
Abstract
The p62/SQSTM1 adapter protein has an important role in the regulation of several key signaling pathways and helps transport ubiquitinated proteins to the autophagosomes and proteasome for degradation. Here, we investigate the regulation and roles of p62/SQSTM1 during acute myeloid leukemia (AML) cell maturation into granulocytes. Levels of p62/SQSTM1 mRNA and protein were both significantly increased during all-trans retinoic acid (ATRA)-induced differentiation of AML cells through a mechanism that depends on NF-κB activation. We show that this response constitutes a survival mechanism that prolongs the life span of mature AML cells and mitigates the effects of accumulation of aggregated proteins that occurs during granulocytic differentiation. Interestingly, ATRA-induced p62/SQSTM1 upregulation was impaired in maturation-resistant AML cells but was reactivated when differentiation was restored in these cells. Primary blast cells of AML patients and CD34+ progenitors exhibited significantly lower p62/SQSTM1 mRNA levels than did mature granulocytes from healthy donors. Our results demonstrate that p62/SQSTM1 expression is upregulated in mature compared with immature myeloid cells and reveal a pro-survival function of the NF-κB/SQSTM1 signaling axis during granulocytic differentiation of AML cells. These findings may help our understanding of neutrophil/granulocyte development and will guide the development of novel therapeutic strategies for refractory and relapsed AML patients with previous exposure to ATRA.
Similar content being viewed by others


Main
p62 or sequestosome 1 (p62/SQSTM1) is a scaffold protein, implicated in a variety of biological processes including those that control cell death, inflammation, and metabolism.1, 2 Through its multi-domain structure, p62/SQSTM1 interacts specifically with key signaling proteins, including atypical PKC family members, NF-κB, and mTOR to control cellular responses.3, 4, 5, 6, 7 p62/SQSTM1 functions also as a key mediator of autophagy. Through its interaction with LC3, an essential protein involved in autophagy, p62/SQSTM1 selectively directs ubiquitinated substrates to autophagosomes leading to their subsequent degradation in lysosomes.8, 9 At the molecular level, p62/SQSTM1 acts as a pro-tumoral molecule by ensuring efficient and selective activation of cell signaling axes involved in cell survival, proliferation, and metabolism (i.e., NF-κB, mTOR, and Nrf-2 pathways).3, 5, 6, 7, 10, 11, 12, 13 p62/SQSTM1 can also signal anti-tumoral responses either by inactivating the pro-oncogenic signaling through BCR-ABL14 and Wnt pathways15, 16 or by inducing the activation of caspase 8, a pro-death protein.17, 18 Interestingly, in response to stress, autophagy promotes the degradation of p62, thus limits the activation of p62-regulatory pathways that control tumorigenesis.10 In addition, p62/SQSTM1 controls pathways that modulate differentiation of normal and cancerous cells. For example, p62/SQSTM1 has been shown to antagonize basal ERK activity and adipocyte differentiation.19 In contrast, p62/SQSTM1 favors differentiation of osteoclasts,20 osteoblasts,21 neurons,22 megakaryocytes23 and macrophages.24 The role and regulation of p62/SQSTM1 during leukemia cell differentiation has been poorly documented.
Acute myeloid leukemia (AML) is a hematological disease characterized by multiple deregulated pathways resulting in a blockade of myeloid precursors at different stages of maturation.25, 26 Acute promyelocyte leukemia (APL) is the M3 type of AML characterized by an arrest of the terminal differentiation of promyelocytes into granulocytes and frequently associated with the expression of the oncogenic PML-RAR alpha fusion gene.27, 28 All-trans retinoic acid (ATRA), a potent activator of cellular growth arrest, differentiation, and death of APL cells, has been shown to effectively promote complete clinical remission of APL when combined with chemotherapy.29, 30, 31 Despite the success of this treatment, some APL patients are refractory to ATRA treatment or relapse owing to the development of resistance to ATRA in leukemia cells.32, 33, 34
Our previous results revealed that autophagy flux is activated during granulocyte differentiation of myeloid leukemia cell lines induced by ATRA.35 In the present study, we observed that p62/SQSTM1, an autophagic substrate, is markedly upregulated at both mRNA and protein levels during the granulocytic differentiation process. Here, we investigated the regulation and the function of p62/SQSTM1 during AML cells differentiation into neutrophils/granulocytes.
Results
p62/SQSTM1 is upregulated during ATRA-induced granu-locytic differentiation of AML cells
To address whether p62/SQSTM1 has a role during the maturation of myeloid leukemia cells, we first examined how its expression is modulated during the granulocyte differentiation of the NB4 cell line, a model of APL.36 As shown in Figure 1a (upper panel), ATRA induces upregulation of p62/SQSTM1 protein in NB4 cells that is associated with an increase in the level of LC3-II. These responses occur in parallel to the granulocyte differentiation of NB4 cells as evidenced by the presence of cells with lobed nuclei and the increased expression of the cell surface marker CD11c (Figure 1a, lower panel). Immunofluorescence analysis confirmed the accumulation of p62/SQSTM1 and suggests that most p62/SQSTM1 proteins colocalize with LC3 upon ATRA treatment of APL cells (Figure 1b). The accumulation of p62/SQSTM1 in NB4 cells is not due to a defect in its clearance by lysosomal or proteasomal pathways, as levels were enhanced when the lysosomal proteases or proteasome activities were inhibited after treatment with E64d and Pepstatin A or MG132, respectively (Figure 1c) confirming once again our previous data showing that autophagy is functionally activated during APL cells maturation.35 We also used the HL60 cell line, which is an in vitro model of myeloid leukemia cells that undergo granulocyte differentiation during ATRA treatment (as revealed by the increase in the expression of CD11c, Figure 1d, right panel). As shown in Figure 1d, p62/SQSTM1 and LC3-II proteins accumulated during ATRA-induced differentiation of the HL60 cells, supporting the hypothesis that the effect of ATRA on p62/SQSTM1 expression and autophagy is not restricted to the NB4 promyelocytic leukemia cells (Figure 1d). Interestingly, p62/SQSTM1 upregulation associated with LC3-II accumulation (Figure 1e, upper panel) was also observed when NB4 cells underwent monocyte maturation after treatment with Phorbol 12–myristate 13-acetate (Figure 1e, lower panel), as evidenced by the increased expression of CD11b and CD14, two cell surface hallmarks of monocyte maturation. This supports the hypothesis that the p62/SQSTM1 upregulation represents a general phenomenon that occurs during myeloid cell lineage differentiation.

