
Abstract
CCAAT/enhancer-binding protein (C/EBP) β is required for both mitotic clonal expansion (MCE) and terminal adipocyte differentiation of 3T3-L1 preadipocytes. Although the role of C/EBPβ in terminal adipocyte differentiation is well defined, its mechanism of action during MCE is not. In this report, histone demethylase Kdm4b, as well as cell cycle genes Cdc45l (cell division cycle 45 homolog), Mcm3 (mini-chromosome maintenance complex component 3), Gins1 (GINS complex subunit 1) and Cdc25c (cell division cycle 25 homolog c), were identified as potential C/EBPβ target genes during MCE by utilizing promoter-wide chromatin immunoprecipitation (ChIP)-on-chip analysis combined with gene expression microarrays. The expression of Kdm4b is induced during MCE and its induction is dependent on C/EBPβ. ChIP, Electrophoretic Mobility Shift Assay (EMSA) and luciferase assay confirmed that the promoter of Kdm4b is bound and activated by C/EBPβ. Knockdown of Kdm4b impaired MCE. Furthermore, Kdm4b interacted with C/EBPβ and was recruited to the promoters of C/EBPβ-regulated cell cycle genes, including Cdc45l, Mcm3, Gins1, and Cdc25c, demethylated H3K9me3 and activated their transcription. These findings suggest a novel feed forward mechanism involving a DNA binding transcription factor (C/EBPβ) and a chromatin regulator (Kdm4b) in the regulation of MCE by controlling cell cycle gene expression.
Similar content being viewed by others


Main
Obesity is a major risk factor for type 2 diabetes, hypertension, hyperlipidemia, and arteriosclerosis.1 Both the increase of adipocyte size (hypertrophy) and number (hyperplasia)2 are major contributors to the development of obesity. The hyperplasia of adipocytes is mimicked during the adipogenic differentiation program of 3T3-L1 preadipocytes.3 Immediately after the treatment with differentiation inducers, growth-arrested 3T3-L1 preadipocytes re-enter the cell cycle, a process referred to as mitotic clonal expansion (MCE) in which cell number increases ∼4-fold during differentiation. The gene expression program leading to terminal adipocyte differentiation is initiated during and after the MCE period. Although there is some controversy,4 numerous experimental observations support the view that MCE is a necessary step for the terminal adipocyte differentiation of 3T3-L1 preadipocytes. The extracellular signal-regulated kinase kinase (MEK) inhibitor U0126 and cyclin-dependent kinase inhibitor roscovitine, which inhibit the cell cycle at different points, block MCE, as well as adipogenesis.5 Moreover, the DNA synthesis inhibitor aphidicolin and the anti-proliferation reagent rapamycin also block MCE and 3T3-L1 preadipocyte differentiation.6, 7
CCAAT/enhancer-binding protein β (C/EBPβ) is an important transcription factor for the initiation of 3T3-L1 preadipocyte differentiation. C/EBPβ is induced very early in adipocyte differentiation. Then it activates the expression of CCAAT enhancer-binding protein α (C/EBPα) and peroxisome proliferator-activated receptor γ (PPARγ), two critical pro-adipogenic transcription factors, by binding to their promoters.8 Previous studies have shown that the expression and the activation of C/EBPβ are required for MCE. When subjected to the same differentiation protocol as 3T3-L1 preadipocytes, a subset of mouse embryo fibroblasts (MEFs) undergo MCE and terminal differentiation into adipocytes. MEFs from C/EBPβ (−/−) mice, however, neither undergo MCE nor differentiate into adipocytes.9 Furthermore, knockdown of C/EBPβ by RNA interference (RNAi) in 3T3-L1 preadipocytes prevents MCE, as well as adipocyte differentiation.10 Additionally, overexpression of a dominant-negative C/EBPβ (A-C/EBP) that blocks C/EBPβ DNA binding by dimerizing through its leucine zipper, also disrupts MCE and adipogenesis of 3T3-L1 cells.11 C/EBPβ also has important roles in the proliferation of certain other cell types such as lobuloalveolar cells, osteoblasts, keratinocytes and augments Ha-ras-induced transformation of NIH 3T3 cells.12, 13, 14, 15 Despite the important role of C/EBPβ in growth stimulation, the mechanism underlying MCE is not fully understood.
The methylation of lysine residues in histones is a central epigenetic modification in the regulation of eukaryotic gene expression. While methylation at H3K4 and H3K36 primarily transduces activating signals, methylation at H3K9, H3K27, and H4K20 is associated primarily with repressed chromatin. Kdm4b is a Jmjc-domain containing histone demethylase for H3K9me3. Recent studies have shown that Kdm4b is required for estrogen receptor α (ERα)-regulated breast cancer progression and mammary epithelial cells (MECs) proliferation.16, 17, 18 In the present study, Kdm4b was identified as a target gene of C/EBPβ that functions as a co-factor of C/EBPβ to demethylate H3K9me3 in the regulatory regions of C/EBPβ-regulated cell cycle genes Cdc45l (cell division cycle 45 homolog), Mcm3 (mini-chromosome maintenance complex component 3), Gins1 (GINS complex subunit 1) and Cdc25c (cell division cycle 25 homolog c), thereby promoting their expression and MCE. This explains why C/EBPβ is required for MCE during adipocyte differentiation.
Results
Identification of C/EBP β binding sites during the MCE of 3T3-L1 preadipocyte differentiation
A promoter-wide chromatin immunoprecipitation (ChIP)-on-chip analysis was employed on 3T3-L1 cells harvested at 20 h after hormonal induction when the cells are in the middle S phase during the first round of MCE.5, 19 Nine-hundred and forty-one significant C/EBPβ binding regions were identified (Supplementary Table S1). To validate the ChIP-on-chip data, C/EBPβ enrichment was measured by ChIP quantitative PCR (ChIP-qPCR) at 30 novel binding regions and 29 of these sites were positive (Supplementary Figure S1a). Fifteen of these sites were also tested by ChIP-qPCR with another C/EBPβ antibody and a similar result was obtained as with the original antibody used for the ChIP-on-chip assays (Supplementary Figure S1b).
Distribution analysis of the binding sites relative to the TSS showed that the majority of C/EBPβ binding regions were located at distances within 2 kb from the nearest annotated transcription start sites (TSSs) (Figure 1a). Further analysis revealed that ∼30% of C/EBPβ binding sites were at proximal promoters (Figure 1b, TSS to −1 kb), and ∼30% of the sites fell within distal promoters, defined as >1 kb upstream from TSS (Figure 1b, >1 kb), while many other sites (26.84%) were located in introns (Figure 1b). Evolutionary conservation of the C/EBPβ binding regions was examined to show that there is a high degree of conservation of C/EBPβ binding sites among higher eukaryotes (Figure 1c), suggesting that these binding sites are likely to be functional transcriptional regulatory regions across species.

