were transiently transfected with plasmid DNA using X-tremeGENE HP (Roche, Mannheim, Germany) according to the manufacturer’s protocols. SiRNA was transfected into cells using Lipofectamine RNAiMax (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocols. Control siRNA and Bclaf1 siRNA target sequences were designed and synthesized from Genechem (Shanghai, China) as follows: siCtrl, 5′-UUCUCCGAACGUGUCACGU-3′; siBclaf1#1, 5′-GGTTCACTTCGTATCAGAA-3′; siBclaf1#2, 5′-TTCTCAGAATAGTCCAATT-3′. The siRNA target sequences of Atm, Atr, p53, Nemo, p65, c-Rel, and C/EBPβ have been previously reported.41, 42, 43, 44, 45, 46, 47
Real-time PCR
Total RNA was extracted using TRIzol (Invitrogen) following the manufacturer’s protocol. Briefly, 0.8 μg of total RNA from different treatments was reverse transcribed using M-MLV reverse transcriptase (Promega) with an oligo (dT) 18 primer. Real-time PCR was performed using UltraSYBR Mixture (Beijing CoWin Biotech, Beijing, China) and a ViiA 7 real-time PCR system (Applied Biosystems, Foster City, CA, USA). The following primers for Bclaf1 were used: Bclaf1 forward, 5′-CCGCGATTCGGCGTGTCAGG-3′; Bclaf1 reverse, 5′-GACCCATTTCTTTTCTCCTTGGTT-3′. The primers for IL-6, IL-8 and C/EBPβ have been previously reported.13
ChIP assay
The ChIP assay was performed using a ChIP-IT Express enzymatic system (Active Motif, Carlsbad, CA, USA) following the manufacturer’s instructions. Briefly, HCT116 cells were crosslinked with 1% formaldehyde and neutralized with 0.125 M glycine. Purified chromatin was digested to ~500 bp by enzymatic shearing. Anti-Bclaf1, anti-C/EBPβ or control IgG (Santa Cruz) antibodies were used for immunoprecipitation. After reverse crosslinking, DNA samples were analyzed by PCR followed by 2% agarose gel electrophoresis. The PCR primers designed for fragments of the Bclaf1 promoters are as follows: NF-κB forward, 5′-TCACATCTTCCCGCGAGAC-3′ and NF-κB reverse, 5′-CGCTAAATATGCGGGCAAG-3′; NS forward, 5′-ACCAAGCAAAACCAGTCAGGT-3′ and NS reverse, 5′-CGTCTCTTCTAAAAATACAA-3′. The primers for fragments of the C/EBPβ promoters are as follows: p1 forward, 5′-CCTCTCGCTCCCAATCCC-3′ and p1 reverse, 5′-TCTCCTGAGCCCGGTTATTT-3′; p2 forward, 5′-CTGAAACCTCCGCCTCCTC-3′ and p2 reverse, 5′-GATTGGGAGCGAGAGGGG-3′; p3 forward, 5′-GTGGGAGTTTACGGGAGGAA-3′ and p3 reverse, 5′-GAGGAGGCGGAGGTTTCAG-3′. Primers for the amplification of C/EBPβ binding sites in the IL-6 and IL-8 promoters have been previously described.13
Immunoprecipitation and western blot
In total, 5 × 106 cells treated with 50 ng/ml doxorubicin for 3 days were lysed in lysis buffer (50 mM Tris-Cl at pH 8.0, 150 mM NaCl, 1% Triton X-100, 1 mM DTT, 1 complete protease inhibitor cocktail tablet and 10% glycerol). Cell lysates were centrifuged at 12 000 r.p.m. for 20 min. The resulting pellets were extracted with lysis buffer containing 500 mM NaCl. After reducing the salt concentration to normal levels (150 mM NaCl) by adding lysis buffer without NaCl, the cell extracts were subjected to immunoprecipitation with 1 μg of Bclaf1 antibody or control IgG plus protein G Sepharose at 4 °C overnight. Cell fractionation was performed using Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Biotechnology, Shanghai, China).
