
Abstract
Signalling through the janus kinase (JAK)/signal transducer and activator of transcription (Stat) pathway is required at different stages of mammary gland development, and this pathway is frequently hyper-activated in cancer, including tumours of the breast. Stats 3, 5 and 6 have important roles in the differentiation and survival of mammary alveolar cells, but somewhat paradoxically, both Stat3 and 5 can have oncogenic activity in the mammary gland. Constitutive activation of JAK2 could be anticipated to result in hyper-activation of Stats 1, 3, 5 and 6 with concomitant cell transformation, although the outcome is difficult to envisage, particularly since Stats 3 and 5 play opposing roles in normal mammary gland development. Here, we show that expression of a constitutively active JAK2 mutant, JAK2 V617F, leads to hyper-activation of Stat5 in mammary epithelial cells (MECs), and transgenic mice expressing JAK2 V617F specifically in the mammary gland exhibit accelerated alveologenesis during pregnancy and delayed post-lactational regression. Overexpressing JAK2 V617F in MECs in vitro results in elevated proliferation and resistance to cell death. Furthermore, constitutively active JAK2 enhances anchorage-independent cell growth in the presence of a co-operating oncogene and accelerates tumourigenesis in a xenograft model. Taken together, our results provide insights into signalling downstream of constitutively active JAK2 and could be important for understanding the molecular mechanisms of breast tumourigenesis.
Similar content being viewed by others


Main
Mammary gland development during pregnancy is characterised by proliferation of progenitor cells of the alveolar lineage, followed by their terminal differentiation upon lactation and subsequent cell death during post-lactational regression (involution). This cycle is regulated by a sequential cascade of signal transducer and activator of transcription (Stat) factors: Stat6 promotes proliferation of alveolar progenitors, while Stat5 is required both for the generation of alveolar progenitors and their functional differentiation to express milk proteins such as β-casein and whey acidic protein (WAP).1 When these cells become superfluous at the end of lactation, cell death and tissue remodelling are initiated by Stat3 and the gland is returned to a pre-pregnant state in readiness for the next cycle.2 The janus kinase (JAK)/Stat signalling pathway is normally activated in response to ligand engagement of cytokine receptors, which triggers auto-cross-phosphorylation of the receptor-associated JAK kinases and subsequent tyrosine phosphorylation of Stat factors, which dimerise, translocate to the nucleus and activate transcription of target genes.3 In addition to the requirement for specific Stats in adult mammary gland development, JAK2 is essential as its conditional deletion in the mouse results in failed lactation with a phenotype that recapitulates that of Stat5-deficient glands.4 Furthermore, loss of JAK2 protects against the onset of mammary tumours.5, 6
Constitutive activation of the JAK/Stat pathway is a feature of many types of cancer, including breast, prostate, head and neck, and lymphoma.7 Stats 3 and 5 are activated in a high proportion of breast tumours,8, 9 and both have been shown to be mammary oncogenes.10, 11 In physiological conditions, JAK/Stat signalling is exquisitely controlled by a cohort of negative regulators, pre-eminently the suppressor of cytokine signalling (SOCS) family. SOCS proteins are direct transcriptional targets of Stats and negatively regulate the pathway by a variety of mechanisms, including binding to the receptor, the Stat or, more usually, the JAK.12 The consequence of this regulatory feedback loop is that signalling through the JAK/Stat pathway is normally of high intensity and short duration. In contrast, when the pathway is constitutively active, signalling is often low level and prolonged. The consequences of this disparate regulation, and how it can lead to tumourigenesis, are not well understood. Unlike many other oncogenes, mutations in the seven mammalian Stats are very rare. Likewise, mutations in the JAKs (JAK1, 2, 3 and TYK2) are infrequently associated with tumour development. Recently, a new JAK2 mutation (I166T) has been discovered in breast cancer by massively parallel DNA sequencing technologies,13 although its functional importance is unclear. Another striking exception is the association of a somatic point mutation at codon 617 (G to T transversion) in the JAK2 homology domain with several myeloproliferative disorders.14 This mutation changes valine to phenylalanine and creates a constitutively active JAK2 (CAJAK2). Studies in mice have confirmed that JAK2 V617F is indeed an oncogene in haematopoietic cells.15 Interestingly, JAK2 V617F can overcome normal SOCS regulation by hyper-phosphorylating SOCS3, thus rendering it incapable of inhibiting JAK kinases and thereby promoting constitutive Stat activity.16
Previous studies with JAK2 conditional knockout mice highlighted the significant role of JAK2 during pregnancy.4 JAK2 has been shown to be required for the phosphorylation of Stats 3 and 6 in addition to Stat5. We hypothesise that constitutive JAK2 activity will perturb mammary gland development during gestation and involution and sought to address this by utilising mice harbouring the JAK2 V617F mutation expressed from the endogenous JAK2 locus. We investigated the consequences of elevated JAK2 activity during gestation, when alveolar progenitors expand to produce lobuloalveolar structures, and during involution, when these structures are removed by programmed cell death, and found that both these events are perturbed. Furthermore, studies in mammary epithelial cells (MECs) in vitro revealed a specificity of JAK2 V617F for Stat5 and demonstrated that this naturally occurring mutant can function as a co-operating oncogene both in culture and in xenografts.