Bioinformatic analyses of the ChIP-on-chip data. Post-confluent growth-arrested 3T3-L1 preadipocytes were induced to differentiation as described. At 20 h ChIP-on-chip were performed and analyzed. (a) Distribution in 200-bp intervals of C/EBPβ binding regions relative to TSSs of the nearest RefSeq genes. (b) Mapping of C/EBPβ binding sites relative to the nearest RefSeq genes. >1 kb: more than 1 kb upstream from the TSS; Downstream: downstream from the 3′ end of the gene; UTR: untranslated region. (c) Average plot for conservation of the C/EBPβ binding regions among higher eukaryotes. (d) Sequence logo illustrating the top-scoring de novo motif for C/EBPβ-bound regions. (e) TRANSFAC matrices enriched around C/EBPβ-bound regions. (f) Overlap between the genes targeted by C/EBPβ (1039 genes) and the genes whose induction was dependent on C/EBPβ (481 genes)
The sequences of C/EBPβ-bound regions were mined to further identify potential binding motifs for transcription factors. A de novo motif search identified a sequence that strongly resembles the C/EBP recognition element as the top-scoring motif (Figure 1d). Consistently, the highest scoring motifs from the TRANSFAC database represented different matrices for C/EBP proteins (Figure 1e and Supplementary Table S2). Gene expression microarrays were further conducted for both control RNAi-treated cells and C/EBPβ RNAi-treated cells to confirm the functional relationship between C/EBPβ binding and gene transcription. Four-hundred and eighty-one genes were identified to be induced (20 h versus 0 h) in a C/EBPβ-dependent manner (Supplementary Tables S3 and S4). When overlapping ChIP-on-chip data and microarray data, 68 potential C/EBPβ target genes were identified (Figure 1f and Supplementary Table S5).
Histone demethylase Kdm4b was identified as one of the C/EBP β target genes required for MCE during 3T3-L1 adipocyte differentiation
Lysine methylation on histones has a pivotal role in transcriptional regulation.20 The histone demethylase Kdm4b (for H3K9me3) was among the 68 potential C/EBPβ target genes identified above. This suggests that C/EBPβ might regulate gene expression through indirect regulation of histone lysine methylation.
A DNA fragment containing the 5′-flanking region of Kdm4b gene was subcloned into the luciferase reporter construct pGL3-basic and transfected into 3T3-L1 preadipocytes with/out the C/EBPβ expression plasmid. Luciferase activity was increased ∼10-fold when co-transfected with C/EBPβ expression plasmid (Figure 2a). Consistently, deletion of the C/EBP binding site (−577 to −564 bp) identified in our ChIP-on-chip analysis almost totally abolished this transactivation. ChIP-qPCR confirmed that the Kdm4b promoter fragment containing the region of −577 bp∼−564 bp was significantly enriched by the C/EBPβ antibody at 16 h and 20 h after adipogenic induction (Figure 2b). The specific binding of C/EBPβ to this region was also confirmed by Electrophoretic Mobility Shift Assay (EMSA) (Supplementary Figure S2). Upon induction of differentiation, Kdm4b messenger RNA (mRNA) level was significantly induced and this upregulation was prevented when C/EBPβ was knocked down by RNAi (Figure 2c). Consistently, Kdm4b protein level also increased post-induction and was impaired in C/EBPβ RNAi-treated cells (Figure 2d). The knockdown efficiency of C/EBPβ RNAi was also confirmed (Figures 2c and d) and its effect on MCE and terminal adipocyte differentiation is consistent with our previous report.10 While the expression of Kdm4b is significantly induced and regulated by C/EBPβ, this is not the case for other Kdm4 family genes (Figure 2e).

Transactivation of Kdm4b by C/EBPβ during MCE of 3T3-L1 preadipocyte differentiation. (a) 3T3-L1 preadipocytes were transfected with control vector or C/EBPβ-expressing vector, together with reporter constructs containing the Kdm4b promoter (−1000 to −1 bp) or mutants. Luciferase activities were measured 36 h later and plotted. (b) 3T3-L1 cells were induced to differentiation and binding of C/EBPβ on the Kdm4b promoter was analyzed by ChIP-qPCR at times indicated. (c) 3T3-L1 cells were treated with control or C/EBPβ RNAi. After induction, the mRNA levels of C/EBPβ and Kdm4b at times indicated were analyzed by RT-qPCR. (d) Cell lysates were harvested at times indicated and subjected to western blotting with antibodies indicated. The C/EBPβ antibody detects two bands: liver-enriched activator protein (LAP, 34 kDa) and liver-enriched inhibitory protein (LIP, 21 kDa, an N-terminally truncated isoform of C/EBPβ). Hsp90 served as a loading control. (e) After induction, mRNA levels of the Kdm4 family members at times indicated were analyzed by RT-qPCR. Data normalized to 0 h time point of each gene. *P<0.05. (f–i) Overexpression of C/EBPβ promotes Kdm4b expression. 3T3-L1 preadipocytes were infected with retrovirus expressing flag-tagged C/EBPβ or with empty vector and induced to differentiation. (f) The overexpression of C/EBPβ (LAP) was confirmed by western blotting at 20 h. (g) At 16 h after induction, cells were labeled with EdU for 2 h and then subjected to FACS analysis. The percentages of cells in S phase were determined. (h) At 8 d after induction, cells were stained with Oil red O. (i) The effect of C/EBPβ overexpression on the expression of Kdm4b was detected by RT-qPCR
To further confirm the role of C/EBPβ in Kdm4b induction, C/EBPβ was overexpressed in 3T3-L1 cells (Figure 2f). As expected, ectopic expression of C/EBPβ promoted MCE as illustrated by increased EdU incorporation (an indicator of DNA synthesis, like BrdU incorporation) (Figure 2g), as well as adipocyte differentiation, as indicated by increased accumulation of cytoplasmic triglyceride (Figure 2h) and enhanced the expression of Kdm4b at both 0 h and 20 h after adipogenic induction (Figure 2i). These results demonstrate that Kdm4b is a bona fide target of C/EBPβ during MCE.
Kdm4b has been shown to regulate cell proliferation, including estrogen-induced breast cancer growth and MECs proliferation.16, 17 Therefore, we hypothesized that Kdm4b might also be involved in regulating MCE during 3T3-L1 preadipocyte differentiation. RNAi oligonucleotide of Kdm4b was transfected into 3T3-L1 preadipocytes. The efficiency and specificity of Kdm4b knockdown were confirmed by western blotting (Figure 3a) and RT-qPCR (Supplementary Figure S3) respectively. The RNAi effect on DNA synthesis was evaluated by using EdU labeling. As shown in Figure 3b, Kdm4b knockdown caused less EdU incorporation. Furthermore, flow cytometry analysis revealed that knockdown of Kdm4b resulted in a significant reduction in the cell population in S phase (from 34 to 19%) with a concomitant increase in the cell population in G1 phase (from 40 to 71%) (Figure 3c). These data suggest that Kdm4b may promote MCE by facilitating G1/S transition. Consistently, cell proliferation was significantly inhibited from 1 to 4 days after induction (Figure 3d). In addition, when the MCE was inhibited after Kdm4b was knocked down, the terminal differentiation of 3T3-L1 cells was also suppressed as indicated by Oil Red O staining (Figure 3e), as well as the adipocyte marker gene expression (Figure 3f). Collectively, these results indicate that Kdm4b is required for MCE.