For western blots, cells were either directly lysed in 2 × sodium dodecyl sulfate (SDS) sample buffer or subjected to cytoplasmic/nuclear fractionation.48 The sliced tumors were minced and lysed in RIPA buffer (0.1% SDS). Subsequent procedures were performed as previously described.43
SA-β-gal and growth curves
HCT116 or MCF-7 cells were transfected with indicated siRNAs and cultured for 24 h. Subsequently, each group of cells were reseeded in 12-well plates and subjected to different treatments. For SA-β-gal staining, cells at 50 to 60% confluency were treated with Dox (50 ng/ml) for the indicated durations and then washed with ice-cold PBS and fixed for 10 min with 3% formaldehyde at room temperature. SA-β-gal staining solution (Beyotime) was prepared before use and incubated with the cells overnight at 37 °C. For growth curves, 0.75 × 104 of either HCT116 or HT-29 cells from each group were plated in 96-well plates in triplicate and treated with 50 ng/ml doxorubicin. The cells were cultured for another 1 to 5 days and monitored by MTT assay.49 The growth curves were generated accordingly.
Luciferase assay
For these experiments, 293T cells either untransfected or transfected with the indicated siRNAs were seeded in 24-well plates and then transfected with 100 ng of luciferase reporter plasmids plus 20 ng of pRL-TK plasmids as an internal control. Luciferase activity was measured using a dual-luciferase reporter assay kit (Promega) 24 h after transfection and then normalized.
Immunofluorescence and immunohistochemistry
Immunofluorescence was performed as previously described.43 For immunohistochemistry, xenografts were fixed in formalin, embedded in paraffin and then sliced. Antigens were retrieved using citric acid and steam and then stained with a Ki67 antibody (1 : 400, Cell Signaling Technology) and hematoxylin.
Enzyme-linked immunosorbent assay (ELISA)
HCT116 cells expressing control shRNA or shBclaf1 treated with Dox for 7 days were reseeded into 6-well plates at a density of 3 × 106 cells per well and cultured with 1 ml fresh median for 24 h. The supernatants were then collected to measure the levels of IL-6 and IL-8 using ELISA kits purchased from Boster (Wuhan, China).
Xenografts
Animal care and protocols were approved by Animal Welfare Committee of China Agricultural University. 3 × 106 HT-29 cells in 100 μl PBS stably expressing either the control shRNA or shBclaf1#1 were injected subcutaneously into the left and right flanks of 6-week-old female athymic BALB/c nu/nu mice (HFK Bioscience, Beijing, China), respectively. When tumors were formed (~7 days), the mice were grouped randomly and given either PBS or 3 mg/kg Dox in PBS intraperitoneally once every 2 days for 6 times. After the treatment was stopped, the mice were allowed to grow for another 10 days before being killed under ether anesthesia by cervical dislocation. The tumors were harvested and weighed, and then were sliced for either Ki67 staining or western blot analysis.
Statistical analysis
Microsoft Excel and GraphPad Prism (La Jolla, CA, USA) were used for statistical analyses. Statistical significance was analyzed by Student’s t-test and expressed as a P-value. P-values of <0.05 were considered statistically significant: *P<0.05; **P<0.01; ***P<0.001.
Abbreviations
- ATM:
-
ataxia telangiectasia mutated protein
- ATR:
-
ataxia telangiectasia and Rad3-related protein
- Bclaf1:
-
BCL2-associated transcription factor 1
- bZIP:
-
basic zipper
- C/EBPβ:
-
CCAAT/enhancer-binding protein-β
- ChIP:
-
chromatin immunoprecipitation
- Chk1:
-
checkpoint kinase 1
- Chk2:
-
checkpoint kinase 2
- DDR:
-
DNA damage response
- Dox:
-
doxorubicin
- ELISA:
-
enzyme-linked immunosorbent assay
- FL:
-
full-length
- IL-6:
-
interleukin-6
- IL-8:
-
interleukin-8
- LAP:
-
liver-enriched transcriptional activator protein
- Nemo:
-
NF-κB essential modulator
- NF-κB:
-
nuclear factor κ-light-chain-enhancer of activated B cells
- SA-β-gal:
-
senescence-associated galactosidase
- SASP:
-
senescence-associated secretory phenotype
- shRNA:
-
short hairpin RNA
- siRNA:
-
small interfering RNA
- TIS:
-
therapeutic drug doxorubicin-induced senescence
References
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS . The essence of senescence. Genes Dev 2010; 24: 2463–2479.
Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM . Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell 2004; 14: 501–513.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW . Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88: 593–602.
d'Adda di Fagagna F . Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 2008; 8: 512–522.
Collado M, Serrano M . Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010; 10: 51–57.
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445: 656–660.
te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP . DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res 2002; 62: 1876–1883.
Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003; 113: 703–716.
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995; 92: 9363–9367.
Fumagalli M, d'Adda di Fagagna F . SASPense and DDRama in cancer and ageing. Nat Cell Biol 2009; 11: 921–923.
Salama R, Sadaie M, Hoare M, Narita M . Cellular senescence and its effector programs. Genes Dev 2014; 28: 99–114.
Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008; 133: 1006–1018.
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008; 133: 1019–1031.
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013; 15: 978–990.
Collado M, Serrano M . The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer 2006; 6: 472–476.
Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 2009; 11: 973–979.
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444: 638–642.
Sulli G, Di Micco R, d'Adda di Fagagna F . Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer 2012; 12: 709–720.
Kasof GM, Goyal L, White E . Btf, a novel death-promoting transcriptional repressor that interacts with Bcl-2-related proteins. Mol Cell Biol 1999; 19: 4390–4404.
McPherson JP, Sarras H, Lemmers B, Tamblyn L, Migon E, Matysiak-Zablocki E et al. Essential role for Bclaf1 in lung development and immune system function. Cell Death Differ 2009; 16: 331–339.
Lamy L, Ngo VN, Emre NC, Shaffer AL 3rd, Yang Y, Tian E et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 2013; 23: 435–449.
Liu H, Lu ZG, Miki Y, Yoshida K . Protein kinase C delta induces transcription of the TP53 tumor suppressor gene by controlling death-promoting factor Btf in the apoptotic response to DNA damage. Mol Cell Biol 2007; 27: 8480–8491.
Sarras H, Alizadeh Azami S, McPherson JP . In search of a function for BCLAF1. ScientificWorldJournal 2010; 10: 1450–1461.
Lee YY, Yu YB, Gunawardena HP, Xie L, Chen X . BCLAF1 is a radiation-induced H2AX-interacting partner involved in gammaH2AX-mediated regulation of apoptosis and DNA repair. Cell Death Dis 2012; 3: e359.
Zhou X, Li X, Cheng Y, Wu W, Xie Z, Xi Q et al. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nat Commun 2014; 5: 4581.
Nardella C, Clohessy JG, Alimonti A, Pandolfi PP . Pro-senescence therapy for cancer treatment. Nat Rev Cancer 2011; 11: 503–511.
Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002; 109: 335–346.
Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF . Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984; 226: 466–468.
Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316: 1160–1166.
Miyamoto S . Nuclear initiated NF-kappaB signaling: NEMO and ATM take center stage. Cell Res 2011; 21: 116–130.
Esteller M . CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene 2002; 21: 5427–5440.
Descombes P, Schibler U . A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 1991; 67: 569–579.
Ossipow V, Descombes P, Schibler U . CCAAT/enhancer-binding protein mRNA is translated into multiple proteins with different transcription activation potentials. Proc Natl Acad Sci USA 1993; 90: 8219–8223.
Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev 2011; 25: 2125–2136.
James LI, Barsyte-Lovejoy D, Zhong N, Krichevsky L, Korboukh VK, Herold JM et al. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat Chem Biol 2013; 9: 184–191.
Bracken CP, Wall SJ, Barre B, Panov KI, Ajuh PM, Perkins ND . Regulation of cyclin D1 RNA stability by SNIP1. Cancer Res 2008; 68: 7621–7628.
