
Abstract
High levels of BCL-2 family proteins are implicated in a failed/ineffective apoptotic programme, often resulting in diseases, including cancer. Owing to their potential as drug targets in cancer therapy, several inhibitors of BCL-2 family proteins have been developed. These primarily target specific members of the BCL-2 family, particularly BCL-2 and BCL-XL but are ineffective against MCL-1. Major efforts have been invested in developing inhibitors of MCL-1, which is commonly amplified in human tumours and associated with tumour relapse and chemoresistance. In this report, the specificity of several BCL-2 family inhibitors (ABT-263, UCB-1350883, apogossypol and BH3I-1) was investigated and compared with putative MCL-1 inhibitors designed to exhibit improved or selective binding affinities for MCL-1 (TW-37, BI97C1, BI97C10, BI112D1, compounds 6 and 7, and MCL-1 inhibitor molecule (MIM-1)). ABT-263, BI97C1, BI112D1, MIM-1 and TW-37 exhibited specificity in inducing apoptosis in a Bax/Bak- and caspase-9-dependent manner, whereas the other agents showed no killing activity, or little or no specificity. Of these inhibitors, only ABT-263 and UCB-1350883 induced apoptosis in a BCL-2- or BCL-XL-dependent system. In cells that depend on MCL-1 for survival, ABT-263 and TW-37 induced extensive apoptosis, suggesting that at high concentrations these inhibitors have the propensity to inhibit MCL-1 in a cellular context. TW-37 induced apoptosis, assessed by chromatin condensation, caspase processing and phosphatidylserine externalisation, in a BAK-dependent manner and in cells that require MCL-1 for survival. TW-37-mediated apoptosis was also partly dependent on NOXA, suggesting that derivatives of TW-37, if engineered to exhibit better selectivity and efficacy at low nanomolar concentrations, may provide useful lead compounds for further synthetic programmes. Expanded medicinal chemistry iteration, as performed for the ABT series, may likewise improve the potency and specificity of the evaluated MCL-1 inhibitors.
Similar content being viewed by others

Main
Most cancer chemotherapeutic agents kill tumour cells by the induction of the intrinsic pathway of apoptosis, resulting from perturbation of mitochondria, release of cytochrome c and caspase activation. Mitochondrial integrity, and hence apoptosis, is primarily regulated by the BCL-2 family of proteins that comprise anti-apoptotic members, including BCL-2, BCL-XL, MCL-1 and BCL2A1, pro-apoptotic molecules, such as the multi-domain BAX and BAK proteins, and BH3-only proteins, including BIM, BAD, PUMA and NOXA.1, 2 Anti-apoptotic BCL-2 family proteins divide into two classes, one comprising BCL-2, BCL-XL and BCL-w, and the other comprising MCL-1 and BCL2A1. Efficient apoptosis generally requires the neutralisation of both these classes.3
As high levels of anti-apoptotic BCL-2 family members are often associated with tumour resistance, there has been a major emphasis to develop inhibitors of the BCL-2 family. Most of these inhibitors have been shown to be rather non-specific with the notable exception of ABT-737 and its orally active analogue ABT-263 (navitoclax), the latter of which has recently entered clinical trials for treating various haematological malignancies.4, 5, 6, 7, 8, 9 Both ABT-737 and ABT-263 inhibit BCL-2, BCL-XL and BCL-w but not MCL-1 or BCL2A1.5, 6 Some tumours are dependent or addicted to a specific anti-apoptotic protein, such as the dependence of chronic lymphocytic leukaemia (CLL) cells for survival on BCL-2. Such cells are rapidly killed by ABT-737 or ABT-263 but these molecules also induce a dose-limiting thrombocytopenia as they inhibit BCL-XL, which is required for survival of platelets. The design of ABT-199, another related analogue that selectively inhibits BCL-2, but not BCL-XL, seems to circumvent this issue.10 Thus, a highly specific inhibitor of one BCL-2 family member may be required for the treatment of some tumours. Of the different BCL-2 family members, as the survival of most tumours is not dependent on a single anti-apoptotic BCL-2 protein, efficient treatment will more commonly require either a pan-BCL-2 family protein inhibitor or a combination of inhibitors that neutralises both classes of anti-apoptotic BCL-2 family proteins. Of the different BCL-2 family members, MCL-1 is commonly amplified in human tumours and is often associated with tumour relapse and chemoresistance, particularly to ABT-737 and ABT-263.7, 11, 12, 13, 14 These findings highlight an urgent need to find either specific MCL-1 inhibitors or pan-BCL-2 family inhibitors, which could be valuable in treating resistant tumours.
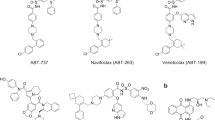
Several putative selective inhibitors of MCL-1, including obatoclax (GX-1570),15 two derivatives of rhodanine (compounds 6 and 7),16 TW-37,17 derivatives of apogossypol/apogossypolone (BI97C1 (commonly referred to as sabutoclax), BI97C10 and BI112D1 (also called BI97D6)18, 19, 20 and MCL-1 inhibitor molecule (MIM-1)21 have been synthesised based on different approaches including modelling studies, in silico and in vitro screens (Figure 1). In this study, we evaluate these compounds either as potentially selective MCL-1 inhibitors or as pan-BCL-2 family inhibitors.