p62/SQSTM1 protein accumulates during terminal differentiation of acute promyelocytic leukemia (APL) cells. (a) The NB4 APL-derived cells were treated with 1 μM ATRA for the times indicated and then subjected to cell lysis. Upper panel: the p62/SQSTM1 and LC3-II protein levels were assessed following immunoblot analysis using antibodies directed against p62/SQSTM1 and LC3. GAPDH was used as a control. Lower panel: NB4 cells were treated with 1 μM ATRA for 4 days, granulocytic differentiation was determined by morphologic changes of cells stained with May-Grunwald Giemsa or by FACS analysis of the expression of the CD11c cell surface marker. (b) NB4 cells were treated with ATRA (1 μM, for 2 days) before immunostaining with p62/SQSTM1 and LC3 antibodies and examined by fluorescence microscopy. Nuclei were counterstained with DAPI (blue). The orange overlay shows the colocalization of p62/SQSTM1 and LC3 proteins. High-magnification images of cells denoted with asterisks are shown in right panel. (c) the NB4 APL-derived cells were treated with 1 μM ATRA for 3 days in the presence and absence of either E64d (10 μg/ml) and methyl ester pepstatin (1 μg/ml) or MG132 (100 nM). The expression levels of p62/SQSTMI and LC3-II proteins were assessed following immunoblot analysis using antibodies directed against p62/SQSTM1 and LC3. (d) The HL60 AML-derived cells were treated with 1 μM ATRA for 4 days and then subjected to either immunoblot analysis of p62/SQSTM1 and LC3 proteins (left panel) or FACS analysis of the expression of the cell surface granulocyte marker, CD11c (right panel). (e) Immunoblot analysis of p62/SQSTM1 and LC3 proteins in the NB4 cells treated with 100 nM PMA for 1 and 3 days (left, panel). Monocyte differentiation was assessed by FACS analysis of the expression of cell surface monocyte markers, CD11b, and CD14 (right panel). For FACS experiments, fluorescence intensity (FI) was measured for each treatment and results were expressed as median fluorescence intensity, MFI. **P<0.01; ***P<0.001 versus untreated cells. For all of the immunoblots, representative examples are shown
ATRA induces p62/SQSTM1 upregulation in differentiation-sensitive NB4 cells but not in differentiation-resistant NB4-LR1 cells
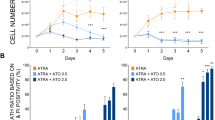
To investigate whether the ATRA-induced accumulation of p62/SQSTM1 protein is the result of the increased levels of the p62/SQSTM1 mRNA, we next examined mRNA levels by performing RT-qPCR. As shown in Figure 2a, p62/SQSTM1 mRNA levels were enhanced during ATRA-induced maturation of NB4 cells (left panel). Interestingly, p62/SQSTM1 mRNA levels were also upregulated upon in vitro granulocytic differentiation of primary CD34+progenitors cells (right panel). To elucidate the relationship between p62/SQSTM1 expression and the differentiation process, we next measured the expression of p62/SQSTM1 protein and the level of mRNA in ATRA-treated NB4-LR1 cells, which are resistant to ATRA-induced maturation.37 Of note, granulocyte differentiation was reestablished in NB4-LR1 when 8-(4-chlorophenylthio), 5′-cyclic adenosine monophosphate (8-CPT-cAMP) was added to ATRA as evidenced by p-nitro-blue tetrazolium (NBT) reduction assay and morphologic changes of cells stained by MGG (Figure 2b). ATRA induced upregulation of p62/SQSTM1 mRNA and protein in NB4 cells (Figures 2a and c) that underwent granulocytic differentiation (Figure 2c). In contrast, no modification of p62/SQSTM1 at the protein and mRNA levels was observed in response to ATRA in maturation-resistant NB4-LR1 cells (Figure 2c). Interestingly, when differentiation was reestablished in NB4-LR1 cells by a combination of ATRA and 8-CPT-cAMP treatment,38 the upregulation of p62/SQSTM1 at both mRNA and protein levels was restored in these cells (Figure 2c upper panel). The level of LC3-II protein was also increased in this condition, which confirms our previous study showing that autophagy is upregulated in mature granulocytes compared with immature ones35 (Figure 2c). Altogether, these results suggest that p62/SQSTM1 expression level and ATRA-induced differentiation are interconnected processes.

ATRA-induced upregulation of p62/SQSTM1 mRNA occurs in NB4 maturation-sensitive cells but not in NB4-LR1 maturation-resistant cells. (a) NB4 cells (left panel) and primary CD34+progenitors cells (right panel) were treated with 1 μM ATRA and 10 ng/ml (hrG-CSF) for the indicated times, respectively. The p62/SQSTM1 mRNA levels were assessed, after RNA extraction, by RT-qPCR as described in Djavaheri-Mergny et al.56 (b and c) NB4-LR1 cells, which are resistant to maturation by ATRA, were treated with 1 μM ATRA and 100-μM 8-CPT-cAMP alone or in combination for 4 days. (b) Granulocytic differentiation was determined by FACS analysis of the expression CD11c or by morphologic changes of cells stained with May-Grunwald Giemsa. (c) Quantification of p62/SQSTM1 mRNA levels by RT-qPCR (upper panel) and immunoblot analysis of p62/SQSTM1 and LC3 proteins (lower panel). The relative expression of p62/SQSTM1 mRNA transcripts to the housekeeping genes, CPB (Figure a, left panel and Figure c) or HMBS (Figure a, right panel) was calculated as 2−ΔΔCT. For each cDNA, results were expressed as fold changes compared with untreated cells. ***P<0.001 versus untreated cells
NF-κB is required for p62/SQSTM1 upregulation during granulocyte differentiation of APL cells
To gain insight into mechanisms underlying p62/SQSTM1 upregulation, we analyzed the region of the p62/SQSTM1 gene, extending into the flanking genes, for transcription factors binding sites (ENCODE data).39 This analysis revealed the existence of several NF-κB binding sites tested by ChIP assay in the proximal promoter region of p62/SQSTM1 (Supplementary Figure 1). Moreover, the Transfac position weight matrix revealed the presence of robust HMR (Human, Mouse, Rabbit) consensus sequence for the NF-κB within this region (Supplementary Figure 1). This analysis, together, with the previous data indicating that NF-κB is activated during ATRA-induced maturation of APL cells,40 prompted us to determine whether NF-κB is responsible for p62/SQSTM1 upregulation by ATRA. We generated NB4 cells (named NB4 MAD cells) stably overexpressing the IκBα (A32/36) mutant, a super repressor form of IκBα that confers inhibition of NF-κB activity by preventing the degradation and phosphorylation of endogenous wild-type (wt) IκBα (Figure 3a and Traenckner et al.41). As shown in Figure 3a, ATRA induced p62/SQSTM1 protein upregulation only in NF-κB competent cells (NB4 control) but not in NF-κB incompetent cells (NB4 MAD cells). Similarly, p62/SQSTM1 mRNA levels were significantly lower in NB4 MAD cells compared with NB4 control cells after treatment with ATRA (Figure 3b). Moreover, NF-κB incompetent cells exhibited significantly lower level accumulation of LC3-II in response to ATRA than did NF-κB competent cells, suggesting that NF-kappaB signaling pathway controls, at least in part, the induction of autophagy during granulocyte differentiation. As previously published by Mathieu J. et al.,40 we found that NF-κB activation is not essential for granulocytic differentiation but prolongs the life span of mature myeloid cells (data not shown). These results indicate a strong dependency on NF-κB for regulation of p62/SQSTM1 expression and LC3-II levels in ATRA-induced granulocytic differentiation of NB4 cells.