Savage KI, Gorski JJ, Barros EM, Irwin GW, Manti L, Powell AJ et al. Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol Cell 2014; 54: 445–459.
Kong S, Kim SJ, Sandal B, Lee SM, Gao B, Zhang DD et al. The type III histone deacetylase Sirt1 protein suppresses p300-mediated histone H3 lysine 56 acetylation at Bclaf1 promoter to inhibit T cell activation. J Biol Chem 2011; 286: 16967–16975.
Haraguchi T, Holaska JM, Yamane M, Koujin T, Hashiguchi N, Mori C et al. Emerin binding to Btf, a death-promoting transcriptional repressor, is disrupted by a missense mutation that causes Emery-Dreifuss muscular dystrophy. Eur J Biochem 2004; 271: 1035–1045.
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B . PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7: 673–682.
Tang J, Qu LK, Zhang J, Wang W, Michaelson JS, Degenhardt YY et al. Critical role for Daxx in regulating Mdm2. Nat Cell Biol 2006; 8: 855–862.
Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8: 37–45.
Cui D, Li L, Lou H, Sun H, Ngai SM, Shao G et al. The ribosomal protein S26 regulates p53 activity in response to DNA damage. Oncogene 2014; 33: 2225–2235.
Petersen SL, Peyton M, Minna JD, Wang X . Overcoming cancer cell resistance to Smac mimetic induced apoptosis by modulating cIAP-2 expression. Proc Natl Acad Sci USA 2010; 107: 11936–11941.
Wan F, Anderson DE, Barnitz RA, Snow A, Bidere N, Zheng L et al. Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell 2007; 131: 927–939.
Refaat A, Zhou Y, Suzuki S, Takasaki I, Koizumi K, Yamaoka S et al. Distinct roles of transforming growth factor-beta-activated kinase 1 (TAK1)-c-Rel and interferon regulatory factor 4 (IRF4) pathways in human T cell lymphotropic virus 1-transformed T helper 17 cells producing interleukin-9. J Biol Chem 2011; 286: 21092–21099.
Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev 2008; 22: 2941–2952.
Rosner M, Hengstschlager M . Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum Mol Genet 2008; 17: 2934–2948.
Geng Y, Wang X, Yang L, Sun H, Wang Y, Zhao Y et al. Antitumor activity of a 5-hydroxy-1H-Pyrrol-2-(5H)-one-based synthetic small molecule in vitro and in vivo. PLoS One 2015; 10: e0128928.
Acknowledgements
We thank Drs. Feng Wenhai and Cong Yusheng for generously providing constructs and reagents, Dr. Tokuko Haraguchi for providing the Bclaf1 expression plasmid and Dr. Sigrun Smola for providing the C/EBPβ plasmids. We also thank Ms. Ruihan Shi and Professor Ruiping She for immunohistological assistance. This work was supported by the National Natural Science Foundation of China (30971487).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Villunger
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Shao, Aw., Sun, H., Geng, Y. et al. Bclaf1 is an important NF-κB signaling transducer and C/EBPβ regulator in DNA damage-induced senescence. Cell Death Differ 23, 865–875 (2016). https://doi.org/10.1038/cdd.2015.150
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.150
This article is cited by
-
BCLAF1 is Expressed as a Potential Anti-oncogene in Bile Duct Cancer
Biochemical Genetics (2024)
-
Exosomal transfer of miR-181b-5p confers senescence-mediated doxorubicin resistance via modulating BCLAF1 in breast cancer
British Journal of Cancer (2023)
-
Bcl-2-associated transcription factor 1 Ser290 phosphorylation mediates DNA damage response and regulates radiosensitivity in gastric cancer
Journal of Translational Medicine (2021)
-
Bclaf1 is a direct target of HIF-1 and critically regulates the stability of HIF-1α under hypoxia
Oncogene (2020)
-
Endogenous interaction profiling identifies DDX5 as an oncogenic coactivator of transcription factor Fra-1
Oncogene (2019)