Chemical structures of different inhibitors used in this study. ABT-263 (navitoclax) is an orally active analogue of ABT-737, developed by Abbott Laboratories (Abbott Park, IL, USA) and designed to mimic the BH3 domain of BAD. ABT-263 binds selectively to the BH3-binding groove of BCL-2, BCL-XL and BCL-w, and is currently in clinical trials for treating several haematological malignancies. UCB-1350883, identified by UCB Pharma Ltd., in a high throughput screen is a moderately potent and selective inhibitor of BCL-2. MIM-1 is a small molecule inhibitor identified by a stapled peptide-based competitive screen, and is found to selectively bind and inhibit the BH3-binding groove of MCL-1. Removal of the toxic aldehyde groups in the naturally occurring polyphenol, gossypol, resulted in the synthesis of apogossypol, which binds to the hydrophobic groove of BCL-2 and BCL-XL. Further substitution of the isopropyl side groups in apogossypol yielded BI97C1 (sabutoclax), which targets multiple anti-apoptotic members, including BCL-2, BCL-XL, MCL-1 and BCL2A1. Apogossypolone is a third-generation gossypol derivative, designed to effectively target MCL-1, with reduced toxicity and non-specific reactivity. Structural derivatives of apogossypolone include BI97C10 and BI112D1. Screening of a small focused library of rhodanine derivatives led to the identification of compounds 6 and 7 molecules that selectively antagonise MCL-1. Compounds 6 and 7 incorporated structural elements of BH3I that was originally designed to inhibit BCL-2 family proteins. TW-37 is a second-generation benzenesulphonyl derivative of gossypol developed through computational screening and NMR that binds to MCL-1 with higher affinity than to BCL-2 and BCL-XL
Results
Putative MCL-1 inhibitors exhibit varied specificity in inducing BAK-dependent apoptosis
BAX and BAK have crucial roles in the release of mitochondrial cytochrome c and activation of apoptosis, and this is counteracted by the anti-apoptotic members.1, 2 Hence, BCL-2 family antagonists are expected to induce cytochrome c release and apoptosis in a BAX/BAK-dependent manner. To test the specificity of the various inhibitors (Figure 1), we used Jurkat-T lymphocytes either deficient in or reconstituted with BAK.9 As Jurkat cells have no endogenous BAX, they rely solely on BAK to initiate cytochrome c release and apoptosis. ABT-263 (navitoclax), a BCL-2 family antagonist that has recently entered clinical trials in patients with B-cell malignancies, was used as a positive control as it induces cell death in a BAX/BAK-dependent manner.6, 22 In agreement, ABT-263 induced a concentration-dependent cell death (assessed by phosphatidylserine (PS) externalisation) in BAK-reconstituted but not in BAK-deficient Jurkat cells (Figure 2). UCB-1350883,23 identified by a high throughput screen as a moderately potent and selective inhibitor of BCL-2, exhibited some selectivity in inducing apoptosis only in the presence of BAK, albeit at high concentrations (30 μM). However, neither ABT-263 nor UCB-1350883 was designed to inhibit MCL-1. In order to specifically target multiple members of BCL-2 family, including MCL-1, several putative broad-spectrum BCL-2 family antagonists with improved specificity to inhibit MCL-1 have been developed.16, 17, 18, 19, 20, 21, 24 One such broad-spectrum BCL-2 family antagonist, apogossypol, demonstrated modest selectivity in killing cells in a BAK-dependent manner, in agreement with our previous report (Figure 2).9 Therefore, we assessed the specificity of different structural derivatives of apogossypol (BI97C1) and apogossypolone (BI97C10 and BI112D1), which were designed to possess enhanced binding affinities to MCL-1, in addition to other BCL-2 family members.18, 19, 20 Of these, BI97C1 exhibited some selectivity at low concentrations (⩽10 μM) but not at higher concentrations (30 μM; Figure 2). BI97C10 was ineffective at lower concentrations (⩽10 μM), and at high concentrations (30 μM) lacked specificity (Figure 2). In marked contrast BI112D1 exhibited marked selectivity, only inducing apoptosis in cells reconstituted with BAK (Figure 2). TW-37, a rationally designed benzenesulphonyl derivative that inhibits BCL-2, BCL-XL and MCL-1,17 exhibited modest selectivity to the BAK-reconstituted Jurkat cells compared with BI97C1 and BI112D1. Neither BH3I-1 nor its structural derivatives, compounds 6 and 7 killed cells even at high concentrations.16, 25 MIM-1, recently reported as a novel MCL-1 inhibitor,21 induced apoptosis only at high concentrations (⩾10 μM), consistent with its reported cellular IC50 and exhibited notable selectivity (Figure 2).