ATRA-induced p62/SQSTM1 upregulation is defective in NB4 cells that overexpress the IκBα (A32/36) mutant, a repressor of NF-κB activity. NB4 cells were transduced with the control lentivirus vector (NB4 control cells) or the lentivirus vector encoding for the IκBα (A32/36) mutant (NB4 MAD cells) and then were incubated for 3 days with 1 μM ATRA. (a) The levels of p62/SQSTM1 protein in control and NB4 MAD cells were assessed following immunoblot analysis using antibodies that directed against either p62/SQSTM1 or LC3-II. Levels of the wild-type (wt) IκBα mutant IκBα (A32/36) and phospho-IκBα proteins were also determined. GAPDH was used as a control. (b) Quantification of the expression of p62/SQSTM1 mRNA levels by RT-qPCR in NB4 control and NB4 MAD cells treated with 1 μM ATRA. ***P<0.001
Functional role of p62/SQSTM1 during granulocyte differentiation of NB4 cells
Given the association between p62/SQSTM1 upregulation and ATRA-induced differentiation of APL cells, we next investigated the function of p62/SQSTM1 in APL cells maturation. To this end, we evaluated differentiation and cell death upon ATRA treatment of APL cells using two different DNA sequences encoding shRNA against p62/SQSTM1. The downregulation of both the basal and ATRA-induced upregulated level of p62/SQSTM1 was verified (Figure 4a). The inhibition of p62/SQSTM1 expression (Figure 4a) did not significantly change the differentiation induced by ATRA as determined by the measurement of CD11c, a cell surface granulocyte marker (Figure 4b), the NBT reduction assay (Figure 4c) or morphologic changes in nuclei of cells stained with MGG (Figure 4d). These results indicate that ATRA-induced p62/SQSTM1 upregulation is dispensable for the induction of neutrophil/granulocyte maturation of APL NB4 cells.

Effect of shRNA-mediated inhibition of p62/SQSTM1 on ATRA-induced neutrophil/granulocyte differentiation of APL cells. NB4 cells expressing either control shRNA or shRNAs that target the p62/SQSTM1 (p62#1 shRNA or p62#2 shRNA) were treated with 1 μM ATRA for 4 days. (a) The p62/SQSTM1 and GAPDH protein levels were assessed following immunoblot analysis. (b–d) Granulocyte differentiation was assessed by measuring: (b) CD11c expression by FACS analysis and results were expressed as median fluorescence intensity, MFI; representative flow cytometry diagrams are shown. Values are means±S.D. (n=3) (c) NBT reduction ability assay, values are means±S.D. (n=3); (d) May-Grunwald Giemsa staining. Representative examples are shown. ns: non-significant
As expected, treatment of NB4 cells with ATRA triggered a moderate amount of cell death as evidenced by the appearance of cleaved forms of caspase 3 and caspase 8 (Figure 5a), the loss of mitochondrial transmembrane potential (Figure 5b), and the increase in the cell plasma membrane permeability (Figure 5c). Interestingly, these features of cell death are all markedly enhanced when p62/SQSTM1 expression was downregulated by using specific shRNAs against p62/SQSTM1 (Figure 5). In accordance with these results, shRNA-mediated knockdown of p62/SQSMT1 leads to a decrease in the number of APL cells that undergo granulocyte differentiation in response to ATRA (data not shown). To further elucidate the role of p62/SQSTM1 in maintaining viability of mature cells, cell death was assessed in NB4-LR1 cells in which differentiation is impaired in response to ATRA but can be reestablished by cotreatement with 8-CPT-cAMP (Figure 2b). As shown in Figure 5d, ATRA promotes a massive cell death in NB4-LR1 cells as revealed by the substantial loss of Δψm (Figure 5d, lower panel), whereas no significant modification of p62/SQSTM1 protein level was observed in this context (Figure 5d, upper panel). Interestingly, the combination of ATRA with 8-CPT-cAMP promotes p62/SQSTM1 upregulation (Figure 5d, upper panel) and protects these cells against death induced by ATRA (Figure 5d, lower panel). These results support the idea that the ability of cells to induce or not p62/SQSTM1 expression may dictate the cell killing effect of ATRA. Altogether, these results clearly indicate that the upregulation of p62/SQSTM1 that occurs during terminal differentiation of APL cells is an essential signal for the survival of APL-derived cells that undergo terminal maturation.