Kdm4b is required for MCE during 3T3-L1 preadipocyte differentiation. 3T3-L1 preadipocytes were infected with empty vector or infected with retrovirus expressing human Kdm4b (hKdm4b). Then cells were transfected with control RNAi or Kdm4b RNAi. After post-confluence, cells were induced to differentiation. (a) Cell lysates were harvested at 20 h and the expression of the indicated genes was detected by western blotting. (b) At 18 h after induction, cells were labeled with EdU for 2 h and then stained with Hoechst. The fluorescence of EdU (red) and Hoechst (blue) was detected with a fluorescence microscope. (c) DNA content was analyzed by PI staining and flow cytometry at 0 h and 20 h time points and analyzed. (d) Cell numbers were determined at times indicated after induction. Data normalized to 0 h time point. *P<0.05. (e) At 8 days after induction, cells were stained with Oil red O. (f) At 8 days after induction, the expression of adipocyte marker genes were detected by western blotting
Rescue experiments were performed to further confirm the above observation. As 3T3-L1 preadipocytes are of mouse origin, human Kdm4b was ectopically expressed in Kdm4b RNAi-treated 3T3-L1 cells by retrovirus transduction. Western blotting analysis demonstrated that human Kdm4b gene is resistant to mouse Kdm4b RNAi knockdown and was expressed at a similar level of total Kdm4b as the parental control cells (Figure 3a). Forced expression of human Kdm4b led to significant rescue of MCE (Figures 3b–d) and terminal adipocyte differentiation (Figures 3e–f). These data reinforce the idea that Kdm4b has an important role in MCE.
Transcriptional activation of cell cycle genes Cdc45l, Mcm3, Gins1 and Cdc25c by C/EBP β during MCE
Gene ontology (GO) analysis revealed that four cell cycle genes (Cdc45l, Mcm3, Gins1 and Cdc25c) were among the 68 potential C/EBPβ target genes (Supplementary Table S5). The expression of Cdc45l, Mcm3, Gins1 and Cdc25c was determined by RT-qPCR and western blotting. After hormonal induction, both the mRNA (Figure 4a) and protein levels (Figure 4b) increased at 8 h. The upregulation of these cell cycle genes occurred subsequent to that of the C/EBPβ gene, which peaked at 4 h after induction (Figures 4a and b). When C/EBPβ expression was knocked down, the induction of these cell cycle genes was significantly reduced as indicated by the reduction of their mRNA and protein levels (Figures 4c and d). These results confirm that C/EBPβ is required for the induction of the four cell cycle genes during MCE in the differentiation program of 3T3-L1 preadipocytes.

Four cell cycle genes Cdc45l, Mcm3, Gins1 and Cdc25c were identified as C/EBPβ target genes. (a, b) 3T3-L1 cells were induced to differentiation. The expression profiles of C/EBPβ and the four cell cycle genes Cdc45l, Mcm3, Gins1 and Cdc25c were analyzed by RT-qPCR (a) and western blotting (b) respectively. (c, d) 3T3-L1 preadipocytes were transfected with control RNAi or C/EBPβ RNAi. After post-confluence, cells were induced to differentiation. The effect of knocking down C/EBPβ on the expression of the four cell cycle genes was detected by RT-qPCR (c) and western blotting (d) respectively
The binding of C/EBPβ to the promoters of the four cell cycle genes was then investigated. The promoter regions of the four genes identified in the C/EBPβ ChIP-on-chip data and the C/EBPβ consensus sequences are illustrated in Supplementary Figure S4. ChIP-qPCR assays were performed using primers covering these C/EBPβ consensus sequences of the four genes.
As Figure 5a shows, C/EBPβ associates with the promoters of the four genes at 16 h and 20 h after induction. In contrast, C/EBPβ occupancy was hardly detectable in quiescent cells before induction (i.e., at 0 h). EMSA was also performed using probes containing the C/EBPβ consensus sequences and nuclear extracts from 3T3-L1 cells at 20 h after induction. As shown in Figure 5b, DNA–protein complexes were formed with these probes (lanes 1, 6, 11, 16). The C/EBPβ containing DNA–protein complexes were verified by the completely supershift of the complexes using C/EBPβ antibody (lanes 3, 8, 13, and 18) compared with that with control IgG (lanes 2, 7, 12, and 17). Moreover, the inhibition of complex formation by unlabeled competitor oligonucleotide, but not by self probes mutated in the C/EBP consensus sequence (Supplementary Figure S4), verified specific binding (compare lanes 4, 9, 14, 19 with lanes 5, 10, 15, 20). Furthermore, promoter-luciferase reporter analysis indicates that C/EBPβ transactivates the promoters of the four cell cycle genes and that mutation of the C/EBPβ binding sites significantly reduces expression (Figure 5c).

C/EBPβ binds to and transactivates the promoters of the four cell cycle genes. (a) Growth-arrested 3T3-L1 preadipocytes were induced to differentiation. At times indicated ChIP was performed to show the binding of C/EBPβ to the promoters of the four cell cycle gene. Data normalized to the IgG controls at each time point. One region of the insulin gene serves as a negative control. (b) EMSA was conducted with nuclear extracts from 3T3-L1 cells at 20 h after induction. Supershift experiments were performed with control IgG or C/EBPβ antibody. For competition analysis, unlabeled competitor probe of the C/EBP element in the C/EBPα gene promoter (Consensus) or unlabeled self probes mutated in the C/EBP element (Mut, shown in Supplementary Figure S4) were used. The positions of the C/EBPβ containing DNA–protein complex and the supershift band (SS) are indicated. (c) 3T3-L1 preadipocytes were transfected with reporter constructs with/out the C/EBP binding sites mutated and with/out the C/EBPβ expression vector. After 48 h, luciferase activity was analyzed and plotted. *P<0.05
The role of the four cell cycle genes in MCE was further examined. Knockdown of the four genes inhibits MCE (Supplementary Figure S5). Additionally, ectopic expression of the four genes together significantly rescues the inhibitory effect of C/EBPβ RNAi on MCE (Supplementary Figure S6). Taken together, the RNAi and overexpression experiments demonstrate that Cdc45l, Mcm3, Gins1 and Cdc25c, which are transactivated by C/EBPβ, have an important role in MCE.
Histone demethylase Kdm4b serves as a co-factor of C/EBP β during MCE
Recent studies have revealed that Kdm4b is a direct target gene of ERα, forms complex with ERα and facilitates ERα-regulated transcription.16, 17 As indicated above, Kdm4b has been shown to be a target gene of C/EBPβ (Figure 2) and like C/EBPβ, Kdm4b is also required for MCE (Figure 3). Thus, we hypothesized that Kdm4 might act as a co-factor of C/EBPβ to promote MCE.