Results
Constitutively active JAK2 accelerates mammary gland development during pregnancy
Although the JAK2 V617F mutation causes several myeloproliferative disorders,14 a role for this mutation in breast tumourigenesis has not been investigated, nor have the consequences of constitutive JAK2 activity in normal mammary gland development been studied. To address these issues, we have used conditional knock-in mice in which expression of the JAK2 V617F mutation is under the control of the endogenous JAK2 promoter and is transcribed upon conditional deletion of a floxed STOP sequence by expression of Cre recombinase.17 These engineered mice have been a valuable tool to study the function of constitutive JAK2 activity in the haematopoietic system. JAK2 V617F mice were crossed with keratin 14 (K14)-Cre mice to drive recombination in the basal mammary epithelium, in the placode during embryonic development and in the stem cells in terminal end buds during puberty.18 The basal epithelium encompasses mammary stem and early progenitor cells that generate both ductal/alveolar luminal and myoepithelial cells.19 Consistently, K14-Cre;ROSA26-LacZ reporter mice show β-galactosidase staining in all basal and luminal epithelial cells.20 We first checked the levels of recombination in the mammary glands of K14-Cre;JAK2 V617F females during different time points of mammary gland development (Supplementary Figure S1A). We observed that the ratio of recombined allele (JAK2R) to targeted (floxed) allele (JAK2F) increased at the time points where the proportion of epithelial cells in the gland was greater, resulting in high levels of recombination during gestation, lactation and after 72 h of involution. Moreover, recombination in isolated epithelial cells, from a 10-day gestation mammary gland, was approaching 100% (Supplementary Figure S1A).
Morphological analysis by whole mount and histology showed that the structure of virgin K14-Cre;JAK2 V617F mammary glands was essentially normal compared to littermate controls (Supplementary Figure S1B), and that constitutive activation of JAK2 did not affect the architecture of mammary ducts and alveoli (Supplementary Figure S1C). However, analysis of whole mounts during gestation revealed a striking increase in the number of side branches in K14-Cre;JAK2 V617F mice compared to controls (Figure 1a). This correlated with a significant increase in the number of mammary structures (ducts and alveoli) counted in hematoxylin and eosin (H&E) sections (Figure 1b). We observed, by immunostaining for the milk protein β-casein, that the additional alveoli present in the K14-Cre;JAK2 V617F mammary glands expressed β-casein in the lumen (Supplementary Figure S2A). The levels of β-casein and WAP, another milk protein that has been defined as a marker of late differentiation in the mammary gland,21 were similar in mutant and control mice (Supplementary Figure S2B) as analysed by western blot. Furthermore, the transcription factor E74-like factor 5 (Elf5), which determines alveolar cell fate,22 was unaffected (Supplementary Figure S2B) and the phenotype of V617F-expressing females during lactation, as determined by whole mounts, histological analysis and milk protein quantification, was normal (data not shown). Thus, expression of JAK2 V617F induces precocious alveologenesis, but does not alter alveolar differentiation. This is consistent with JAK2 V617F acting upstream of Stat5 as Stats 1 and 3 are not normally activated during pregnancy.2

K14-Cre;JAK2 V617F mice show increased pregnancy-induced branching. Representative whole-mount pictures (a) or H&E-stained sections (b) of the abdominal mammary glands of JAK2 V617F (control, left) or K14-Cre;JAK2 V617F (right) females at 10 days gestation. Scale bars: 1 mm (a) and 500 μm (b). In (b), quantification of the number of epithelial structures (ducts and alveoli) per area in the H&E sections of control mice (JAK2 V617F, K14-Cre;JAK2 wt and JAK2 wt, n=12) and K14-Cre;JAK2 V617F mice (n=4) is shown. Data are expressed in arbitrary units (a.u.) and mean±S.D. is represented. *P<0.05 versus control mice
This prompted us to investigate firstly the rate of proliferation using immunostaining for Ki67 and we found that proliferation of MECs in ducts and alveoli was significantly increased in K14-Cre;JAK2 V617F females compared with their littermate controls (Figure 2a). Next, using immunostaining, we observed that the number of phospho-Stat (p-Stat5)-positive cells was significantly increased in the alveoli of tissue from K14-Cre;JAK2 V617F mice compared to controls (Figure 2b), and these elevated levels were maintained until lactation (Figures 2c and d). Western blot analysis revealed that phosphorylation of Stats 3 and 6 was not affected by CAJAK2 (Figure 2c), while p-Stat1 could not be detected at any time point analysed. K14-Cre;JAK2 V617F and control mammary glands showed similar levels of total JAK2 during gestation and lactation (Supplementary Figures S3A and B), indicating that the increased levels of p-Stat5 in K14-Cre;JAK2 V617F females were due to the constitutive activation of JAK2 and not to different expression levels of JAK2.