Effect of shRNA-mediated inhibition of p62/SQSTM1 expression on cell death induced by ATRA. NB4 cells were transduced with control shRNA or shRNAs that target p62/SQSTM1 (p62#1 shRNA and p62#2 shRNA) and then treated with 1 μM ATRA for 4 days. (a–d) Cell death features were characterized by: (a) evaluation of the appearance of the cleaved forms of caspase 3 and caspase 8 by immunoblot assay; (b) measurement of the loss of mitochondrial transmembrane potential by TMRM staining, representative flow cytometry diagrams are shown. Values are means±S.D. (n=4); (c) assessment of the cell plasma membrane permeability by propidium iodide staining, values are means±S.D. (n=4). (d) NB4-LR1 cells, which are resistant to maturation by ATRA, were treated with 1 μM ATRA and 100 μM 8-CPT-cAMP alone or in combination for 4 days and then subjected to either immunoblot analysis of p62/SQSTM1 expression (upper panel) or FACS analysis of the loss of mitochondrial transmembrane potential by TMRM staining (lower panel). M represents the population of cells with low TMRM staining. ***P<0.001
p62/SQSTM1 regulates the levels of ubiquitinated-protein aggregates that accumulate during terminal differentiation of APL cells
Because p62/SQSTM1 is known to interact with ubiquitinated proteins,42, 43, 44, 45 we next examined whether the increase in p62/SQSTM1 protein levels by ATRA treatment of APL NB4 cells was associated with an accumulation of polyubiquitinated proteins. We used western blotting to assess the distribution of polyubiquitinated proteins into the Triton X-100-insoluble protein fractions prepared from NB4 extracts. As shown in Figure 6a, ATRA treatment of NB4 cells promoted the accumulation of polyubiquitinated proteins in Triton X-100-insoluble fraction, indicating that ubiquitinated-protein aggregates accumulate during granulocytic differentiation process. Immunofluorescence analysis in NB4 cells reveals also a robust accumulation of ubiquitinated proteins during the induction of differentiation of NB4 cells by ATRA (Figure 6b). By using a fluorescence-based assay for the detection of aggregated proteins, we confirmed the abundance of aggregated proteins during ATRA-induced differentiation of NB4 cells as well as in NB4 cells treated with MG132, an inducer of aggresome formation (Figure 6c). NB4 cells expressing shRNAs that target the p62/SQSTM1 mRNA (p62#1 shRNA or p62#2 shRNA) manifested a higher accumulation of ubiquitinated-protein aggregates during differentiation process (2.1±0.3 fold and 2.6±0.1 fold, respectively) than did cells expressing control shRNA (1.7 fold ±0.1) as evidenced by ubiquitin immunoblot analysis (Figure 6d). In addition, ATRA induction of ubiquitinated-protein aggregates was enhanced when autophagy flux was inhibited by Bafilomycin A1 (3.0±0.74 fold versus 1.7±0.05 fold in cells treated with ATRA alone) supporting the idea that the autophagy/lysosomal pathway is responsible, at least in part, for the clearance of such proteins (Figure 6d). Moreover, shRNA-mediated knockdown of p62/SQSTM1 expression in NB4 cells resulted in increased aggresome formation during differentiation when compared with control shRNA as revealed by the quantitative fluorescence-based assay for detection of aggresomes (Figure 6e). Altogether, these results support the idea that p62/SQSTM1 and autophagy cooperate to control the levels of aggregated proteins that accumulate during the maturation of APL cells.

p62/SQSTM1 regulated the level of ubiquitinated-protein aggregates during terminal differentiation of APL cells. (a) Triton X-100-insoluble protein fractions of NB4 cells treated with 1 μM ATRA for times indicated. Samples were separated by SDS-PAGE and immunoblots were analyzed with antibodies against ubiquitin. (b) Immunofluorescence analysis of ubiquitin staining in NB4 cells treated in the presence and absence of 1 μM ATRA for 4 days. (c) The abundance of aggregated proteins was measured by flow cytometry analysis using the ProteoStat Aggresome detection kit in NB4 cells treated with either ATRA (1 μM, for 6 days) or MG132 (5 μM, for 8 h), an inducer of aggresome formation. Representative example is shown. The results were presented as histogram overlays. (d and e) NB4 cells were transduced with control shRNA, p62#1 shRNA, or p62#2 shRNA and treated with 1 μM ATRA for 4 days. NB4 cells treated with 1 μM ATRA for 4 days and in the presence and absence of 10-nM Bafilomycin A1. (d) Representative immunoblot of Triton X-100-insoluble protein fractions for ubiquitinated-protein aggregates is shown. Quantification of ubiquitinated aggregates proteins are indicated. Results were means of two independent experiments and were expressed as the ratio of ubiquitinated aggregates proteins in ATRA-treated cells to that in untreated control shRNA NB4 cells (left panel) or untreated NB4 cells (right panel). (e) The levels of aggresomes were determined by the Proteostat aggresome detection kit following FACS analysis. The results were scored as the ratio of MFI of flow cytometery histogram in ATRA-treated cells to that in untreated cells. Values are means±S.D. (n=3). *P<0.05; ***P<0.001
p62/SQSTM1 mRNA expression is dowregulated in AML patient samples
To support our finding that the expression of p62/SQSTM1 is enhanced during granulocytic maturation of AML cell lines, we further investigated whether the p62/SQSTM1 mRNA levels are repressed in primary blast cells from AML patients that are blocked in their maturation at the myeloid stage. We examined the p62/SQSTM1 mRNA levels by quantitative RT-PCR in 98 primary AML patient samples (FAB M0-M4), in 14 granulocytes preparations from healthy donors, and in three samples of CD34+ progenitors cells. p62/SQSTM1 mRNA was detected in 95 of the 98 AML patient samples, in all granulocyte samples from healthy donors and in the CD34+ progenitor cell samples. However, as shown in Figure 7, p62/SQSTM1 mRNA levels were significantly lower in AML patient samples irrespective of their genetic abnormality types and their karyotypes and in CD34+ progenitor cells than in granulocytes from healthy donors. Together, these results clearly show that the p62/SQSTM1 mRNA levels are downregulated in both AML patient samples and primary CD34+ progenitors, which have an immature myeloid phenotype, compared with normal granulocytes. This underscores the tight association between p62/SQSTM1 expression level and maturation phenotype of myeloid cells.