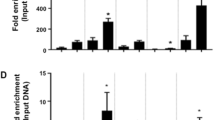
ChIP assays were performed to determine whether Kdm4b interacts with the C/EBPβ-targeted cell cycle gene loci. The results revealed that, similar to C/EBPβ, Kdm4b is recruited to the promoter regions of the four cell cycle genes 20 h after, but not before, induction (Figure 6a). Furthermore, re-ChIP analysis indicated that the promoters of the four genes, which are immunoprecipitated by C/EBPβ antibody, are re-immunoprecipitated by Kdm4b antibody (Figure 6b). The interaction between C/EBPβ and Kdm4b was further confirmed in co-immunoprecipitation experiments. Immunoprecipitation of the nuclear extracts at 20 h post-induction with C/EBPβ antibody pulls down Kdm4b (Figure 6c), and Kdm4b antibody also immunoprecipitates C/EBPβ (Figure 6d). Collectively, these results verify that C/EBPβ and Kdm4b form a complex that targets the promoters of the four cell cycle genes. Furthermore, luciferase assays demonstrate that Kdm4b enhances the ability of C/EBPβ to activate the promoters of the four cell cycle genes (Figure 6e), which indicates that Kdm4b functions as a positive transcriptional co-factor for C/EBPβ to regulate the four cell cycle genes.

Kdm4b acted as a co-factor of C/EBPβ to activate the promoters of the four cell cycle genes. (a) ChIP analysis indicated that Kdm4b was also associated with the promoters of the four cell cycle genes. (b) At 20 h post-induction, soluble chromatin was prepared for ChIPs and Re-ChIPs with the indicated antibodies. Recovery of the four cell cycle gene promoters was analyzed by PCR. (c, d) Interaction between C/EBPβ and Kdm4b. At 20 h post-induction, cell lysates were harvested and immunoprecipitated with antibody against C/EBPβ (c) or Kdm4b (d). Immunoprecipitate complexes were detected by western blotting with the indicated antibodies. The band detected by the C/EBPβ antibody is liver-enriched activator protein (LAP) (34 kDa). (e) 3T3-L1 preadipocytes were transfected with reporter constructs with/out the indicated expression vectors. After 48 h, luciferase activity was analyzed and plotted. *P<0.05
Kdm4b is a histone demethylase that reverses tri-methylation at H3K9. H3K9me3 is generally associated with gene silencing and is enriched in heterochromatin of inactive genes. It was found that H3K9me3 modification of the promoter regions of Cdc45l, Mcm3, Gins1 and Cdc25c is high at 0 h and significantly decreases by 20 h after induction (Figures 7a–d). Consistently, the decrease in H3K9me3 associated with these promoters following adipogenic induction was only observed in control, but not in Kdm4b-depleted cells (Figure 7e). As expected, knockdown of the expression of Kdm4b impaired the induction of the four cell cycle genes (Figure 7f).

Kdm4b regulates the expression of C/EBPβ-targeted cell cycle genes through demethylation of H3K9me3. (a–d) Decrease of H3K9me3 modification level on the promoters of the four cell cycle genes during MCE. Upper panel: Schematic representation of the genomic locus. The regions from ‘a’ to ‘d’ were analyzed by ChIP-qPCR. The TSS is indicated by the arrow. Lower panel: ChIP-qPCR analysis of H3K9me3 occupancy on the sites in the gene loci indicated in the upper panel was performed at 0 h and 20 h after induction. Primers for ChIP-qPCR were available upon requested. (e) 3T3-L1 preadipocytes were treated with control RNAi or Kdm4b RNAi and then induced to differentiation. Cells were collected at times indicated and subjected to ChIP-qPCR analysis of H3K9me3 modification at position ‘c’ of each gene promoter loci. Data were normalized to 0 h time point of control RNAi-treated cells. (f) 3T3-L1 preadipocytes were treated as in (e). The relative mRNA level of C/EBPβ-targeted cell cycle genes at times indicated was analyzed by RT-qPCR
C/EBP β and Kdm4b specifically regulate MCE, but not normal 3T3-L1 preadipocyte proliferation
To determine whether the function of C/EBPβ and Kdm4b is specific to MCE, the effects of knocking down C/EBPβ and Kdm4b on the proliferation of normal 3T3-L1 preadipocyte were examined. Knockdown of C/EBPβ or Kdm4b neither significantly altered the growth rate of pre-confluent 3T3-L1 cell, as assessed by cell number analysis, nor cell cycle phase (Figures 8a and b, respectively). Furthermore, only weak binding of C/EBPβ or Kdm4b on the promoters of the four cell cycle genes was detected in pre-confluent 3T3-L1 cells (Figures 8c and d, respectively). Consistently, the expression of the four cell cycle genes was not affected when C/EBPβ or Kdm4b was knocked down (Figure 8e). These observations indicate that C/EBPβ and Kdm4b has a specific role in MCE, but not in normal 3T3-L1 preadipocyte proliferation.

Knockdown the expression of C/EBPβ and Kdm4b has no effect on pre-confluent 3T3-L1 cell proliferation. (a) 3T3-L1 preadipocytes were plated at 20% confluence, next day the cells were transfected with C/EBPβ or Kdm4b RNAi. Forty-eight hours later cell proliferation was determined by cell number counting at times indicated. (b) Cells were treated as in (a). At 48 h after RNAi transfection, cell cycle phase profiles were analyzed by PI staining followed by flow cytometry. (c, d) ChIP-qPCR analysis of C/EBPβ (c) and Kdm4b (d) binding on the promoters of mouse Cdc45l, Mcm3, Gins1 and Cdc25c was performed in pre-confluent 3T3-L1 cells. (e) Forty-eight hours after transfection of C/EBPβ or Kdm4b RNAi, cell lysates were harvested and the expression of the indicated genes was assessed by western blotting
Discussion
C/EBPβ is a basic leucine zipper transcription factor expressed in various cell types and has been characterized as a regulator of differentiation, such as the differentiation of adipocytes, the female reproductive system, mammary gland and bone.21, 22, 23 While these studies have shed light on its role in differentiation, some other studies also show that C/EBPβ functions in cell proliferation of some experimental and neoplastic models. The mechanistic insight into C/EBPβ action in those types of cell proliferation, however, is limited.