Constitutive activation of JAK2 in the mammary gland increases proliferation during pregnancy and overactivates Stat5 during gestation and lactation. (a) Ki67-positive cells (red) and (b) p-Stat5-positive cells (red) in the mammary glands of JAK2 V617F (control, left) or K14-Cre;JAK2 V617F (right) females at 10 days gestation. E-cadherin staining was used in (a) for visualisation of epithelial cells. Nuclei are in blue. Scale bar: 20 μm. Quantification of Ki67-positive cells (a, n=3 for each genotype) and p-Stat5-positive cells (b, n=5 for each genotype) in ducts and alveoli are shown in the corresponding graphs. (c) Western blot analysis of p-Stat5, p-Stat3 and p-Stat6 in the mammary glands of K14-Cre;JAK2 V617F and control mice (JAK2 V617F and K14 Cre;JAK2 wt) at 24 h lactation (first day post-partum). Total Stats were used for normalisation. Quantification of p-Stat5 signal compared to total Stat5 is shown in (d). Results are mean±S.D. *P<0.05, **P<0.01 versus control mice
Constitutive activation of JAK2 delays involution
Involution is marked by extensive cell death and tissue remodelling. Striking differences between K14-Cre; JAK2 V617F and control females were observed during involution (Figure 3). Although sections from control glands after 72 h of involution showed extensive tissue remodelling, characterised by the presence of adipose tissue stroma and collapsed alveoli, sections from K14-Cre;JAK2 V617F glands resembled lactating mammary tissue, characterised by prominent and expanded milk-producing alveoli surrounded by few adipocytes (Figure 3a), and the expression of higher levels of β-casein, as determined by real-time quantitative PCR (RTQ-PCR) (Figure 3b). Increased levels of cleaved caspase 3 activity, a surrogate marker for cell death, at 72 h involution were abrogated in K14-Cre;JAK2 V617F glands (Figure 3c), suggesting that the induction of cell death after cessation of the suckling stimulus is altered by persistent JAK2 activity. Tissue remodelling is a hallmark of the second, and irreversible, phase of involution and matrix metalloproteinases (MMPs) have been shown to be involved in this process.23 Control mammary glands showed an increase in MMP-2 and -9 mRNA expression after 72 h involution compared to lactation (Figure 3d). This increase was significantly less in K14-Cre;JAK2 V617F glands (Figure 3d), indicating that the activation of MMPs during involution is also altered. Thus, both epithelial cell death and tissue remodelling were altered by constitutive JAK2 activity. The fact that we still see an increase in the expression of MMPs, particularly MMP9 (Figure 3d), in the JAK2 V617F-expressing glands suggests that CAJAK2 cannot abolish involution, but can delay the process. This is most likely due to the role of p-Stat5 as a survival factor, counteracting Stat3 activity.24 This delay in mammary gland involution in K14-Cre;JAK2 V617F females was a consequence of constitutive JAK2 activation and not due to different levels of JAK2 expression, as demonstrated by western blot analysis (Supplementary Figure S3C).

Constitutive activation of JAK2 leads to delayed involution. (a) Representative H&E-stained sections of the abdominal mammary glands of JAK2 V617F (control, left) or K14-Cre;JAK2 V617F (right) females at 72 h involution. Scale bars: 1 mm (top) and 200 μm (bottom). (b) and (d) β-Casein (b) and MMP-2 and -9 (d) mRNA expression in control and K14-Cre;JAK2 V617F mammary glands at 72 h involution (b) or at 24 h lactation and 72 h involution (d) determined by real-time quantitative PCR. (c) Caspase 3 activity in control and K14-Cre;JAK2 V617F glands at 24 h lactation and 72 h involution. Results are expressed in arbitrary units (a.u.) and mean±S.D. is represented. Six control mice (JAK2 V617F, K14-Cre;JAK2 wt and JAK2 wt) and five K14-Cre;JAK2 V617F mice were used for all the experiments. *P<0.05, ***P<0.001 versus control mice
JAK2 V617F affects proliferation and differentiation of MECs in vitro
To further study signalling downstream of CAJAK2 in MECs, we infected KIM-2 MECs with retrovirus harbouring JAK2 wt or JAK2 V617F. KIM-2 cells model mammary gland development and can be induced to differentiate, express milk proteins and undergo cell death upon lactogenic hormone withdrawal.25 Stable overexpression of JAK2 wt and JAK2 V617F was confirmed by immunofluorescence (Figure 4a) and western blot (Figure 4b). Expression of JAK2 V617F resulted in elevated phosphorylation of Stats 5 and 1 (Figure 4b). This was abolished when the cells were treated with the specific JAK2 inhibitor TG101348 (Supplementary Figure S4), indicating that JAK2 alone is responsible for the activation of Stats 5 and 1 in this cell line. The phosphorylation of other Stat family members, which play important roles in mammary gland development, such as Stats 3 and 6,26, 27 was not significantly affected by the expression of JAK2 V617F (Figure 4b). Stat proteins act predominantly in the nucleus to regulate gene expression and p-Stat5 was found mainly in the nuclear fraction of KIM-2 cells transduced with JAK2 V617F, confirming functional activation by constitutive JAK2 activity (Figure 4c). Similar results were obtained with another MEC line, EpH4 (Supplementary Figure S5). Therefore, we have demonstrated that the primary target of JAK2 V617F in MECs both in vivo and in vitro is Stat5.

Constitutively active JAK2 in mammary epithelial cells induces Stat5 activation. (a) Representative immunofluorescence staining for JAK2 (red) in KIM-2 cells overexpressing JAK2 wt or JAK2 V617F. MIG (MSCV-IRES-GFP) represents cells carrying the empty vector. Nuclei are in blue. Scale bar: 50 μm. (b) Western blot analysis of JAK2 and the phosphorylation of different Stats in KIM-2 cells harbouring MIG, JAK2 wt or JAK2 V617F. β-Actin and total Stats were used for normalisation. (c) Western blot of JAK2, Stat5 and p-Stat5 in cytosolic and nuclear protein extracts in MIG-, JAK2 wt- and JAK2 V617F-KIM-2 cells. α-tubulin and lamin A/C were used as markers of cytosolic and nuclear fractions, respectively. Experiments were carried out at least twice for two independent sets of retroviral infections
Having demonstrated the functional consequences of JAK2 V617F expression, we sought to clarify the mechanism behind the observed phenotypes in vivo. As K14-Cre;JAK2 V617F females showed increased pregnancy-induced branching and proliferation, we first determined whether JAK2 V617F could also affect proliferation of MECs in vitro. Using Trypan blue staining and Matrigel colony assays, we showed that undifferentiated KIM-2 cells harbouring JAK2 V617F proliferated at a faster rate (Figure 5a) and generated larger colonies in Matrigel (Figure 5b) than cells overexpressing JAK2 wt or transduced with empty vector.