Putative MCL-1 inhibitors induce BAK-dependent apoptosis to different extents. Jurkat-T cells either deficient in (dotted lines) or reconstituted with BAK (BAK-R, continuous bold lines) were exposed for 24 h to different concentrations of the indicated inhibitors and the extent of apoptosis assessed by PS externalisation, using Annexin-FITC and PI staining. Error bars represent the mean±S.E.M. from at least three independent experiments. In some cases, the error bar is not visible because it is smaller than the symbol on the graph. *P<0.05 using a paired t-test. Western blots in the inset indicate the expression level of BAK in the reconstituted and deficient cells compared with Jurkat A3 cells used as a positive control
MCL-1 inhibitors induce apoptosis primarily in a caspase-9-dependent manner
Cytochrome c, released from the mitochondria in a BAX/BAK-dependent manner, results in formation of the Apaf-1 apoptosome followed by activation of caspase-9, the apical caspase in the intrinsic pathway of apoptosis. To investigate whether the putative MCL-1 inhibitors induced apoptosis in a caspase-9-dependent manner, we used Jurkat-T lymphocytes that are either deficient in or reconstituted with caspase-9. Surprisingly, the extent of apoptosis observed in the wild-type (caspase-9 reconstituted) Jurkat cells, following exposure to most putative MCL-1 inhibitors, was significantly diminished compared with the BAK-reconstituted Jurkat cells (Supplementary Figure S1). Examination of the expression levels of different pro- and anti-apoptotic proteins in these cells revealed very low levels of BAK in both the caspase-9-reconstituted and -deficient cells compared with normal Jurkat cells (Supplementary Figure S1), which most probably resulted in diminished apoptosis (Supplementary Figure S1). To overcome this problem and to ascertain whether the inhibitors induced apoptosis in a caspase-9-dependent manner, we next used mouse embryonic fibroblasts (MEFs) that are either deficient in caspase-9 (caspase-9 null) or deficient in Bax and Bak (DKO). As BH3I-1, compounds 6 and 7, and UCB-1350883 showed either little or no activity at concentrations <30 μM in Jurkat cells (Figure 2), it was decided to carry out only limited studies of these compounds. Exposure of the MEFs to ABT-263 resulted in a concentration-dependent apoptosis, which was predominantly both Bax/Bak- and caspase-9-dependent (Figure 3). The structural derivatives of apogossypol (BI97C1 and BI112D1) and TW-37 were more potent than ABT-263 and also resulted in a Bax/Bak- and caspase-9-dependent apoptosis in MEFs (Figure 3). However, such specificity was generally lost when these inhibitors were used at high concentrations (>10 μM), possibly owing to off-target effects (Figure 3). Of the different inhibitors tested, MIM-1 was more modest in inducing apoptosis in these cells, consistent with our previous findings in Jurkat cells (Figures 2 and 3 and Supplementary Figure 1). Taken together, these data indicate that the putative MCL-1 inhibitors primarily induce the intrinsic pathway of apoptosis.

Effects of putative MCL-1 inhibitors on Bax/Bak- and caspase-9-deficient MEFs. MEFs deficient in either Bax and Bak (dotted lines) or caspase-9 (dashed lines) along with their wild-type counterparts (continuous bold lines) were exposed for 24 h to different concentrations of the indicated inhibitors and the extent of apoptosis assessed by PS externalisation. Error bars represent the mean±S.E.M. from at least three independent experiments. In some cases, the error bar is not visible because it is smaller than the symbol on the graph. Western blots in the inset indicate the expression levels of Bax, Bak and caspase-9 in the different cells used
ABT-263, but not other putative BCL-2 family antagonists, specifically inhibits BCL-2 and BCL-XL
Although ABT-263 is selective against certain members of the BCL-2 family (BCL-2, BCL-XL and BCL-w), it does not inhibit MCL-1,6 which is a major anti-apoptotic protein contributing to tumour relapse and chemoresistance. Unlike ABT-263, the structural derivatives of apogossypol (BI97C1 and BI112D1), and TW-37, exhibit increased in vitro binding affinities to multiple members of the BCL-2 family, including MCL-1, and hence have the potential to be either pan-BCL-2 family inhibitors or to more specifically inhibit MCL-1. Initially, we examined whether these inhibitors were selective against BCL-2 or BCL-XL, using IL-3-dependent FL5.12 cells, stably transfected with either BCL-2 or BCL-XL. Deprivation of IL-3 results in rapid apoptosis in control FL5.12 cells (vector control), which is rescued by the expression of BCL-2 or BCL-XL.26 Thus, in the absence of IL-3 these cells solely depend on overexpressed BCL-2 or BCL-XL for survival, which can be exploited to assess the specificity of the inhibitors. As expected, ABT-263 did not induce apoptosis in control FL5.12 cells in the presence of IL-3. However, following removal of IL-3, when the cells were dependent for survival on BCL-2 or BCL-XL, ABT-263 selectively targeted these anti-apoptotic members and induced a concentration-dependent apoptosis (Figures 4 and 5). To further validate this model, we used ABT-199, a first-in-class selective inhibitor of BCL-2.10 In agreement with previous observations,10 ABT-199 was selective in inducing apoptosis in the FL5.12 cells overexpressing BCL-2 (Figure 4a) and was much less potent in the BCL-XL-dependent system (Figure 5a). UCB-1350883 induced a concentration-dependent apoptosis in a BCL-2-dependent system (Figure 4a); however, at high concentrations it also induced apoptosis in a BCL-XL-dependent system (Figure 5a), suggesting that it shows some selectivity to BCL-2. In contrast, none of the other inhibitors tested (BI97C1, BI112D1 and TW-37) exhibited selectivity in either the BCL-2- or BCL-XL-dependent systems (Figures 4a and 5a). First, all these inhibitors induced enhanced cell death in control FL5.12 cells even in the presence of IL-3, thus suggesting that they may induce cell death by other mechanisms in this model system. However, the reduction in the induction of apoptosis induced by these inhibitors in FL5.12 cells expressing BCL-2 or BCL-XL suggested that neither BCL-2 nor BCL-XL was efficiently inhibited by low concentrations of these inhibitors. Furthermore, MIM-1 did not induce apoptosis in these BCL-2 or BCL-XL addictive systems, (data not shown), consistent with its reported binding specificity for MCL-1.21