Kdm4b was originally shown to antagonize H3K9me3 at pericentric heterochromatin in mammalian cells.24 Recent reports reveal that Kdm4b also functions in regulating gene expression. In ER signaling, Kdm4b demethylates H3K9me3 in the regulatory regions of ER responsive genes, thereby releasing a transcriptional block and promoting breast cancer cell proliferation and mammary gland development.16, 17, 18 Our findings have identified a specific role for Kdm4b in MCE during the proliferative phase of adipocyte differentiation. Kdm4b was found to be a bona fide target gene of C/EBPβ during MCE. C/EBPβ upregulates Kdm4b expression (Figure 2), which in turn demethylates H3K9me3 associated with C/EBPβ-regulated cell cycle genes, thereby activating the expression of these genes (Figure 7). Specifically, C/EBPβ drives the expression of the four cell cycle genes via two pathways, one through transcriptional activation and the other through an epigenetic pathway (Supplementary Figure S7). C/EBPβ requires Kdm4b to reverse H3K9me3 modification thereby enhancing its transcriptional activity. Moreover, Kdm4b requires C/EBPβ to be transcriptionally induced. This finding provides new insights into potential roles of C/EBPβ in epigenetic and transcriptional regulation during MCE of preadipocyte differentiation. Of interest, C/EBPβ is also required for mammary gland development. Deletion of C/EBPβ results in retarded proliferation of MECs and severe inhibition of lobuloalveolar development,25 which is similar to the phenotype of conditional deletion of Kdm4b in mammary epithelium.17 Our findings suggest a collaboration between C/EBPβ and Kdm4b during mammary gland development.
In the current study, four cell cycle genes were identified as direct targets of C/EBPβ during MCE. Mcm3 is a component of MCM2-7 (mini-chromosome maintenance proteins 2–7) complex while Gins1 is a subunit of GINS (go-ichi-ni-san) complex.26, 27 Cdc45l, MCM2-7 and GINS form a large complex referred to as CMG, which is found at the sites of DNA unwinding during replication and contains an intrinsic helicase activity.28 The CMG complex is involved in the regulation of eukaryotic chromosomal DNA replication and is required for the initiation and elongation steps, as well as the co-ordination of DNA replication.29 Cdc25c is a phospho-tyrosine phosphatase belonging to the Cdc25 phosphatase family. It directs dephosphorylation of cyclin B-bound Cdc2 and triggers entry into mitosis.30 A study indicates that Cdc25c not only has a role in mitosis, but also contributes to S-phase entry.31 Our results verify that the four cell cycle genes are downstream target genes of C/EBPβ during MCE (Figures 4, 5; Supplementary Figure S5–S6). Knockdown of the four genes impairs MCE, followed by the inhibition of terminal adipocyte differentiation (Supplementary Figure S5). This observation lends additional credence to the idea that MCE is an early essential step in the adipocyte differentiation program of 3T3-L1 cells.
Propidium iodide (PI) staining and EdU labeling experiments indicate that the inhibition of MCE by the knockdown of Kdm4b and the four cell cycle genes is followed by a blockade of the G1/S transition (Figures 3b and c; Supplementary Figure S5b–S5c). These findings are also consistent with the role of C/EBPβ in MCE, as functional disruption of C/EBPβ blocks G1/S transition followed by the inhibition of MCE.5, 11 Histone acetyltransferase binding to ORC1 (HBO1) is known to positively regulate the initiation of DNA replication.32 Knockdown of HBO1 impairs the ability of 3T3-L1 preadipocytes to differentiate into mature adipocytes by inhibiting DNA replication and MCE.33 In addition, aphidicolin and roscovitine, which are S-phase inhibitors, block DNA synthesis, MCE and adipogenesis in the process of 3T3-L1 adipocyte differentiation.5, 6 Therefore, our present results together with previous reports, suggest that the regulation of G1/S transition is a key step in MCE. This hypothesis supports the notion that DNA replication during MCE increases the accessibility of promoter or enhancer elements to factors required for transcription of genes involved in the initiation of differentiation.34
As proposed in this study, Kdm4b serves as a co-factor for C/EBPβ to regulate cell cycle genes. To get a global sense of the potential collaboration between C/EBPβ and Kdm4b during MCE, promoter-wide search of Kdm4b binding sites was performed. This analysis identified 379 Kdm4b binding regions at 20 h after 3T3-L1 preadipocytes were induced to differentiation (Supplementary Table S6). By integrating the C/EBPβ and Kdm4b ChIP-on-chip data, 101 regions, which correspond to 119 genes, were found to be co-occupied by C/EBPβ and Kdm4b (Supplementary Figure S8 and Table S7). Furthermore, 30 genes of the 68 potential C/EBPβ target genes were also targeted by Kdm4b, including the four cell cycle genes described in this study (Supplementary Table S8). We have previously reported that histone H4, which is involved in chromatin assembly and is required for normal progression of S phase, is transactivated by C/EBPβ during MCE.10 Intriguingly, histone H4 is also targeted by Kdm4b, as illustrated in the ChIP-on-chip data (Supplementary Table S6 and Table S8). Consistently, our study verifies that Kdm4b positively regulates the expression of histone H4 through demethylation of H3K9me3 on its promoter (Supplementary Figure S9). Thus, C/EBPβ and Kdm4b co-regulate the expression of cell cycle genes, as well as chromatin assembly genes, such as histone H4, to efficiently promote MCE.
Knockdown of C/EBPβ or Kdm4b does not appear to impair pre-confluent 3T3-L1 preadipocytes proliferation (Figure 8). This suggests that C/EBPβ and Kdm4b are not general regulators of cell proliferation, but have specific functions in certain types of cell proliferation including MCE. Unlike the strong promoter bindings during MCE, C/EBPβ and Kdm4b bind weakly to the promoters of the four cell cycle genes in pre-confluent 3T3-L1 cells (Figures 8c and d). It is possible that additional co-factors or specific post-translational modifications of C/EBPβ, which might be absent in pre-confluent 3T3-L1 cells, are required for efficient binding of C/EBPβ and Kdm4b to these promoters during MCE.
In conclusion, our data indicate that C/EBPβ transactivates Kdm4b, which regulates the epigenetic signature in C/EBPβ binding regions leading to the induction of cell cycle genes and chromatin assembly genes and thereby promoting MCE (Supplementary Figure S7). It is conceivable that through upregulation of Kdm4b, C/EBPβ and Kdm4b form a feed forward regulatory loop to facilitate MCE. This suggests a profound role for C/EBPβ in the epigenetic control of MCE and offers new insights into the critical action of C/EBPβ in the proliferation of certain specific cell types.
Materials and Methods
Cell culture, induction of differentiation
3T3-L1 preadipocytes were propagated and maintained in DMEM containing 10% calf serum and HEK293T cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). To induce differentiation, 2-day post-confluent 3T3-L1 preadipocytes (designated 0 day or 0 h) were fed DMEM containing 10% FBS, 1 μg/ml insulin, 1 μM dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine (MIX) until day 2. Cells were then fed DMEM supplemented with 10% FBS and 1 μg/ml insulin for 2 days, after which they were fed every other day with DMEM containing 10% FBS. Adipocyte gene expression and acquisition of the adipocyte phenotype begins on day 3 and is maximal by day 8. Cell number was determined by hemocytometer.