JAK2 V617F increases proliferation of mammary epithelial cells in two- and three-dimensional cultures. (a) Growth curve determined by direct counting of KIM-2 cells transduced with empty vector (MIG), JAK2 wt or JAK2 V617F. Results are means±S.D. of four independent experiments. **P<0.01 versus KIM-2 JAK2 V617F cells. (b) Representative pictures of Matrigel colony assays of MIG-, JAK2 wt- and JAK2 V617F-KIM-2 cells. Scale bar: 2 mm. n=4 independent experiments
The differentiation of KIM-2 cells was also affected by constitutive activation of JAK2. Transient or stable overexpression of JAK2 V617F was sufficient to induce the expression of β-casein mRNA, without the addition of any lactogenic stimulus (Figure 6a). However, we could not detect any change in the expression of WAP mRNA (Figure 6a) and the increase in β-casein mRNA in undifferentiated KIM-2 JAK2 V617F-expressing cells was not translated into differences in protein levels (Figure 6b, first 3 lanes), possibly because other lactogenic factors are necessary to stabilise β-casein mRNA.28 In support of this notion, when differentiated with lactogenic hormones, KIM-2 cells harbouring JAK2 V617F expressed more β-casein protein than the control cells (Figure 6b), and this was even more apparent when the levels of WAP were measured (Figure 6c). Prolactin (PRL)-induced expression of milk proteins during lactation is dependent on Stat5 activation.29 Interestingly, despite this increase in milk protein expression, p-Stat5 levels in differentiated KIM-2 JAK2 V617F cells were similar to the levels in JAK wt and control cells (Figure 6d). The transcription factor Elf5 has been shown to be regulated by the PRL receptor/JAK2/Stat5 pathway, among others.30 On the other hand, the Stat5a promoter has an Elf5 binding site,20 suggesting that Stat5 and Elf5 can regulate each other's expression. Overexpression of JAK2 wt and JAK2 V617F led to a decrease in the levels of Elf5 in undifferentiated and differentiated KIM-2 cells (Figure 6d), suggesting that the JAK2/Stat5 pathway can activate milk protein expression independently of Elf5 in our model. It is also possible that Elf5 has a peak of expression in progenitor cells to drive alveolar specification and differentiation, and then it is switched off for terminal differentiation.

Constitutively active JAK2 increases the expression of differentiation markers in mammary epithelial cells in vitro. (a) β-Casein and whey acidic protein (WAP) mRNA expression in cells transiently or stably transduced with empty vector (MIG), JAK2 wt or JAK2 V617F was determined by RT-PCR (representative experiment, n=2). Cyclophilin A was used for normalisation. (b–d) Western blot analysis of β-casein (b), WAP (c), p-Stat5 and Elf5 (d) in KIM-2 cells harbouring MIG, JAK2 wt or JAK2 V617F and incubated with lactogenic hormones for the indicated times (representative images, n=3). β-Actin or total Stat5 were used for normalisation
Constitutive activation of JAK2 induces resistance to cell death
K14-Cre;JAK2 V617F females exhibited a delay in involution and reduced caspase 3 activity (Figure 3). We sought to determine if constitutive activation of JAK2 could also induce resistance to cell death in MECs in vitro. KIM-2 cells normally undergo apoptosis when cultured in low (3%) serum medium.25 However, KIM-2 cells expressing JAK2 V617F did not die after being cultured in low serum-containing medium for 48 h (Figure 7a). Moreover, the growth rate was similar in both conditions (Figure 7b), suggesting that CAJAK2 induces not only resistance to cell death, but also insensitivity to anti-growth signals. Differentiated KIM-2 cells can also undergo cell death upon removal of lactogenic hormones,25 mimicking more accurately the involution process in vivo. Differentiated KIM-2 JAK2 V617F cells were also resistant to this mode of cell death induction, as demonstrated by decreased levels of cleaved caspase 3 compared to the corresponding control (empty vector and JAK2 wt) cells (Figure 7c).

Constitutive activation of JAK2 in mammary epithelial cells promotes resistance to cell death. Cell viability (a) or cell growth related to day 0 (b) assessed by Trypan blue staining of MIG-, JAK2 wt- and JAK2 V617F-KIM-2 cells cultured for 48 h in normal (10%) or reduced (3%) serum. Results are means±S.D. of four independent experiments. *P<0.05 versus JAK2 V617F-KIM-2 cells; #P<0.05 versus 10% foetal bovine serum (FBS)-treated cells. (c) Western blot analysis of cleaved caspase 3 in KIM-2 cells transduced with MIG, JAK2 wt or JAK2 V617F, cultured with lactogenic hormones for 12 days and then induced to undergo apoptosis by removal of these hormones for the indicated times (representative images, n=2); h represent hours. β-actin was used for normalisation
Constitutively active JAK2 acts as a co-operating oncogene in MECs
KIM-2 cells expressing JAK2 V617F cells showed, after a few passages in culture, an elongated spindle-like morphology (Figure 8a) characteristic of transformed cells. In addition, CAJAK2 induced some of the hallmarks of cancer,31 such as increased proliferation, resistance to cell death and insensitivity to anti-growth signals. We thus hypothesised that expression of JAK2 V617F could contribute to MEC transformation. One characteristic of transformed cells is their ability to grow in an anchorage-independent manner, so we carried out soft agar colony assays. Expression of JAK2 V617F alone was insufficient to induce colonies in soft agar at the normal culture temperature of 37 °C (Figure 8b). However, KIM-2 cells express a temperature-sensitive variant of simian virus-40 large T antigen and become transformed at 33 °C.25 At this permissive temperature, KIM-2 cells formed colonies in soft agar and expression of JAK2 V617F increased the number of these colonies by fivefold (Figure 8b), suggesting that CAJAK2 may contribute to MEC transformation as a co-operating oncogene.