ABT-263, ABT-199 and UCB-1350883 but not BI97C1, BI112D1 and TW-37 are selective inhibitors of BCL-2. (a) FL5.12 cells overexpressing BCL-2 were incubated without IL-3 for 24 h before exposure to different concentrations of ABT-263, ABT-199, UCB-1350883, BI97C1, BI112D1 or TW-37 for a further 24 h and analysed for apoptosis by PS externalisation. (b) CLL cells freshly isolated from the peripheral blood of patients were exposed to the indicated concentrations of ABT-263, ABT-199, UCB-1350883, BI97C1, BI112D1 or TW-37 for 4 or 24 h and cell death was assessed by PS externalisation. Error bars represent the mean±S.E.M from at least three independent experiments

Only ABT-263 is a potent and selective inhibitor of BCL-XL (a) FL5.12 cells overexpressing BCL-XL were incubated without IL-3 for 24 h before exposure to different concentrations of ABT-263, ABT-199, UCB-1350883, BI97C1, BI112D1 or TW-37 for a further 24 h and analysed for apoptosis by PS externalisation. (b) H1299 cells reverse-transfected with MCL-1 siRNA for 24 h were exposed to the indicated concentrations of ABT-263, ABT-199, BI97C1, BI112D1 or TW-37 for a further 4 or 24 h and cell death was assessed by PS externalisation. Error bars represent the mean±S.E.M. from at least three independent experiments
In addition to using the IL-3-dependent system for assessing selectivity, we also used freshly isolated primary CLL cells from patients. CLL cells depend on BCL-2 for survival and can be considered BCL-2-addicted.27, 28 Both ABT-263 and ABT-199 induced a rapid concentration-dependent apoptosis in primary CLL cells, which was evident both at early times (4 h) and at low nanomolar concentrations in agreement with previous studies.10, 29 The only other inhibitor to induce a similar rapid concentration-dependent induction of apoptosis in primary CLL cells was UCB-1350883, although it was much less potent than either ABT-263 or ABT-199 (Figure 4b). None of the other inhibitors tested (BI97C1, BI112D1 and TW-37) exhibited rapid apoptosis even at high concentrations (30 μM; Figure 4b). However, prolonged exposure of CLL cells to BI97C1 and to a lesser extent BI112D1 resulted in apoptosis, whereas TW-37 appeared inactive (Figure 4b). Thus, BI97C1 and to a lesser extent BI112D1 induced apoptosis in a BCL-2-dependent system, albeit to a very modest extent in comparison with ABT-263, ABT-199 and UCB-1350883.
For a model of BCL-XL-addicted cells, we used H1299 cells, a non-small cell lung carcinoma line. As survival of these cells depends exclusively on both BCL-XL and MCL-1, depletion of endogenous MCL-1 should result in these cells being dependent solely on BCL-XL for survival.14 This seemed to be the case as ABT-263, an inhibitor of both BCL-2 and BCL-XL, induced a rapid (4 h) concentration-dependent apoptosis, which was evident even at low nanomolar concentrations (Figure 5b). ABT-199 induced a similar concentration-dependent induction of apoptosis in these cells, although it required much higher concentrations than ABT-263, demonstrating its higher selectivity for inhibiting BCL-2 compared with BCL-XL (Figure 5b). In these BCL-XL-addicted H1299 cells, BI97C1, BI112D1 and TW-37 had little, if any, effect at early times but induced a concentration-dependent apoptosis at later times (24 h; Figure 5b). Taken together, our data indicate that the relative potencies of these compounds to inhibit BCL-2 was ABT-199>ABT-263>>UCB-1350883>>BI197C1>BI112D1>TW-37 and to inhibit BCL-XL was ABT-263>>ABT-199>>BI197C1∼BI112D1∼TW-37.
ABT-263 and TW-37, but not other putative BCL-2 family inhibitors, induce apoptosis in an MCL-1-dependent system
To assess the ability of these inhibitors to induce apoptosis in an MCL-1-dependent system, we used H23 cells, a non-small cell lung carcinoma line that expresses no BCL-2, barely detectable amounts of BCL-XL and depend solely on MCL-1 for survival.14 None of the putative MCL-1 inhibitors induced apoptosis at early times (4 h), with the possible exception of BI97C1 (Figure 6a). However, after longer exposure (24 h), ABT-263, BI97C1 and TW-37 induced significant apoptosis, whereas BI112D1 and MIM-1 showed little or no activity (Figure 6a). The ability of ABT-263, BI97C1 and TW-37 to induce apoptosis in H23 cells lent some support to the suggestion that this may be related to their ability to inhibit MCL-1. To confirm and extend these findings to another cellular system, we used H1299 cells, which are dependent for survival on BCL-XL and MCL-1.14, 30 To render the survival of these cells dependent solely on MCL-1, a low concentration of ABT-737 (1 μM) was used to inhibit BCL-XL. Silencing of MCL-1 using RNA interference alone had little effect on the viability of these cells, whereas it induced extensive apoptosis in the presence of ABT-737, supporting the validity of this approach (Figure 6b). Inhibition of BCL-XL with ABT-737 did not enhance the sensitivity of these cells to apoptosis induced by BI97C1, BI112D1, MIM-1, or compounds 6 and 7 (Figure 6b). Surprisingly, the only compound to exhibit increased sensitivity to apoptosis was TW-37 (Figure 6b), further supporting the notion that TW-37 inhibits MCL-1 in H1299 cells. Taken together, these data suggest that TW-37 inhibits MCL-1 in a cellular context, as evidenced by its ability to induce apoptosis in two different MCL-1-dependent cell lines.