Oil red O staining
Oil red O staining was conducted as described.19 Cells were washed three times with PBS and then fixed for 10 min with 3.7% formaldehyde. Oil red O (0.5% in isopropanol) was diluted with water (3 : 2), filtered through a 0.45 μm filter, and incubated with the fixed cells for 1 h at room temperature. Cells were washed with water, and the stained fat droplets in the cells were visualized by light microscopy and photographed.
ChIP and Re-ChIP
Cells were fixed with 1% formaldehyde for 10 min at room temperature. Glycine was added to a final concentration of 0.125 M, and the incubation was continued for additional 5 min. Cells were then washed twice with ice-cold PBS, harvested by scraping, pelleted, and resuspended in 1 ml SDS lysis buffer containing 50 mM Tris-HCl pH 8.0, 1% SDS, 10 mM EDTA, and protease inhibitors. Samples were sonicated eight times for 30 s each with an interval of 30 s with a Bioruptor sonicator (Diagenode, Liège, Belgium). Samples were centrifuged at 14 000 × g at 4 °C for 10 min. After removal of an input aliquot (whole-cell extract), supernates were diluted 10-fold in ChIP dilution buffer containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% TritonX-100, and complete protease inhibitor tablets. Samples were immunoprecipitated using antibodies against C/EBPβ (sc-150 and sc-7962, Santa Cruz Biotechnologies, Santa Cruz, CA, USA), Kdm4b (A301-477A, Bethyl, Montgomery, AL, USA), H3K9me3 (ab8898, Abcam, Cambridge, MA, USA) or the non-specific IgG control (Abcam). Immunoprecipitated samples were eluted and reverse cross-linked by incubation overnight at 65 °C. Genomic DNA was then extracted using a PCR purification kit (Qiagen, Valencia, CA, USA). Primers for ChIP-qPCR were in the Supplementary Table S9. ChIP data were normalized to control IgG or expressed as percentage of input. Re-ChIPs were done essentially the same as primary ChIPs. Beads from the first ChIP were incubated with 10 mM DTT at 37 °C for 30 min and the eluted immunocomplex were diluted 1 : 50 in dilution buffer followed by re-immunoprecipitation with the second antibodies.
ChIP-on-chip arrays and data analysis
ChIP-on-chip analysis was performed as described.35 C/EBPβ antibody (sc-150, Santa Cruz Biotechnologies) and Kdm4b antibody (A301-477A, Bethyl) were used for the ChIPs. ChIP samples were then amplified by ligation-mediated PCR, labeled and hybridized to Affymetrix mouse promoter 1.0 oligonucleotide tiling arrays (Affymetrix, Santa Clara, CA, USA, for C/EBPβ) or Nimblegen mouse 385 K RefSeq promoter arrays (Roche NimbleGen, Madison, WI, USA, for Kdm4b). ChIP-on-chip assays were analyzed with MAT, with probes remapped to the February 2006 Mouse Genome Assembly (mm8). The threshold cutoffs for binding regions were FDR≤1%. For each binding region, the nearest gene was determined, and the distance from the center of the binding region to the TSSs of the gene was calculated using CEAS35, 36 according to gene coordinates in the February 2006 Mouse Genome Assembly (mm8). For conservation analysis, the ChIP regions were aligned at the center and uniformly expanded to 1500 bp at each end, and at each position the average phastCons score retrieved from UCSC Genome Browser (http://genome.ucsc.edu) was generated for the average conservation plot.
Analysis of the enriched motifs
An enumeration-based de novo motif discovery method was performed on the ChIP-on-chip data.37 Z-score was used to assess the significance of motif enrichment. Z−score=(C−E)/sqrt (E), quantifying the significance of enrichment. Here, C and E represent the observed and expected numbers of instances in ChIP regions, respectively. Background regions were randomly selected from design regions of Affymetrix mouse Promoter array. Enrichment of known transcription factor motifs in the TRANSFAC database was evaluated by Z-scores. Using a matrix similarity threshold of 0.8, the numbers of instances in ChIP and background regions were counted for 554 annotated vertebrate PWMs.
GO analysis
GO analysis was performed using DAVID (http://david.abcc.ncifcrf.gov), by examining the biological processes in which these genes are involved, and using RefSeq mRNA IDs with the entire mouse genome as a background model.35, 38 P-values were calculated using Fisher Exact statistics.
RNA interference
Synthetic RNAi oligonucleotides specific for regions in the C/EBPβ, Kdm4b, Cdc45l, Mcm3, Gins1, Cdc25c mRNAs were designed and synthesized by Invitrogen (Carlsbad, CA, USA) Stealth RNAi. The silencing effects of several RNAi oligonucleotides were screened and tested initially for their ability to knock down the expression of these genes by RT-qPCR. The sequences for successful RNAi knockdown were listed in the Supplementary Table S10. Stealth RNAi Negative Control Duplexes with a similar GC content were used as a negative control. 3T3-L1 cells were transfected at ∼50% confluence with RNAi oligonucleotides using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions.
RNA isolation, qPCR analysis, and gene expression profiling
Total RNAs were extracted using Trizol (Invitrogen). First-strand cDNAs were synthesized using the PrimeScript reverse transcriptase and Random primers (Takara Bio, Otsu, Japan). qPCR was performed using primers as described. Power SYBR Green PCR Mastermix (Applied Biosystems, Carlsbad, CA, USA) and the PRISM 7300 instrument (Applied Biosystems) were used. Analysis was performed using the standard curve method and normalization of all genes of interest to the control gene 18S rRNA. Primers for RT-qPCR were available in the Supplementary Table S9. RT-qPCR data were normalized to 0 h time point of control RNAi-treated cells. Gene expression profiling was carried out by KangChen Bio-tech (Shanghai, China). At 0 h or 20 h after induction, total RNA from 3T3-L1 cells was extracted, amplified, labeled and hybridized using the NimbleGen system (Roche NimbleGen).
Western blotting
Cells were lysed with lysis buffer containing 2% SDS, 10 mM dithiothreitol, 50 mM Tris-HCl, pH 6.8, 10% glycerol, 0.002% bromphenol blue, 1 × protease inhibitor mixture. Equal amounts of protein were separated by SDS-PAGE, and transferred to PVDF membrane (Millipore, Billerica, MA, USA), immunoblotted with antibodies. Antibodies recognizing the following proteins were used: PPARγ (Santa Cruz Biotechnologies), C/EBPα (Santa Cruz Biotechnologies), 422/ap2 (prepared in Dr. Lane’s lab), Cdc45l (GeneTex, Irvine, CA, USA), Mcm3 (GeneTex), Gins1 (Abgent, San Diego, CA, USA), Cdc25c (Santa Cruz Biotechnologies), C/EBPβ (sc-150, Santa Cruz Biotechnologies), Kdm4b (A301-478A, Bethyl) and Hsp90 (Abcam).