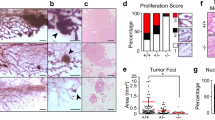
Constitutively active JAK2 enhances mammary tumourigenesis. (a) Morphology of KIM-2 cells stably expressing empty vector (MIG), JAK2 wt or JAK2 V617F. Scale bar: 100 μm. (b) Number of colonies in soft agar formed by KIM-2 cells harbouring MIG, JAK2 wt or JAK2 V617F after 3 weeks at the indicated temperatures. Bars represent mean±S.D. of three independent experiments. **P<0.01 versus JAK2 V617F-KIM-2 cells. (c) JAK2 mRNA expression in MCF-7 cells stably transduced with empty vector (MIG), JAK2 wt or JAK2 V617F was determined by RT-PCR (representative experiment, n=2). GAPDH mRNA expression was used for normalisation. (d) Western blot analysis of the phosphorylation of Stat5 in MCF-7 cells harbouring MIG, JAK2 wt or JAK2 V617F (representative images, n=2). Total Stat5 was used for normalisation. (e) Final tumour volume of MIG-, JAK2 wt- and JAK2 V617F-MCF-7-derived xenografts. Mean of each experimental group (n=5) is represented with a horizontal bar and P-values versus JAK2 V617F-MCF-7 xenografts are indicated. (f) Images of MIG-MCF-7- and JAK2 V617F-MCF-7-derived xenografts 6 weeks after the injection of the cells. (g) Representative H&E sections from MIG-MCF-7- and JAK2 V617F-MCF-7-derived xenografts showing well-defined tumour boundary in MIG-MCF-7-derived tumours and invasive clusters of neoplastic cells (marked by an arrow) in JAK2 V617F-MCF-7-derived tumours. Dashed lines indicate tumour margins. Scale bar: 300 μm
Spontaneous tumours were not observed in K14-Cre;JAK2 V617F females up to 3–4 months old, suggesting that JAK2 V617F is not a potent oncogene per se and may require the presence of a co-operating oncogenic input to induce breast tumours. As KIM-2 cells do not grow as xenografts, we elected to express JAK2 V617F in the luminal breast cancer cell line MCF-7 and observed that this led to Stat5 overactivation (Figures 8c and d). Injection of MCF-7 cells expressing control vector, JAK2 wt or JAK2 V617F into mammary fat pads of nude mice resulted in tumour growth in some of the mice. Tumours generated after the injection of JAK2 V617F-MCF-7 cells showed a shorter onset and were larger than those derived from control cells (Figures 8e and f). Specifically, 15 days following the injection of the cells, tumours were observed in four out of five JAK2 V617F-MCF-7 transplants, while none of the JAK2 wt and two of the five MSCV-IRES-GFP (MIG) transplants gave rise to tumours. The third tumour in the MIG group appeared after 30 days. Interestingly, the JAK2 V617F-MCF-7 tumours displayed a more invasive phenotype with clusters of neoplastic cells observed distant from the main tumour mass (Figure 8g). Although the numbers of tumours are small, and are just approaching significance, these data indicate that CAJAK2 is a co-operating breast oncogene.
Discussion
JAK2 is essential for mammary gland development as conditional deletion results in severe lactation failure.4 On the other hand, the JAK2/Stat pathway is overactivated in a subset of breast tumours32 and JAK2 inhibitors are promising drugs for the treatment of several solid tumours, including breast cancer.33 However, little is known about the consequences of constitutive activation of JAK2 in MECs or in involution. We demonstrate herein that expression of the constitutively active mutant JAK2 V617F in the mammary glands of genetically altered mice and in MECs in vitro (a) results in precocious alveologenesis and delayed involution in vivo and induces MEC proliferation and resistance to cell death in vitro; (b) contributes to mammary tumourigenesis; and (c) has Stat5 as its principal target. In KIM-2 and EpH4 MEC lines, expression of JAK2 V617F was sufficient to induce the phosphorylation of Stat5 in the absence of the lactogenic hormone PRL, normally required for Stat5 phosphorylation. Stats 3 and 6 do not seem to be targets of JAK2 V617F, as we could not detect any change in their phosphorylation status following JAK2 V617F expression in vitro and in vivo. Stat5 is also the main target of JAK2 V617F expression in blood cells.17 Recently, it has been shown that Stat1 also plays a role in the pathogenesis of JAK2 V617F-positive myeloproliferative disorders and that the balance between Stat5 and Stat1 activation determines the clinical phenotype of the patients.34 Interestingly, expression of JAK2 V617F in KIM-2 cells also induces Stat1 phosphorylation, but we did not detect p-Stat1 in either the transgenic mice or in EpH4 cells.
Our data demonstrate that transgenic mice expressing CAJAK2 in mammary gland have accelerated development during pregnancy and delayed involution. This phenotype is very similar to that induced by constitutive activation of Stat5 in mammary gland.11, 35 Furthermore, caveolin-1 (Cav-1)-deficient mice, which show overactivation of JAK2/Stat5 signalling, exhibit accelerated mammary gland development during pregnancy.36 These results resonate well with our data, suggesting that Stat5 is the mediator of constitutive JAK2 activity in mammary gland.