TW-37 kills MCL-1-dependent cancer cells in a NOXA-dependent manner. (a) H23 cells exposed for 4 or 24 h to the indicated concentrations of the different inhibitors and cell death assessed by PS externalisation. (b) H1299 cells reverse-transfected with MCL-1 siRNA for 24 h were exposed to 1 μM of ABT-737 for a further 20 h. Western blots in the inset demonstrate the transfection efficiency of the siRNA oligoduplex. Alternatively, H1299 cells exposed for 4 h to 10 μM of BI97C1, BI112D1, TW-37 or MIM-1 followed by a further 20 h exposure to 1 μM of ABT-737 were assessed for cell death by PS externalisation. (c) H1299 cells reverse-transfected with control or NOXA siRNA for 24 h were exposed for 4 h to 10 μM of TW-37, followed by 20 h of 10 μM ABT-737 and cell death assessed by PS externalisation. *P<0.05 using a paired t-test. Blots show the efficiency of knockdowns. In a–c, error bars represent the mean±S.E.M. from at least three independent experiments
MCL-1/NOXA axis has a critical role in sensitisation to apoptosis
MCL-1 is a short-lived protein with a complex regulation, including transcriptional and translational control, as well as the involvement of different E3 ligases and deubiquitinating enzymes.13, 31 It may also be regulated by its interaction with some BH3-only proteins, particularly NOXA.32 We therefore exposed H1299 cells to TW-37 in the presence of ABT-737, which resulted in the induction of NOXA but not BIM or PUMA (Figure 6c and data not shown). Moreover, knockdown of NOXA using RNA interference resulted in marked but incomplete inhibition of TW-37/ABT-737-induced apoptosis (Figure 6c), implying an important role for NOXA in the sensitivity of H1299 cells to this combination.
TW-37 induces all the hallmarks of apoptosis
As high concentrations (>10 μM) of TW-37 were required to induce cell death (Figure 6), we wished to exclude the possibility that TW-37 could result in other forms of cell death due to off-target effects. Ultrastructural examination of H23 cells exposed to TW-37 revealed early indications of apoptosis, characterised by chromatin condensation (8 h), and by 16–24 h extensive blebbing and secondary necrosis was observed (Figure 7a). Pretreatment with a broad-spectrum caspase inhibitor, Z-VAD.fmk, completely blocked the induction of apoptosis (Figure 7a). TW-37 also resulted in the activation of caspase-9 and the cleavage of poly(ADP-ribose) polymerase (PARP; Figure 7b). Moreover, PARP cleavage, caspase-9 activation and PS externalisation, as well as the ultrastructural changes were significantly inhibited in BAK-deficient Jurkat cells (Figures 7c and d). Taken together, our data indicate that derivatives of TW-37, if engineered to exhibit better selectivity and efficacy, may provide useful lead compounds for further synthetic programmes.

Cell death induced by TW-37 exhibits characteristic hallmarks of apoptosis. (a) H23 cells exposed for 0–24 h to TW-37 (10 μM) with and without Z-VAD.fmk (50 μM) were monitored for apoptotic morphology using electron microscopy (scale bar, 5 μm). % PS positive cells indicate the percentage of apoptotic cells, characterised by PS externalisation. (b) Whole-cell lysates of H23 cells exposed for 0–24 h to TW-37 (10 μM) were probed with antibodies against PARP and caspase-9. The appearance of both the p89 processed form of PARP and the p35 form of caspase-9 were characteristic of the intrinsic pathway of apoptosis. (c) Whole-cell lysates of BAK-reconstituted and -deficient Jurkat cells exposed for 0–24 h to TW-37 (10 μM) were probed as in B. % PS positive cells indicate the percentage of apoptotic cells, characterised by PS externalisation. (d) Electron micrographs of control and TW-37 (10 μM for 24 h); treated BAK-reconstituted and -deficient Jurkat cells reveal a dependence on BAK for TW-37-mediated apoptosis (scale bar, 5 μm)
Discussion
ABT-263, which inhibits BCL-2, BCL-XL and BCL-w,6 seems ideal to treat BCL-2-addicted tumours, such as CLL.27, 28 However, a dose-limiting thrombocytopenia is observed owing to inhibition of BCL-XL, the primary survival factor for platelet viability.33 This has resulted in the synthesis of ABT-199, a BCL-2-specific inhibitor, which appears to act in vivo to specifically inhibit BCL-2 without affecting BCL-XL, and thus platelets are spared.10 However, the survival of only a minority of tumours is dependent on a single anti-apoptotic BCL-2 family member, whereas most tumours are addicted to at least two anti-apoptotic BCL-2 proteins. Therefore, development of small molecule inhibitors that selectively target a specific BCL-2 family member would rely on combination therapy to efficiently inhibit multiple BCL-2 family members. The BCL-2-specific inhibitors, ABT-263 and ABT-199, are ineffective against other anti-apoptotic BCL-2 family members, such as MCL-1 and BCL2A1, which are often associated with chemoresistance.7, 12, 13, 14
Many attempts have been made to inhibit MCL-1 by alteration of its transcriptional, translational or post-translational regulatory mechanisms.12, 13, 31 In this study, we have evaluated the specificity of putative MCL-1 inhibitors to inhibit MCL-1. Although MCL-1 is structurally similar to other BCL-2 family members, there are differences in its BH3-binding grove, resulting in a lower affinity of commonly available BCL-2 antagonists. Although obatoclax was reported to be a broad-spectrum BCL-2 family inhibitor that antagonised MCL-1,15 it did not act as such but rather behaved like a mitochondrial toxin.4, 8, 9 In an attempt to design a specific MCL-1 inhibitor, the structural features of BH3I-1, as an inhibitor of BCL-2 proteins, were incorporated into the synthesis of compounds 6 and 7.16, 25 On the basis of isothermal calorimetry data, compound 7 was reported to bind exclusively to MCL-1, whereas compound 6 although binding more tightly to MCL-1 also bound to