p62/SQSTM1 mRNA expression is downregulated in both primary AML patient samples and CD34+ progenitors cells. p62/SQSTM1 mRNA levels were quantified by RT-qPCR in primary CD34+ progenitor cells, in granulocytes from healthy donors and in AML subtypes FAB M0-M4 harboring the following molecular lesions: (inv(16), t(8;21), t(15;17)) K= Karyotype Federzoni et al.53 Ct values were normalized to HMBS and ABL1 (−ΔCt) and represent log2 expression levels. Mann–Whitney U-test *P<0.05; **P<0.01; ***P<0.001
Discussion
The cell survival function of p62/SQSTM1 has been shown in various cellular contexts but little is known about the role of this protein in cell viability during differentiation processes.1, 46, 47 In fact, in several settings, p62/SQSTM1 alleviates, the cytotoxic effects of aggregated proteins by inducing their degradation.42, 43, 44, 45, 46 We established, here, that p62/SQSTM1 operates as a cell survival signal for AML cells that undergo granulocyte differentiation. Furthermore, we showed that p62/SQSTM1 mitigates the effects of accumulation of aggregated proteins during ATRA-induced differentiation of APL cells. Whether p62/SQSTM1 is involved in the degradation of a bulk aggregate-prone proteins degradation system or allows the clearance of specific proteins remains an open question. Along this line, autophagy has been shown to contribute to the degradation of PML-RARα, a leukemogenic protein, through a mechanism that involved the binding of p62/SQSTM1 to PML-RARα.48, 49 Yet, how PML-RARα is recognized and directed to autophagosomes by p62/SQSTM1 is not elucidated. Interestingly, PML-RARα is, in turn, involved in the activation of constitutive autophagy activity implying a mutual regulation between PML-RARα and autophagy pathways.50 Further investigation is required to determine whether and how p62/SQSTM1 protects cells against proteotoxic stress during granulocyte differentiation process.
In the present study, we found that p62/SQSTM1 expression is regulated through an NF-κB-dependant mechanism during granulocyte differentiation of AML cells.40 Interestingly, the activity of NF-κB is, in turn, regulated by p62/SQSTM1.1, 3, 4, 47 This suggests that NF-κB-mediated p62/SQSTM1 upregulation may represent a feed-forward signal for prolongation of NF-κB activity and thus cell survival during granulocytic maturation. In line with this assumption, Ling et al.51 showed that the NF-κB pathway is activated by Kras (G12D) in pancreatic ductal adenocarcinoma through p62/SQSTM1 and IL-1 alpha feed-forward loops. In fact, the mutual regulation between p62/SQSTM1 and NF-κB pathways warrants further investigation. Moreover, our results corroborate a recent study that showed that miR-17, -20, -93, and -106, which target p62/SQSTM1, are expressed at higher levels in mouse and human hematopoietic blast cells than in mature myeloid cells.52 Other transcriptional factors and miRNAs may also impact on p62/SQSTM1 expression.
We showed that the levels of p62/SQSTM1 mRNA and protein were both significantly increased during ATRA-induced differentiation of both the APL and AML cells (Figures 1a and d). In agreement with these results, we found an upregulation of p62/SQSTM1 upon in vitro granulocytic differentiation of primary CD34+ progenitors cells. Accordingly, primary blast cells of AML patients exhibited significantly lower p62/SQSTM1 mRNA levels than did granulocytes from healthy donors. p62/SQSTM1 upregulation was also shown during maturation of AML cells into either monocytes (Figure 1e) or megakaryocytes (data not shown).23 Of note, our findings differ from those reported by Wang et al.49 who showed that p62/SQSTM1 is downregulated upon ATRA treatment of AML cells and contributes to their differentiation. To explain this discrepancy, we compared the expression of p62/SQSTM1 following cell lysis in two different lysis buffers and by using two different antibodies against p62/SQSTM1 (detailed information in Materials and Methods). In conclusion, using two different anti-p62 antibodies and two different procedures of protein extraction, we reproducibly confirmed a significant upregulation of p62/SQSTM1 protein in ATRA-treated NB4 cells.
Taken together, these results support the idea that p62/SQSTM1 upregulation may represent a general physiological response that occurs during terminal differentiation of myeloid cells towards a specific cell lineage. These findings also suggest that p62/SQSTM1 upregulation may serve as a therapeutic marker, which may allow for early detection of refractory and relapsed AML patients with previous exposure to ATRA. Further studies are needed to assess for the validity of this hypothesis.
In conclusion, our data demonstrated that the p62/SQSTM1 is downregulated in cells that harbors an immature myeloid phenotype (i.e. AML cell lines, primary CD34+ progenitors cells, and primary blasts from AML patients) as compared with terminally differentiated cells. Moreover, our results revealed that NF-κB-mediated p62/SQSTM1 upregulation has a critical role in the viability of AML cells that undergo granulocytic differentiation. Additional studies are needed to establish the physiological and physiopathological significance of the pro-survival function of p62/SQSTM1 in neutrophil/granulocyte development and in the outcome of AML patients, in particular, those who exhibit relapse disease after previous exposure to ATRA.
Materials and Methods
Reagents
ATRA (#R2625), E64d (#516485), Bafilomycin A1, Triton X-100 (#T8532), 8-(4-chlorophenylthio) 5′-cyclic adenosine monophosphate (8-CPT-cAMP) (#C3912), MG132 (#M7449), NBT chloride (NBT chloride (#298-83-9), and Puromycin (#P8833) Phorbol 12–myristate 13-acetate (#16561-29-8) were purchased from Sigma-Aldrich (Saint-Quentin Fallavier, France). Pepstatin A methyl ester (#516485) was obtained from Millipore SAS (Molsheim, France). Tetramethylrhodamine methyl ester (TMRM) (#T-668) and propidium Iodide (# P3566) were purchased from Life Technologies (Saint-Aubin, France). Antibodies against the following proteins were used: p62/SQSTM1 (#610832), p62 (#SC28359) Santa Cruz Biotechnology (Dallas TX, USA), BD Biosciences (Rungis, France); Caspase 3 (#9665), Cell Signaling Technology (Danvers, MA, USA); Caspase 8 (# 21115), Millipore SAS; LC3 (#L7543), Sigma-Aldrich; Ubiquitin (#sc-8017), GAPDH (#sc-51907) and IκBα (#sc-847) Santa Cruz Biotechnology; CD11c (#333145) R&D Systems (Lille, France). The ProteoStat Aggresome detection kit (ENZ-51035-K100) was purchased from Enzo life sciences (Villeurbanne, France). The RNeasy Plus Mini kit (#74134) was obtained from Qiagen (Hilden, Germany). The SuperscriptIII reverse transcriptase (#10432292), the pDONR201 plasmid (#430234) and cell culture media were purchased from Life Technologies-Invitrogen (Saint-Aubin, France).