Implications for breast cancer progression
The JAK/Stat pathway is deregulated in many cancers, although JAK2 mutations or amplifications are very rare in breast tumours, with the exception of the newly identified I166T JAK2 mutation.13 Although Stat5a/b-activating mutations or amplification of their encoding genes have not been found, Stat5 seems to be active in a subset of breast tumours, where it is associated with good prognosis.9 This overactivation of the JAK2/Stat pathway could occur by a variety of mechanisms. Firstly, the levels of SOCS proteins can be altered and SOCS2 expression has been inversely correlated with the histological grade of breast tumours and correlated with higher survival rates.37 The microRNA miR-155 is overexpressed in breast cancer cell lines and in primary breast tumours, and its expression is inversely correlated with SOCS1 expression.38 Secondly, high levels of circulating PRL increase the risk of developing breast tumours, and recently, a constitutively active variant of the PRL receptor has been identified in breast cancer patients.39 Another possible mechanism could involve Cav-1, and a dominant-negative mutation in this gene has been found in up to 16% of human breast cancers.40
The JAK2/Stat5 pathway has a complex role in breast cancer. Although it promotes tumourigenesis in mouse models, it is a marker of good prognosis in human breast tumours and inhibits the metastatic behaviour of human breast cancer cell lines.32 It seems, therefore, that this signalling pathway may have a dual role whereby Stat5 promotes the earlier steps of tumour progression, but maintains the differentiation status of established breast cancers, inhibiting their ability to metastasise. Our results show that expression of CAJAK2 in MECs induces proliferation and resistance to cell death, but also enhances the expression of differentiation markers in vitro. In addition, JAK2 V617F is not a potent oncogene in mammary cells as its expression per se is not sufficient to induce MEC transformation. Our soft agar results with KIM-2 cells cultured at the permissive temperature, and the MCF-7 xenografts show that JAK2 V617F can be considered as a co-operating oncogene. Our studies thus provide new insights into signalling downstream of a CAJAK2/Stat5 pathway and could be important for understanding the molecular mechanisms of breast tumourigenesis.
Materials and Methods
Animals
JAK2 V617F mice were generated as described previously17 and have a C57Bl/6 background. K14-Cre mice were kindly donated by Dr. Michaela Frye (Centre for Stem Cell Research, Cambridge, UK), were originally from The Jackson Laboratories (Bar Harbor, ME, USA), and have a mixed C57Bl/6 × CBA background. JAK2 V617 mice were crossed with K14-Cre mice and K14-Cre;JAK2 V617F, and control littermates (JAK2 V617F, K14-Cre;JAK2 wt and JAK2 wt) were used for all the experiments to avoid strain differences. Eight-week-old virgin female mice were mated and males were subsequently removed mid-gestation. Samples were collected at the indicated time points. For involution studies, pups were force-weaned at 10 days of lactation. The abdominal mammary glands were used for all the experiments, except for the analysis of Cre recombination (Supplementary Figure S1A) for which the upper mammary glands were taken. For xenograft experiments, 8-week old nude female mice (Charles River, Wilmington, MA, USA) were implanted subcutaneously with 17β-estradiol pellets (Innovative Research of America, Sarasota, FL, USA), and 4 days later, 2.5 × 106 MIG-, JAK2 wt- or JAK2 V617F-MCF-7 cells resuspended in 25% Matrigel (BD Biosciences, San Jose, CA, USA) were inoculated into one of the abdominal mammary glands. Mice were palpated twice weekly until tumours were detected and thereafter tumours were measured with callipers. Tumour volume was calculated as (4π/3) × (width/2)2 × (length/2). Animals were killed 6 weeks following cell transplantation and tumours were collected. All mice were kept in specific pathogen-free conditions and all procedures were performed according to the United Kingdom Home Office regulations with local ethical approval.
Cell culture
KIM-2 cells25 were grown in 1 : 1 Dulbecco's modified Eagle's medium (DMEM):F12 (Invitrogen, Carlsbad, CA, USA) media containing 10% FCS (Sigma, St. Louis, MO, USA), 0.8 mM insulin (Sigma), 0.8 mM epidermal growth factor (EGF) (Sigma) and 17 mM linoleic acid (Sigma). EpH4 cells were kindly given by Dr. Beug (Institute of Molecular Pathologie, Vienna, Austria) and were grown in DMEM medium (Invitrogen) containing 10% FCS. MCF-7 cells (ATCC-LGC, Middlesex, UK) were cultured in minimum essential medium (MEM) (Invitrogen) supplemented with 10% FCS and 1.6 mM insulin. Stable expression of JAK2 wt and JAK2 V617F in KIM-2, EpH4 and MCF-7 cells was achieved by retroviral infection. Cells were transduced for 48 h with supernatants obtained from Phoenix cells previously transfected with retroviral vectors carrying mouse JAK2 wt, JAK2 V617F or the corresponding empty construct (MSCV-IRES-GFP, MIG). All these constructs were kindly provided by Dr. Göttgens (Department of Haematology, Cambridge, UK). Green fluorescent protein (GFP)-positive infected cells were selected by cell sorting using a MoFlo XDP sorter (Beckman Coulter, Brea, CA, USA). Unless specified, all the experiments were carried out in stably transduced cells at the first 10 passages. For transient gene expression analysis (Figure 6a), RNA was extracted directly from GFP-positive infected KIM-2 just after their isolation by cell sorting. Pictures of the morphology of KIM-2 were carried out with an Olympus CKX41 microscope (Essex, UK). Cell viability and cell growth were determined by trypsinising and counting cells with the vital stain Trypan blue (Sigma). For differentiation studies, KIM-2 cells were grown to confluence and then incubated, during the indicated times, in differentiation medium containing lactogenic hormones: 1 : 1 DMEM : F12, 10% FCS, 0.8 mM insulin, 0.2 mM PRL (Sigma), 1 mM dexamethasone (Sigma) and 17 mM linoleic acid. Cell death in differentiated cells was induced by the removal of lactogenic hormones after 12 days of culture in differentiation medium. The specific JAK2 inhibitor TG101348 (Axon Medchem BV, Groningen, The Netherlands) was used at the indicated concentrations in Supplementary Figure S4.
Mammary epithelial primary cells were isolated from JAK2 V617F mice as follows. The mammary gland of a K14-Cre;JAK2 V617F female at 10 days of gestation was collected and digested overnight at 37 °C with 1 mg/ml collagenase IA (Roche Applied Science, Penzberg, Germany) in complete medium (CM) containing 10% FCS, 5 μg/ml insulin and 10 ng/ml EGF. The following day, tissues were shaken and spun for 5 min at 1000 r.p.m. The supernatant and fat were removed, and the pellet containing epithelial cells was washed twice, resuspended in CM and plated into cell culture flasks. After 1–2 days in culture, clusters of epithelial cells (called organoids) began to settle and expand as a monolayer. Fibroblasts were removed every 2–3 days with differential trypsinisation.