BCL-XL.16 However, these inhibitors failed to induce apoptosis in the present study. Using NMR data, fluorescence polarisation assays and molecular docking studies, structural derivatives of apogossypol/apogossypolone (BI97C1, BI112D1 and BI97C10) were synthesised with a higher MCL-1 selectivity.18, 19, 20 Both BI97C1 and BI112D1 were selective in killing cells in a BAX/BAK- and caspase-9-dependent manner. However, the inhibitors lacked the potency to induce similar extents of death in BCL-2-, BCL-XL- or MCL-1-dependent cells (Figures 2, 4 and 6). Recent studies have suggested that BCL-2 inhibitors may inhibit myeloid leukaemic stem cells.34, 35, 36 Specifically, sabutoclax (BI97C1) was proposed to act as a pan-BCL-2 inhibitor in sensitising bone marrow-resident leukaemic cells to tyrosine kinase inhibition.34 However, in our study, sabutoclax was not an effective inhibitor of BCL-2, as evidenced by its inability to induce rapid apoptosis in either a BCL-2- or BCL-XL-addicted system (Figures 4b and 5b). Furthermore, it exhibited varied sensitivity in inhibiting MCL-1 in different MCL-1-addicted cells (Figure 6). These results suggest that sabutoclax (BI97C1) is not a pan-BCL-2 inhibitor in all cellular systems, and it seems to exert other effects unrelated to BCL-2 family inhibition. Surprisingly, ABT-263, in addition to killing BCL-2- and BCL-XL-dependent cells (Figures 4 and 5), induced cell death in an MCL-1-dependent cell line, suggesting that at high concentrations it may inhibit MCL-1 in a cellular context (Figure 6), although these high concentrations may exert other non-specific toxicities. However, in favour of a potential specific effect of ABT-263, a recent study has shown that its structurally related analogue, ABT-737, induced a caspase-dependent induction of NOXA, which can inhibit MCL-1.37
Using a library of stabilised alpha-helix of BCL-2 domains, the BH3 helix of MCL-1 on its own was identified as a potent and exclusive MCL-1 inhibitor.38 Such stapled peptides exhibit selectivity in disrupting specific BH3-mediated interactions in vitro, and their sequence-dependent pro-apoptotic activity has been documented in vivo.39, 40 The interaction of an MCL-1-stapled peptide with the BH3-binding groove was employed in a competitive screen to identify a novel MIM-1 that selectively targets the BH3-binding groove of MCL-1.21 Whereas MIM-1 exhibits BAK-dependent apoptotic activity, its potency may be limited and cell-type dependent, as it failed to induce apoptosis in two MCL-1-dependent cell lines (Figure 6). Using a structure-based approach with gossypol as the starting material, TW-37, which bound more avidly to MCL-1 than BCL-2 or BCL-XL, was synthesised.17 Of the inhibitors examined, TW-37 possibly offered the most promise in inducing apoptosis in a NOXA-dependent manner in MCL-1-dependent cells (Figure 6).3, 32 As knockdown of NOXA led to incomplete inhibition of TW-37-induced apoptosis, other mechanisms, such as direct inhibition of MCL-1 and/or displacement of bound BH3-only proteins, including NOXA, may also be involved. Although high concentrations of TW-37 were required to induce cell death, the ultrastructural changes observed were characteristic of apoptosis (Figure 7), thus confirming that TW-37 did not exert non-specific toxicity to cells, unlike some of the putative inhibitors previously reported.9 Thus, TW-37 may provide a useful lead compound for further synthetic programmes to develop more specific and potent small molecule inhibitors of MCL-1.
Our data demonstrate that most of the putative MCL-1 inhibitors tested in this study are either insufficiently potent or not especially selective in inducing apoptosis of the selected MCL-1-dependent cancer cell lines. Clearly, the design of such specific inhibitors is complicated by the complex regulation of MCL-1, the nature of the BH3-binding groove and the identification of other isoforms of MCL-1 that may have other cellular functions, including ER membrane reorganisation and mitochondrial fission–fusion dynamics.41, 42, 43 Thus, inhibiting MCL-1 may also interfere with its other ‘day-jobs’, in addition to its anti-apoptotic role, and so could be detrimental to normal cells. Further understanding of these processes together with innovative synthetic approaches such as that used in the reverse engineering of ABT-263 to yield ABT-199,10 a BCL-2-specific inhibitor, and additional medicinal iteration of the evaluated molecules may ultimately yield potent and specific MCL-1 inhibitors amenable to clinical translation.
Materials and Methods
Cell culture
Jurkat-T lymphocytes (deficient and reconstituted with either BAK or caspase-9) from Dr. K. Schulze-Osthoff (University of Tubingen, Tubingen, Germany), non-small cell lung cancer cell lines H1299 from ATCC (Middlesex, UK) and H23 from Professor C. Pritchard (University of Leicester, Leicester, UK) were cultured in RPMI 1640 medium supplemented with 10% foetal calf serum (FCS) and 5 mM L-glutamine (all from Life Technologies Inc, Paisley, UK). Wild-type and Bax/Bak DKO MEFs from Dr. A. Strasser (WEHI, Melbourne, Australia) and caspase-9 null MEFs from Professor T. Mak (University of Toronto, Toronto, Canada) were cultured in DMEM medium supplemented with 5 mM L-glutamine and 10% FCS. Lymphocytes purified from blood samples of patients with CLL were cultured as previously described.28 FL5.12 cells overexpressing BCL-2 or BCL-XL were kindly provided by Dr. A. Letai (Dana-Farber Cancer Institute, Boston, MA, USA) and Dr. L. Boise (Emory University, Atlanta, GA, USA), respectively, and cultured in RPMI 1640 supplemented with 10% FCS, 2 mM L-glutamine and 10% of IL3-containing WEHI-3 cell culture supernatant.26 To assess their sensitivity to BCL-2 inhibitors, FL5.12 cells were cultured for 24 h in the absence of IL-3 before exposure to the BCL-2 inhibitors for an additional 24 h.