Cell culture conditions
The acute promyelocytic leukemia-derived cell lines, NB4 (maturation-sensitive), and NB4-LR1 (maturation-resistant) carrying the reciprocal chromosomal translocation t(15 : 17) were obtained from Dr. M Lanotte.36, 37 The myeloid leukemia cell line HL60 was purchased from the ATCC. All cells were cultivated in a RPMI 1640 medium supplemented with 2 mM L-glutamine, 50 units/ml penicillin, 50 μg/ml streptomycin, and 10% decomplemented fetal calf serum. Cells were grown in a humidified incubator at 37 °C with 5% CO2 and 95% air.
Isolation of primary AML blast cells, CD34+ progenitors, and granulocytes
A cohort of 98 samples from patients with a diagnosis of primary AML (FAB M0-M4) were enrolled on HOVON/SAKK (Dutch-Belgian Hematology-Oncology/Swiss Group for Clinical Cancer Research Cooperative group) protocols -04, -04A, -29, and -42 (available at www.hovon.nl) between 1987 and 2006.53 Isolation of primary cells and in vitro differentiation of CD34+ progenitor cells was done as described earlier.54 The details of patients examined in the study are indicated in Supplementary Table.
Immunofluorescence study
Cells were fixed and permeabilized with ice-cold methanol at −20 °C for 4 min before incubation with the primary antibody diluted in blocking solution (PBS supplemented with 1% bovine serum albumin). Cells were washed with PBS supplemented with 0.1% Tween 20 before incubation with the secondary antibody. Cells were mounted on slides with mounting medium containing DAPI to counterstain nuclei. For polyubiquitinated proteins staining, cells were fixed in 4% paraformaldeyde and permeabilized in 0,3% Trition X-100 prior incubation with anti-ubiquitin antibody. Immunofluorescence and confocal microscopy were performed with a × 60 objective using either an Olympus iX80 microscope or a Zeiss LSM 510 Meta microscope.
Lentivirus construction, preparation and cell transduction
The IκBα (A32/A36: serine-to-alanine mutations at residues 32 and 36) vector encoding a super repressor form of IκBα was previously described41 and was kindly provided from Dr. N Faumont (CNRS-6101, Limoges, France). The MAD lentivirus vector was constructed by using Gateway technology after cloning into pDONR201 plasmid. The MAD cDNA was subsequently transferred by LR recombination into a Gateway adapted vector pER-60 plasmid that contains the human phosphoglycerate kinase (PGK) promoter, a Gateway attR cassette, the mouse PGK promoter, and the puromycin acetyltransferase gene cloned into pRRLhPGK.GFP.SIN to give the MAD lentivirus vector.55 pER-60 was self-ligated to give the control lentivirus vector. The shRNA lentivirus transduction approach was used to silence the p62/SQSTM1 expression gene. Three pLKO1 lentiviral vectors expressing DNA sequences encoding shRNA p62#0 shRNA (TRCN0000007234), p62#1 shRNA (TRCN0000007235), and p62#2 shRNA (TRCN0000007236) were purchased from Open Biosystems (Huntsville, AL, USA). The pXS68 non-targeting shRNA was used as lentivirus cell transduction control. Lentiviruses were produced by calcium phosphate transfection of HEK 293T cells.55 Virus-containing supernatants were collected 48 h after transfection, concentrated by centrifugal filters Amicon Ultra 50K (Millipore SAS). The titer of each lentiviral batch was determined on NB4 cells. At 48 h after infection, transduced cells were selected by treatment with puromycin (2.5 μg/ml) for 3 days.
Isolation of RNA and real-time quantitative PCR
Total RNA extraction and cDNA generation were performed as previously described.56 The sequences of forward and reverse oligonucleotide primers, specific to the chosen candidate and housekeeping genes, were designed using Primer3 software: http://primer3.sourceforge.net/. PCR primers used for amplification of p62/SQSTM1 were (forward) TACGACTTGTGTAGCGTCTGC and (reverse) GTGTCCGTGTTTCACCTTCC and the primers used to amplify the housekeeping gene cyclophilin B (CPB) were (forward) ACTTCACCAGGGGAGATGG and (reverse) AGCCGTTGGTGTCTTTGC). Real-time quantitative PCR (RT-qPCR) was performed in a LightCycler (Roche, Meylan, France) thermal cycler as previously described.56 Threshold Ct values, which correlate inversely with the target mRNA levels, were calculated for each cDNA. The p62/SQSTM1 mRNA values were normalized to the expression level of the housekeeping gene, CPB. Results were expressed as n-fold variations of p62/SQSTM1 mRNA expression compared with untreated cells by using the 2−ΔΔCT method of relative gene expression quantification.57 All experiments were performed in triplicate. Patient data represent values normalized to the expression levels of the housekeeping genes HMBS (hydroxymethylbilane synthase) and ABL1 (Abelson murine leukemia viral oncogene homolog1).
Preparation of whole-cell extracts and Triton X100 -soluble and -insoluble protein fractions
Whole-cell extracts were prepared from 3–5 × 106 cells subjected to lysis in either Tris/SDS buffer (10 mM Tris, pH 7.4, 1% SDS, 1 mM sodium orthovanadate)56 or RIPA buffer. Note, a significant p62/SQSTM1 upregulation was observed upon ATRA treatment of NB4 cells irrespective of cell lysis buffers used (data not shown). For some experiments, Triton X100-soluble and -insoluble protein fractionation were performed following cell lysis in 20 mM NaCl, 20 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 0.1 mM EDTA, 0.1% Triton X-100, and protease cocktail, for 30 min at 4 °C. Then, cell extracts were centrifuged for 15 min at 15 000 g to separate soluble (supernatant) and insoluble (pellet) fractions.58
Protein gel blot analysis
Fractions (30 μg to 50 μg) of cellular extract were subjected to SDS-polyacrylamide gel electrophoresis using a Tris/glycine buffer system based on the method of Laemmli as previously described.56 ‘The p62/SQSTM1 protein levels were assessed following immunoblot analysis by using two different anti-p62/SQSTM1 antibodies: one from (BD Transduction Laboratory, #610832) and one from Santa Cruz biotechnology (#SC28359), which was used by Wang et al.49
Determination of aggregated proteins
Expression of aggregated proteins was determined by (i) immunoblot analysis of Triton X-100 using an antibody against polyubiquitinated proteins (ii) immunofluorescence analysis of polyubiquitinated proteins in cells , and (iii) measurement of aggresome formation using the ProteoStat Aggresome detection kit. Briefly, cells are incubated with the ProteoStat dye that becomes brightly fluorescent upon binding to aggregated proteins and processed for flow cytometeric analysis for determination of aggresomes in cells. MG132 is used as an inducer of aggresome formation.