Whole mounts and histology
For whole-mount analysis, abdominal mammary glands were spread out on a glass slide, soaked overnight in Carnoy's fixative, stained with Carmine Alum and cleared in ethanol and xylene. Pictures were taken with a MZ75 light microscope (Leica, Wetzlar, Germany). For histology studies, tissue samples of the abdominal gland or MCF-7 xenografts were fixed in 4% formaldehyde in PBS for 24 h and stored in 70% ethanol at 4 °C until further processing. Tissue was embedded in paraffin and sections of 5 μm were prepared and used for immunofluorescence studies or stained with H&E. H&E sections were observed with a DM LB light microscope (Leica) and pictures were acquired with a DC500 digital camera (Leica).
Immunofluorescence analysis
For immunohistochemistry, paraffin-embedded mammary sections were de-paraffinised and antigen retrieval was performed using boiling 10 mM tri-sodium citrate buffer, pH 6.0, for 10 min. Sections were blocked in 10% normal goat serum (Dako, Glostrup, Denmark) for 1 h at room temperature, incubated with primary antibody overnight at 4 °C and detected using the corresponding secondary antibodies for 1 h at room temperature. Cell nuclei were stained with Hoechst 33 342 (Sigma). For immunocytochemistry, cells were grown in culture glass slides (BD Biosciences), fixed in 4% paraformaldehyde, permeabilised with 0.25% Triton X-100 and immunostained as described above. Primary antibodies against E-cadherin (BD Biosciences), Ki67 (Leica), p-Stat5 and JAK2 (Cell Signaling Technology, Danvers, MA, USA), CK-14 (Abcam, Cambridge, UK), CK18 (Progen, Heidelberg, Germany) and β-casein (kindly donated by Bert Binas, Texas A & M University, College Station, TX, USA) were used. Secondary antibodies used were Cy3 anti-rabbit IgG (Sigma) and Alexa Fluor 488 anti-mouse IgG (Invitrogen). Pictures were acquired on a Zeiss Axioplan 2 microscope (Carl Zeiss, Thornwood, NY, USA).
Western blot analysis
Cell lysates from abdominal mammary glands and cell lines were obtained as described previously,27 subjected to SDS-PAGE and proteins transferred onto polyvinylidene fluoride membranes. The following primary antibodies from Cell Signaling Technology were used: cleaved caspase 3, JAK2, lamin A/C, p-Stats 1, 3 and 5 and Stats 1 and 3. The following primary antibodies were from Abcam: α-tubulin, β-actin and p-Stat6. The following primary antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA) were used: Elf5, Stat6 and WAP. Stat5 antibody was from BD Biosciences and β-casein antibody was a gift from Bert Binas. Secondary HRP-conjugated antibodies were from Dako. Luminograms were obtained with the Amersham Enhanced Chemiluminescence Detection Kit (GE Healthcare, Uppsala, Sweden) or with the Millipore Immobilon Western System (Billerica, MA, USA).
Real-time quantitative-PCR and reverse transcriptase-PCR
RNA was extracted from frozen tissues or cell lines using TRIzol reagent (Invitrogen) and RNeasy Mini Kit (Qiagen, Hilden, Germany). cDNA was prepared using Transcriptor Reverse Transcriptase (Roche Applied Science, Penzberg, Germany). The primers used for reverse transcriptase-PCR (RT-PCR) were: mouse β-casein, 5′-GGCACAGGTTGTTCAGGCTT-3′ and 5′-AAGGAAGGGTGCTACTTGCTG-3′; mouse JAK2, 5′-CATTCTTACCAAAGTGCGTTCG-3′ and 5′-CAAACTGTTCTGTGTAGAAGGCA-3′; and mouse WAP, 5′-CGCTCAGAACCTAGAGGAACA-3′ and 5′-CGGGTCCTACCACAGGAAAC-3′. Mouse cyclophilin A (5′-CCTTGGGCCGCGTCTCCTT-3′ and 5′-CACCCTGGCACATGAATCCTG-3′) and human GAPDH (5′-GGGAAGCTCACTGGCATGGCCTTCC-3′ and 5′-CATGTGGGCCATGAGGTCCACCAC-3) were used as housekeeping genes.
Real-time quantitative PCR (RTQ-PCR) was run in the 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Pre-developed Taqman primers specific for mouse β-casein, MMP-2, MMP-9 and 18S were purchased from Applied Biosystems. Results were normalised according to 18S quantification in the same sample reaction.
Caspase 3 activity
Caspase 3 activity was measured in mammary tissue lysates using the Caspase 3/CPP32 Colorimetric Assay Kit (BioVision, Mountain View, CA, USA). Mammary gland was homogenised in lysis buffer, incubated on ice for 10 min and centrifuged at 10 000 × g for 15 min at 4 °C. Supernatants from tissue homogenates (100 μg of protein) were used to measure caspase 3 activity following the manufacturer's instructions.
Matrigel colony assays
Cells were trypsinised, counted and mixed with cold Matrigel (5000 cells in 70 μl). After the Matrigel became gel at 37 °C, KIM-2 maintenance medium was added to the plate and changed every week. Colonies were visualised after 2 weeks using a Leica MZ75 light microscope.