Reagents and plasmids
ABT-263 and TW-37 were obtained from Selleck Chemicals Co. (Houston, TX, USA) and BH3I-1 was from Calbiochem (Nottingham, UK). UCB-1350883 was a kind gift from Dr. A. Payne (UCB Pharma Ltd., Slough, UK). Apogossypol, BI97C1, BI97C10 and BI112D1 were synthesised as described.18, 19, 20 Compounds 6 and 7 were synthesised as previously described.16 MIM-1 was synthesised as previously described.21 Antibody against murine caspase-9 was obtained from Abcam (Cambridge, UK) and antibodies against NOXA and tubulin from Calbiochem, MCL-1 from Santa Cruz Biotechnology (Santa Cruz, CA, USA), BAX and BAK from Millipore (Watford, UK), PARP from Enzo Life Sciences (Exeter, UK) and human caspase-9 antiserum (a gift from Dr. X. Sun, MRC Toxicology Unit, Leicester, UK) were used. Z-VAD.fmk was obtained from MP Biomedicals (Cambridge, UK). All other reagents, unless mentioned otherwise, were from Sigma-Aldrich Co. (St. Louis, MO, USA).
siRNA knockdowns and western blotting
Cells were reverse-transfected with 10 nM of MCL-1 (ID# s8583) or NOXA (ID# L-005275) oligoduplexes (Life Technologies or Thermo Scientific, Waltham, MA, USA) using Interferin Reagent (Polyplus Transfection Inc., New York, USA), according to the manufacturer’s protocol and processed 72 h after transfection. Western blots were carried out according to standard protocols.9 Briefly, 50 μg of total protein lysate was subjected to SDS-PAGE. Subsequently, proteins were transferred to nitrocellulose membrane and protein bands visualised with ECL reagents (GE Healthcare, Bucks, UK).
Flow cytometry and microscopy
Cell death was assessed by PS externalisation and staining with AnnexinV-FITC as described previously.28 For electron microscopy, cells were fixed and processed as described previously.28 Ultrathin sections were stained with lead citrate and recorded using a Megaview 3 digital camera and iTEM software (Olympus Soft Imaging Solutions GmbH, Münster, Germany) in a Jeol 100-CXII electron microscope (Jeol UK Ltd, Welwyn Garden City, UK).
Abbreviations
- CLL:
-
chronic lymphocytic leukaemia
- MEFs:
-
mouse embryonic fibroblasts
- PARP:
-
poly(ADP-ribose) polymerase
- SAHBs:
-
stabilised alpha-helix of BCL-2 domains
References
Adams JM, Cory S . The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007; 26: 1324–1337.
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 2005; 19: 1294–1305.
Lessene G, Czabotar PE, Colman PM . BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov 2008; 7: 989–1000.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681.
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008; 68: 3421–3428.
van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10: 389–399.
Vogler M, Dinsdale D, Dyer MJ, Cohen GM . Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ 2009; 16: 360–367.
Vogler M, Weber K, Dinsdale D, Schmitz I, Schulze-Osthoff K, Dyer MJ et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ 2009; 16: 1030–1039.
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013; 19: 202–208.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 463: 899–905.
Dai Y, Grant S . Targeting multiple arms of the apoptotic regulatory machinery. Cancer Res 2007; 67: 2908–2911.
Gores GJ, Kaufmann SH . Selectively targeting Mcl-1 for the treatment of acute myelogenous leukemia and solid tumors. Genes Dev 2012; 26: 305–311.
Zhang H, Guttikonda S, Roberts L, Uziel T, Semizarov D, Elmore SW et al. Mcl-1 is critical for survival in a subgroup of non-small-cell lung cancer cell lines. Oncogene 2011; 30: 1963–1968.
Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA 2007; 104: 19512–19517.
Bernardo PH, Sivaraman T, Wan KF, Xu J, Krishnamoorthy J, Song CM et al. Structural insights into the design of small molecule inhibitors that selectively antagonize Mcl-1. J Med Chem 2010; 53: 2314–2318.
Wang Z, Song W, Aboukameel A, Mohammad M, Wang G, Banerjee S et al. TW-37, a small-molecule inhibitor of Bcl-2, inhibits cell growth and invasion in pancreatic cancer. Int J Cancer 2008; 123: 958–966.
Wei J, Kitada S, Rega MF, Stebbins JL, Zhai D, Cellitti J et al. Apogossypol derivatives as pan-active inhibitors of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J Med Chem 2009; 52: 4511–4523.
Wei J, Stebbins JL, Kitada S, Dash R, Placzek W, Rega MF et al. BI-97C1, an optically pure Apogossypol derivative as pan-active inhibitor of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J Med Chem 2010; 53: 4166–4176.
Wei J, Stebbins JL, Kitada S, Dash R, Zhai D, Placzek WJ et al. An optically pure apogossypolone derivative as potent pan-active inhibitor of anti-apoptotic bcl-2 family proteins. Front Oncol 2011; 1: 28.
Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SR, Koss B et al. A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem Biol 2012; 19: 1175–1186.
Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol 2012; 30: 488–496.