Assessment of granulocyte differentiation
Granulocyte differentiation in the NB4 cells was determined by (i) morphologic assessment of cells stained with May-Grunwald Giemsa (MGG), (ii) analysis of the expression of the CD11c cell surface marker, and (iii) the nitroblue tetrazolium (NBT) reduction assay, as previously described.40, 35 Monocyte differentiation in the NB4 cells was assessed by analysis of the expression of the CD14 and CD11b monocyte cell surface markers. The isolation of CD34+ progenitors and their in vitro granulocytic differentiation was performed following incubation of CD34+ progenitor cells with 10 ng/ml human recombinant granulocytic colony-stimulating factor as previously described.54
Evaluation of cell death processes
Cell death modalities were characterized by (i) measurement of the mitochondrial transmembrane potential (ΔΨm) as previously described35, 56 using TMRM dye, (ii) evaluation of plasma cell membrane permeability as previously described using propidium iodide dye or (iii) western blotting analyses of the cleaved forms of caspases.
Statistical analysis
Unless otherwise stated, the ANOVA Tukey test was used for calculation of P-values for comparisons between two groups using GraphPad Prism software. For the analysis of the p62/SQSTM1 mRNA data in primary blast cells from AML patients, the non-parametic Mann–Whitney U-test was used for calculation of P-values. P-values<0.05 were considered to be statistically significant. Results are given as means plus or minus S.D. of three independent experiments. For western blotting experiments, data are representative of at least two independent experiments.
Abbreviations
- ABL1:
-
Abelson murine leukemia viral oncogene homolog 1
- AML:
-
acute myeloid leukemia
- APL:
-
acute promyelocytic leukemia
- ATRA:
-
all-trans retinoic acid
- ChIP:
-
chromatin immunoprecipitation
- 8-CPT-cAMP:
-
8-(4-chlorophenylthio), 5’-cyclic adenosine monophosphate
- CPB:
-
cyclophilin B
- HMBS:
-
hydroxymethylbilane synthase
- LC3-II:
-
microtubule-associated protein 1 light chain 3-II
- MGG:
-
May-Grunwald Giemsa
- MFI:
-
Median fluorescence intensity
- NF-κB:
-
nuclear factor-κB
- Nrf-2:
-
NF-E2 related factor E2
- NBT:
-
p-nitro-blue tetrazolium
- PKC:
-
protein kinase C
- PMA:
-
Phorbol 12–myristate 13-acetate PML
- PML:
-
promyelocytic leukemia
- RAR-α:
-
Retinoic acid receptor alpha
- RT-qPCR:
-
real-time quantitative polymerase chain reaction
- TMRM:
-
tetramethylrhodamine methyl ester
- shRNA:
-
small hairpin RNA
- SQSTM1:
-
sequestosome 1
References
Komatsu M, Kageyama S, Ichimura Y . p62/SQSTM1/A170: physiology and pathology. Pharmacol Res 2012; 66: 457–462.
Geetha T, Vishwaprakash N, Sycheva M, Babu JR . Sequestosome 1/p62: across diseases. Biomarkers 2012; 17: 99–103.
Wooten MW, Seibenhener ML, Mamidipudi V, Diaz-Meco MT, Barker PA, Moscat J . The atypical protein kinase C-interacting protein p62 is a scaffold for NF-kappaB activation by nerve growth factor. J Biol Chem 2001; 276: 7709–7712.
Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008; 13: 343–354.
Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 2011; 44: 134–146.
Nezis IP, Stenmark H . p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxid Redox Signal 2012; 17: 786–793.
Puissant A, Fenouille N, Auberger P . When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res 2012; 2: 397–413.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282: 24131–24145.
Lin X, Li S, Zhao Y, Ma X, Zhang K, He X et al. Interaction domains of p62: A bridge between p62 and selective autophagy. DNA Cell Biol 2013; 32: 220–227.
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137: 1062–1075.
Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 2010; 285: 22576–22591.
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010; 12: 213–223.
Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 2010; 30: 3275–3285.
Goussetis DJ, Gounaris E, Wu EJ, Vakana E, Sharma B, Bogyo M et al. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012; 120: 3555–3562.
Petherick KJ, Williams AC, Lane JD, Ordonez-Moran P, Huelsken J, Collard TJ et al. Autolysosomal beta-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J 2013; 32: 1903–1916.
Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T et al. Autophagy negatively regulates Wnt signalling by promoting dishevelled degradation. Nat Cell Biol 2010; 12: 781–790.
Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009; 137: 721–735.
Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem 2012; 287: 12455–12468.
Lee SJ, Pfluger PT, Kim JY, Nogueiras R, Duran A, Pages G et al. A functional role for the p62-ERK1 axis in the control of energy homeostasis and adipogenesis. EMBO Rep 2010; 11: 226–232.
Duran A, Serrano M, Leitges M, Flores JM, Picard S, Brown JP et al. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell 2004; 6: 303–309.
Waters S, Marchbank K, Solomon E, Whitehouse CA . Autophagic receptors Nbr1 and p62 coregulate skeletal remodeling. Autophagy 2010; 6: 981–983.
Samuels IS, Seibenhener ML, Neidigh KB, Wooten MW . Nerve growth factor stimulates the interaction of ZIP/p62 with atypical protein kinase C and targets endosomal localization: evidence for regulation of nerve growth factor-induced differentiation. J Cell Biochem 2001; 82: 452–466.
Colosetti P, Puissant A, Robert G, Luciano F, Jacquel A, Gounon P et al. Autophagy is an important event for megakaryocytic differentiation of the chronic myelogenous leukemia K562 cell line. Autophagy 2009; 5: 1092–1098.
Chang CP, Su YC, Hu CW, Lei HY . TLR2-dependent selective autophagy regulates NF-kappaB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ 2013; 20: 515–523.
Licht JD . Acute promyelocytic leukemia—weapons of mass differentiation. N Engl J Med 2009; 360: 928–930.
Becker H, Bloomfield CD . Acute myeloid leukaemia in 2012: En route to improved treatment options. Nat Rev Clin Oncol 2013; 10: 76–79.
de The H, Chomienne C, Lanotte M, Degos L, Dejean A . The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 1990; 347: 558–561.