EdU labeling, microscopic fluorescence, PI staining, and flow cytometry analysis
EdU labeling was performed as previously described.39 Cells were induced to differentiation with standard protocol. Sixteen hours after induction, cells were labeled for 2 h with 10 μM EdU before fixation with 4% paraformaldehyde. EdU was stained by incubating for 20 min with 100 mM Tris (from 2 M stock, pH 8.5), 0.5 mM CuSO4, 10 μM fluorescent azide (from 100 mM stocks in DMSO), and 50 mM ascorbic acid (added last to the mix from a 0.5 M stock in water). The staining mix was prepared fresh each time and used for staining cells immediately after addition of ascorbate. Cells were then washed and counterstained with Hoechst. The incorporation of EdU was visualized by fluorescence microscopy. At each step described above, the cells were washed with PBS three times after treatment. For FACS analysis, EdU-labeled cells were trypsinized, treated with EdU-staining chemicals, washed and subjected to flow cytometry with FACScan (Becton Dickinson, Franklin Lakes, NJ, USA). For PI staining, cells were trypsinized, fixed with 2% (wt/vol) paraformaldehyde in 1 × PBS. Then they were treated with 0.5 mg/ml RNase A for 1 h at room temperature and incubated with 0.1 mg/ml PI (Sigma, St. Louis, MO, USA) for 45 min at 37 °C. DNA content was determined by flow cytometry analysis.
Electrophoretic Mobility Shift Assay
Nuclei were isolated and nuclear extracts prepared using the buffer containing 0.3 M NaCl, 1 M urea, 1% Nonidet P-40, 25 mM HEPES (pH 7.9), and 1 mM DTT. Protein concentration was determined by the Bradford method (Bio-Rad, Hercules, CA, USA). EMSA was performed as follows. Reaction mixtures containing ∼0.25 ng of 32P-labeled oligonucleotide probe, 2 μg of poly [d(I-C)], and 10 μg of nuclear extract protein in 30 μl of buffer (10 mM HEPES, 0.1 mM EDTA, 5% glycerol, 100 mM NaCl, 0.3 M urea, 0.3% NP-40) were incubated on ice for 15 min, at room temperature for 15 min, and then were separated electrophoretically on 5% polyacrylamide gels 0.5 × TBE [44.5 mM Tris, 44.5 mM boric acid, 1 mM EDTA (pH 8.3)]. For the competition assays, 100-fold excess of unlabeled probes were added to the binding mixtures. In all, 2 μg of antibodies against C/EBPβ (sc-7962, Santa Cruz Biotechnology) were used in the supershift analyses. Oligonucleotides used are listed in the Supplementary Table S11.
Luciferase reporter assays
Promoter regions of mouse Kdm4b (−1000 to −1 bp), Cdc45l (−1000 to +208 bp), Mcm3 (−500 to +23 bp), Gins1 (−258 to +105 bp), Cdc25c (−500 to +145 bp) were amplified via PCR from genomic DNA of 3T3-L1 (primer sequences are available upon request) and cloned into PGL3-basic (Promega, Madison, WI, USA). Cells were transfected by Lipofectamine 2000 (Invitrogen). Luciferase activity was measured using Dual Luciferase Reporter Assay (Promega), normalizing firefly luciferase to renilla activity.
Immunoprecipitation assay
Cells were washed with PBS, scraped off and collected by centrifugation. Then cells were suspended in RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) in the presence of protease inhibitors (Roche) for 3 h at 4 °C. After centrifugation, the supernates were incubated with the indicated antibodies at 4 °C overnight. The next day, protein A-agarose beads (Invitrogen) was added. After 3 h incubation, the beads were washed with TBS-T (TBS+0.05% Tween-20). The immunoprecipitates were separated by SDS-PAGE and subjected to western blotting.
Statistical analysis
Results are expressed as mean±S.E. Comparisons between groups were made by unpaired two-tailed Student’s t-test, where P<0.05 was considered as statistically significant. All experiments were repeated at least three times and representative data are shown.
Abbreviations
- Cdc25c:
-
cell division cycle 25 homolog c
- Cdc45l:
-
cell division cycle 45 homolog
- C/EBPβ:
-
CCAAT/enhancer-binding protein β
- C/EBPα:
-
CCAAT enhancer-binding protein α
- ChIP:
-
chromatin immunoprecipitation
- EMSA:
-
Electrophoretic Mobility Shift Assay
- ER:
-
estrogen receptor
- Gins1:
-
GINS complex subunit 1
- LAP:
-
liver-enriched activator protein
- LIP:
-
liver-enriched inhibitory protein
- MCE:
-
mitotic clonal expansion
- Mcm3:
-
mini-chromosome maintenance complex component 3
- MECs:
-
mammary epithelial cells
- PPARγ:
-
peroxisome proliferator-activated receptor γ
- RNAi:
-
RNA interference
- TSS:
-
transcription start site
References
Haslam DW, James WP . Obesity. Lancet 2005; 366: 1197–1209.
Shepherd PR, Gnudi L, Tozzo E, Yang H, Leach F, Kahn BB . Adipose cell hyperplasia and enhanced glucose disposal in transgenic mice overexpressing GLUT4 selectively in adipose tissue. J Biol Chem 1993; 268: 22243–22246.
Green H, Kehinde O . An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell 1975; 5: 19–27.
Qiu Z, Wei Y, Chen N, Jiang M, Wu J, Liao K . DNA synthesis and mitotic clonal expansion is not a required step for 3T3-L1 preadipocyte differentiation into adipocytes. J Biol Chem 2001; 276: 11988–11995.
Tang QQ, Otto TC, Lane MD . Mitotic clonal expansion: a synchronous process required for adipogenesis. Proc Natl Acad Sci USA 2003; 100: 44–49.
Reichert M, Eick D . Analysis of cell cycle arrest in adipocyte differentiation. Oncogene 1999; 18: 459–466.
Yeh WC, Bierer BE, McKnight SL . Rapamycin inhibits clonal expansion and adipogenic differentiation of 3T3-L1 cells. Proc Natl Acad Sci USA 1995; 92: 11086–11090.
Farmer SR . Transcriptional control of adipocyte formation. Cell Metab 2006; 4: 263–273.
Tang QQ, Otto TC, Lane MD . CCAAT/enhancer-binding protein beta is required for mitotic clonal expansion during adipogenesis. Proc Natl Acad Sci USA 2003; 100: 850–855.
Zhang YY, Li X, Qian SW, Guo L, Huang HY, He Q et al. Transcriptional activation of histone H4 by C/EBPbeta during the mitotic clonal expansion of 3T3-L1 adipocyte differentiation. Mol Biol Cell 2011; 22: 2165–2174.
Zhang JW, Tang QQ, Vinson C, Lane MD . Dominant-negative C/EBP disrupts mitotic clonal expansion and differentiation of 3T3-L1 preadipocytes. Proc Natl Acad Sci USA 2004; 101: 43–47.
Zhu S, Yoon K, Sterneck E, Johnson PF, Smart RC . CCAAT/enhancer binding protein-beta is a mediator of keratinocyte survival and skin tumorigenesis involving oncogenic Ras signaling. Proc Natl Acad Sci USA 2002; 99: 207–212.