Porter J, Payne A, de Candole B, Ford D, Hutchinson B, Trevitt G et al. Tetrahydroisoquinoline amide substituted phenyl pyrazoles as selective Bcl-2 inhibitors. Bioorg Med Chem Lett 2009; 19: 230–233.
Dash R, Azab B, Quinn BA, Shen X, Wang XY, Das SK et al. Apogossypol derivative BI-97C1 (Sabutoclax) targeting Mcl-1 sensitizes prostate cancer cells to mda-7/IL-24-mediated toxicity. Proc Natl Acad Sci USA 2011; 108: 8785–8790.
Lugovskoy AA, Degterev AI, Fahmy AF, Zhou P, Gross JD, Yuan J et al. A novel approach for characterizing protein ligand complexes: molecular basis for specificity of small-molecule Bcl-2 inhibitors. J Am Chem Soc 2002; 124: 1234–1240.
Gottschalk AR, Boise LH, Oltvai ZN, Accavitti MA, Korsmeyer SJ, Quintans J et al. The ability of Bcl-x(L) and Bcl-2 to prevent apoptosis can be differentially regulated. Cell Death Differ 1996; 3: 113–118.
Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A . Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest 2007; 117: 112–121.
Vogler M, Dinsdale D, Sun XM, Young KW, Butterworth M, Nicotera P et al. A novel paradigm for rapid ABT-737-induced apoptosis involving outer mitochondrial membrane rupture in primary leukemia and lymphoma cells. Cell Death Differ 2008; 15: 820–830.
Vogler M, Furdas SD, Jung M, Kuwana T, Dyer MJ, Cohen GM . Diminished sensitivity of chronic lymphocytic leukemia cells to ABT-737 and ABT-263 due to albumin binding in blood. Clin Cancer Res 2010; 16: 4217–4225.
Song L, Coppola D, Livingston S, Cress D, Haura EB . Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther 2005; 4: 267–276.
Warr MR, Shore GC . Unique biology of Mcl-1: therapeutic opportunities in cancer. Curr Mol Med 2008; 8: 138–147.
Craxton A, Butterworth M, Harper N, Fairall L, Schwabe J, Ciechanover A et al. NOXA, a sensor of proteasome integrity, is degraded by 26S proteasomes by an ubiquitin-independent pathway that is blocked by MCL-1. Cell Death Differ 2012; 19: 1424–1434.
Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S et al. Programmed anuclear cell death delimits platelet life span. Cell 2007; 128: 1173–1186.
Goff DJ, Recart AC, Sadarangani A, Chun HJ, Barrett CL, Krajewska M et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 2013; 12: 316–328.
Hogdal LJ, Letai A . BCL-2 inhibition: stemming the tide of myeloid malignancies. Cell Stem Cell 2013; 12: 269–270.
Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013; 12: 329–341.
Bertin-Ciftci J, Barre B, Le Pen J, Maillet L, Couriaud C, Juin P et al. pRb/E2F-1-mediated caspase-dependent induction of Noxa amplifies the apoptotic effects of the Bcl-2/Bcl-xL inhibitor ABT-737. Cell Death Differ 2013; 20: 755–764.
Stewart ML, Fire E, Keating AE, Walensky LD . The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol 2010; 6: 595–601.
Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004; 305: 1466–1470.
LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest 2012; 122: 2018–2031.
Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol 2012; 14: 575–583.
Varadarajan S, Bampton ET, Smalley JL, Tanaka K, Caves RE, Butterworth M et al. A novel cellular stress response characterised by a rapid reorganisation of membranes of the endoplasmic reticulum. Cell Death Differ 2012; 19: 1896–1907.
Varadarajan S, Butterworth M, Wei J, Pellecchia M, Dinsdale D, Cohen GM . Sabutoclax (BI97C1) and BI112D1, Putative Inhibitors of MCL-1, Induce Mitochondrial Fragmentation Either Upstream of or Independent of Apoptosis. Neoplasia 2013; 15: 568–578.
Acknowledgements
We thank Professors Boise, Letai, Mak, Pritchard, Schulze-Osthoff and Strasser for cells. We thank Drs. Maurizio Pellecchia and Jun Wei (Sanford-Burnham Institute, La Jolla, CA, USA) for apogossypol, BI97C1, BI97C10 and BI112D1 and Dr. Christina Chai (Agency for Science Technology and Research, Singapore) for compounds 6 and 7. We also thank Dr. A Payne (UCB Celltech, Slough, UK) for UCB-1350883, Tim Smith and Maria Guerra Martin for technical assistance and the Medical Research Council for core support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by C Borner
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Varadarajan, S., Vogler, M., Butterworth, M. et al. Evaluation and critical assessment of putative MCL-1 inhibitors. Cell Death Differ 20, 1475–1484 (2013). https://doi.org/10.1038/cdd.2013.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2013.79
Keywords
This article is cited by
-
BH3-mimetics: recent developments in cancer therapy
Journal of Experimental & Clinical Cancer Research (2021)
-
BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics
Cell Death & Differentiation (2020)
-
Mcl-1 Inhibitor Induces Cells Death in BRAF-Mutant Amelanotic Melanoma Trough GSH Depletion, DNA Damage and Cell Cycle Changes
Pathology & Oncology Research (2020)
-
Confounding off-target effects of BH3 mimetics at commonly used concentrations: MIM1, UMI-77, and A-1210477
Cell Death & Disease (2019)
-
BH3-only proteins are dispensable for apoptosis induced by pharmacological inhibition of both MCL-1 and BCL-XL
Cell Death & Differentiation (2019)

{kind=link}