Dyck JA, Maul GG, Miller WH Jr, Chen JD, Kakizuka A, Evans RM . A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein. Cell 1994; 76: 333–343.
Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988; 72: 567–572.
Castaigne S, Chomienne C, Daniel MT, Ballerini P, Berger R, Fenaux P et al. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood 1990; 76: 1704–1709.
Mi JQ, Li JM, Shen ZX, Chen SJ, Chen Z . How to manage acute promyelocytic leukemia. Leukemia 2012; 26: 1743–1751.
Gallagher RE . Retinoic acid resistance in acute promyelocytic leukemia. Leukemia 2002; 16: 1940–1958.
Petrie K, Zelent A, Waxman S . Differentiation therapy of acute myeloid leukemia: past, present and future. Curr Opin Hematol 2009; 16: 84–91.
Kamimura T, Miyamoto T, Harada M, Akashi K . Advances in therapies for acute promyelocytic leukemia. Cancer Sci 2011; 102: 1929–1937.
Trocoli A, Mathieu J, Priault M, Reiffers J, Souquere S, Pierron G et al. ATRA-induced upregulation of Beclin 1 prolongs the life span of differentiated acute promyelocytic leukemia cells. Autophagy 2011; 7: 1108–1114.
Lanotte M, Martin-Thouvenin V, Najman S, Balerini P, Valensi F, Berger R . NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3). Blood 1991; 77: 1080–1086.
Duprez E, Ruchaud S, Houge G, Martin-Thouvenin V, Valensi F, Kastner P et al. A retinoid acid 'resistant' t(15;17) acute promyelocytic leukemia cell line: isolation, morphological, immunological, and molecular features. Leukemia 1992; 6: 1281–1287.
Zhao Q, Tao J, Zhu Q, Jia PM, Dou AX, Li X et al. Rapid induction of cAMP/PKA pathway during retinoic acid-induced acute promyelocytic leukemia cell differentiation. Leukemia 2004; 18: 285–292.
Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57–74.
Mathieu J, Giraudier S, Lanotte M, Besancon F . Retinoid-induced activation of NF-kappaB in APL cells is not essential for granulocytic differentiation, but prolongs the life span of mature cells. Oncogene 2005; 24: 7145–7155.
Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA . Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J 1995; 14: 2876–2883.
Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 2002; 160: 255–263.
Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW . Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 2004; 24: 8055–8068.
Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171: 603–614.
Lamark T, Kirkin V, Dikic I, Johansen T . NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009; 8: 1986–1990.
Ichimura Y, Komatsu M . Selective degradation of p62 by autophagy. Semin Immunopathol 2010; 32: 431–436.
Moscat J, Diaz-Meco MT . p62: a versatile multitasker takes on cancer. Trends Biochem Sci 2012; 37: 230–236.
Isakson P, Bjoras M, Boe SO, Simonsen A . Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010; 116: 2324–2331.
Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao Y et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARalpha oncoprotein. Autophagy 2011; 7: 401–411.
Huang Y, Hou JK, Chen TT, Zhao XY, Yan ZW, Zhang J et al. PML-RARalpha enhances constitutive autophagic activity through inhibiting the Akt/mTOR pathway. Autophagy 2011; 7: 1132–1144.
Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012; 21: 105–120.
Meenhuis A, van Veelen PA, de Looper H, van Boxtel N, van den Berge IJ, Sun SM et al. MiR-17/20/93/106 promote hematopoietic cell expansion by targeting sequestosome 1-regulated pathways in mice. Blood 2011; 118: 916–925.
Federzoni EA, Valk PJ, Torbett BE, Haferlach T, Lowenberg B, Fey MF et al. PU.1 is linking the glycolytic enzyme HK3 in neutrophil differentiation and survival of APL cells. Blood 2012; 119: 4963–4970.
Britschgi C, Jenal M, Rizzi M, Mueller BU, Torbett BE, Andres AC et al. HIC1 tumour suppressor gene is suppressed in acute myeloid leukaemia and induced during granulocytic differentiation. Br J Haematol 2008; 141: 179–187.
Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D et al. A third-generation lentivirus vector with a conditional packaging system. J Virol 1998; 72: 8463–8471.
Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem 2006; 281: 30373–30382.
Livak KJ, Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25: 402–408.
Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES . Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J Cell Biol 1999; 146: 1239–1254.
Acknowledgements
This work was supported by funds from the Institut National de la Santé et de la Recherche Médicale (INSERM), the University of Bordeaux, the Institut Bergonié, the ‘Conseil Regional d’Aquitaine’, the Ligue contre le Cancer Comité de la Gironde (to MDM), the ‘ INCa-DGOS-Inserm 6046 ‘ (to MDM), the Ligue contre le Cancer Comité Ile de France (to E S-B) and by grants from Swiss National Science Foundation (31003A_143739 to MPT), the Werner and Hedy Berger-Janser Foundation of Cancer Research (to MPT), and the Bern University Research Foundation (to MPT) AT is supported by a PhD fellowship from the Conseil Régional Aquitaine/INSERM (to M.D.M.). We gratefully acknowledge Dr. PJM Valk and Dr. B Löwenberg and the HOVON (Dutch-Belgian Hematology-Oncology) cooperative group for providing primary AML patient samples. We thank Prof. Richard Iggo for constructive advice and Pascal Correia (institut Bergonié, unité de biologie moléculaire, Bordeaux) for his technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Baehrecke
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Trocoli, A., Bensadoun, P., Richard, E. et al. p62/SQSTM1 upregulation constitutes a survival mechanism that occurs during granulocytic differentiation of acute myeloid leukemia cells. Cell Death Differ 21, 1852–1861 (2014). https://doi.org/10.1038/cdd.2014.102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2014.102
This article is cited by
-
Sphingolipid-mediated inflammatory signaling leading to autophagy inhibition converts erythropoiesis to myelopoiesis in human hematopoietic stem/progenitor cells
Cell Death & Differentiation (2019)
-
WX20120108, a novel IAP antagonist, induces tumor cell autophagy via activating ROS-FOXO pathway
Acta Pharmacologica Sinica (2019)
-
5-Aminoimidazole-4-carboxamide ribonucleoside-induced autophagy flux during differentiation of monocytic leukemia cells
Cell Death Discovery (2017)

{kind=link}
{kind=link}