Iyer VV, Kadakia TB, McCabe LR, Schwartz RC . CCAAT/enhancer-binding protein-beta has a role in osteoblast proliferation and differentiation. Exp Cell Res 2004; 295: 128–137.
Seagroves TN, Krnacik S, Raught B, Gay J, Burgess-Beusse B, Darlington GJ et al. C/EBPbeta, but not C/EBPalpha, is essential for ductal morphogenesis, lobuloalveolar proliferation, and functional differentiation in the mouse mammary gland. Genes Dev 1998; 12: 1917–1928.
Shuman JD, Sebastian T, Kaldis P, Copeland TD, Zhu S, Smart RC et al. Cell cycle-dependent phosphorylation of C/EBPbeta mediates oncogenic cooperativity between C/EBPbeta and H-RasV12. Mol Cell Biol 2004; 24: 7380–7391.
Shi L, Sun L, Li Q, Liang J, Yu W, Yi X et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc Natl Acad Sci USA 2011; 108: 7541–7546.
Kawazu M, Saso K, Tong KI, McQuire T, Goto K, Son DO et al. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One 2011; 6: e17830.
Yang J, Jubb AM, Pike L, Buffa FM, Turley H, Baban D et al. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res 2010; 70: 6456–6466.
Tang QQ, Lane MD . Activation and centromeric localization of CCAAT/enhancer-binding proteins during the mitotic clonal expansion of adipocyte differentiation. Genes Dev 1999; 13: 2231–2241.
Peterson CL, Laniel MA . Histones and histone modifications. Curr Biol 2004; 14: R546–R551.
Sterneck E, Tessarollo L, Johnson PF . An essential role for C/EBPbeta in female reproduction. Genes Dev 1997; 11: 2153–2162.
Robinson GW, Johnson PF, Hennighausen L, Sterneck E . The C/EBPbeta transcription factor regulates epithelial cell proliferation and differentiation in the mammary gland. Genes Dev 1998; 12: 1907–1916.
Gutierrez S, Javed A, Tennant DK, van Rees M, Montecino M, Stein GS et al. CCAAT/enhancer-binding proteins (C/EBP) beta and delta activate osteocalcin gene transcription and synergize with Runx2 at the C/EBP element to regulate bone-specific expression. J Biol Chem 2002; 277: 1316–1323.
Fodor BD, Kubicek S, Yonezawa M, O'Sullivan RJ, Sengupta R, Perez-Burgos L et al. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev 2006; 20: 1557–1562.
Grimm SL, Rosen JM . The role of C/EBPbeta in mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia 2003; 8: 191–204.
Lin DI, Aggarwal P, Diehl JA . Phosphorylation of MCM3 on Ser-112 regulates its incorporation into the MCM2-7 complex. Proc Natl Acad Sci USA 2008; 105: 8079–8084.
Nagahama Y, Ueno M, Miyamoto S, Morii E, Minami T, Mochizuki N et al. PSF1, a DNA replication factor expressed widely in stem and progenitor cells, drives tumorigenic and metastatic properties. Cancer Res 2010; 70: 1215–1224.
Aparicio T, Ibarra A, Mendez J . Cdc45-MCM-GINS a new power player for DNA replication. Cell Div 2006; 1: 18.
Tercero JA, Labib K, Diffley JF . DNA synthesis at individual replication forks requires the essential initiation factor Cdc45p. EMBO J 2000; 19: 2082–2093.
Aressy B, Ducommun B . Cell cycle control by the CDC25 phosphatases. Anticancer Agents Med Chem 2008; 8: 818–824.
Turowski P, Franckhauser C, Morris MC, Vaglio P, Fernandez A, Lamb NJ . Functional cdc25C dual-specificity phosphatase is required for S-phase entry in human cells. Mol Biol Cell 2003; 14: 2984–2998.
Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W et al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell 2006; 21: 51–64.
Johmura Y, Osada S, Nishizuka M, Imagawa M . FAD24 acts in concert with histone acetyltransferase HBO1 to promote adipogenesis by controlling DNA replication. J Biol Chem 2008; 283: 2265–2274.
Cornelius P, MacDougald OA, Lane MD . Regulation of adipocyte development. Annu Rev Nutr 1994; 14: 99–129.
Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev 2008; 22: 2941–2952.
Ji X, Li W, Song J, Wei L, Liu XS . CEAS: cis-regulatory element annotation system. Nucleic Acids Res 2006; 34: W551–W554.
Wang K, Wang P, Shi J, Zhu X, He M, Jia X et al. PML/RARalpha targets promoter regions containing PU.1 consensus and RARE half sites in acute promyelocytic leukemia. Cancer Cell 2010; 17: 186–197.
Steger DJ, Grant GR, Schupp M, Tomaru T, Lefterova MI, Schug J et al. Propagation of adipogenic signals through an epigenomic transition state. Genes Dev 2010; 24: 1035–1044.
Aparicio T, Guillou E, Coloma J, Montoya G, Mendez J . The human GINS complex associates with Cdc45 and MCM and is essential for DNA replication. Nucleic Acids Res 2009; 37: 2087–2095.
Acknowledgements
This research is supported partially by National Key Basic Research Project Grant 2011CB910201 and 2009CB825604, The State Key Program of National Natural Science Foundation 31030048C120114, Shanghai Key Science and Technology Research Project 10JC1401000 (For QQT); National Natural Science Foundation Grant 30870510 (For XL), National Natural Science Foundation Grant 31000603 (For LG); The Department is supported by Shanghai Leading Academic Discipline Project B110 and 985 Project 985III-YFX0302. We thank KangChen Bio-tech and Dr. Kan-kan Wang for ChIP-on-chip assays.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by RA Knight
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Guo, L., Li, X., Huang, JX. et al. Histone demethylase Kdm4b functions as a co-factor of C/EBPβ to promote mitotic clonal expansion during differentiation of 3T3-L1 preadipocytes. Cell Death Differ 19, 1917–1927 (2012). https://doi.org/10.1038/cdd.2012.75
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.75
Keywords
This article is cited by
-
Deubiquitinase USP1 enhances CCAAT/enhancer-binding protein beta (C/EBPβ) stability and accelerates adipogenesis and lipid accumulation
Cell Death & Disease (2023)
-
MYOD1 inhibits avian adipocyte differentiation via miRNA-206/KLF4 axis
Journal of Animal Science and Biotechnology (2021)
-
Mechanism of the fungal-like particles in the inhibition of adipogenesis in 3T3-L1 adipocytes
Scientific Reports (2021)
-
HMGB2 orchestrates mitotic clonal expansion by binding to the promoter of C/EBPβ to facilitate adipogenesis
Cell Death & Disease (2021)
-
Histone demethylase KDM4D cooperates with NFIB and MLL1 complex to regulate adipogenic differentiation of C3H10T1/2 mesenchymal stem cells
Scientific Reports (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}