Soft agar colony assays
Cells were trypsinised, counted and resuspended in KIM-2 maintenance medium containing 0.2% agarose at a concentration of 1000 cells per ml. The mixture was allowed to set at room temperature and then incubated at the indicated temperatures for 3 weeks. Colonies were stained with MTT (Sigma) according to the manufacturer's instructions for better visualisation and counted using with a Leica MZ75 light microscope.
Statistical analysis
ANOVA with a post hoc analysis by the Student–Newman–Keuls' test was routinely used. Unless otherwise stated, results are expressed as mean±S.D.
Abbreviations
- JAK2:
-
janus kinase 2
- Stat:
-
signal transducer and activator of transcription
- p-Stat:
-
phospho-Stat
- WAP:
-
whey acidic protein
- SOCS:
-
suppressor of cytokine signalling
- K14:
-
keratin 14
- CK:
-
cytokeratin
- Elf5:
-
E74-like factor 5
- MECs:
-
mammary epithelial cells
- MMP:
-
matrix metalloproteinase
- PRL:
-
prolactin
- Cav-1:
-
caveolin-1
- MIG:
-
MSCV-IRES-GFP
- CM:
-
complete medium
- CAJAK2:
-
constitutively active JAK2
- GFP:
-
green fluorescent protein
- DMEM:
-
Dulbecco's modified Eagle's medium
- EGF:
-
epidermal growth factor
- H&E:
-
hematoxylin and eosin
- RT-PCR:
-
reverse transcriptase PCR
- RTQ-PCR:
-
real-time quantitative PCR
References
Yamaji D, Na R, Feuermann Y, Pechhold S, Chen W, Robinson GW et al. Development of mammary luminal progenitor cells is controlled by the transcription factor STAT5A. Genes Dev 2009; 23: 2382–2387.
Watson CJ, Neoh K . The Stat family of transcription factors have diverse roles in mammary gland development. Semin Cell Dev Biol 2008; 19: 401–406.
O'Shea JJ, Gadina M, Schreiber RD . Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell 2002; 109 (Suppl): S121–S131.
Wagner KU, Krempler A, Triplett AA, Qi Y, George NM, Zhu J et al. Impaired alveologenesis and maintenance of secretory mammary epithelial cells in Jak2 conditional knockout mice. Mol Cell Biol 2004; 24: 5510–5520.
Sakamoto K, Lin WC, Triplett AA, Wagner KU . Targeting janus kinase 2 in Her2/neu-expressing mammary cancer: implications for cancer prevention and therapy. Cancer Res 2009; 69: 6642–6650.
Sakamoto K, Triplett AA, Schuler LA, Wagner KU . Janus kinase 2 is required for the initiation but not maintenance of prolactin-induced mammary cancer. Oncogene 2010; 29: 5359–5369.
Yu H, Jove R . The STATs of cancer – new molecular targets come of age. Nat Rev Cancer 2004; 4: 97–105.
Diaz N, Minton S, Cox C, Bowman T, Gritsko T, Garcia R et al. Activation of stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin Cancer Res 2006; 12: 20–28.
Cotarla I, Ren S, Zhang Y, Gehan E, Singh B, Furth PA . Stat5a is tyrosine phosphorylated and nuclear localized in a high proportion of human breast cancers. Int J Cancer 2004; 108: 665–671.
Barbieri I, Pensa S, Pannellini T, Quaglino E, Maritano D, Demaria M et al. Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res 2010; 70: 2558–2567.
Vafaizadeh V, Klemmt P, Brendel C, Weber K, Doebele C, Britt K et al. Mammary epithelial reconstitution with gene-modified stem cells assigns roles to Stat5 in luminal alveolar cell fate decisions, differentiation, involution, and mammary tumor formation. Stem Cells 2010; 28: 928–938.
Croker BA, Kiu H, Nicholson SE . SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol 2008; 19: 414–422.
Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010; 464: 999–1005.
Levine RL, Pardanani A, Tefferi A, Gilliland DG . Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer 2007; 7: 673–683.
Bumm TG, Elsea C, Corbin AS, Loriaux M, Sherbenou D, Wood L et al. Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Res 2006; 66: 11156–11165.
Hookham MB, Elliott J, Suessmuth Y, Staerk J, Ward AC, Vainchenker W et al. The myeloproliferative disorder-associated JAK2 V617F mutant escapes negative regulation by suppressor of cytokine signaling 3. Blood 2007; 109: 4924–4929.
Li J, Spensberger D, Ahn JS, Anand S, Beer PA, Ghevaert C et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood 2010; 116: 1528–1538.
Dunbar ME, Dann P, Brown CW, Van Houton J, Dreyer B, Philbrick WP et al. Temporally regulated overexpression of parathyroid hormone-related protein in the mammary gland reveals distinct fetal and pubertal phenotypes. J Endocrinol 2001; 171: 403–416.
Woodward WA, Chen MS, Behbod F, Rosen JM . On mammary stem cells. J Cell Sci 2005; 118 (Part 16): 3585–3594.
Choi YS, Chakrabarti R, Escamilla-Hernandez R, Sinha S . Elf5 conditional knockout mice reveal its role as a master regulator in mammary alveolar development: failure of Stat5 activation and functional differentiation in the absence of Elf5. Dev Biol 2009; 329: 227–241.
Li S, Rosen JM . Nuclear factor I and mammary gland factor (STAT5) play a critical role in regulating rat whey acidic protein gene expression in transgenic mice. Mol Cell Biol 1995; 15: 2063–2070.
Oakes SR, Naylor MJ, Asselin-Labat ML, Blazek KD, Gardiner-Garden M, Hilton HN et al. The Ets transcription factor Elf5 specifies mammary alveolar cell fate. Genes Dev 2008; 22: 581–586.
Uria JA, Werb Z . Matrix metalloproteinases and their expression in mammary gland. Cell Res 1998; 8: 187–194.
Clarkson RW, Boland MP, Kritikou EA, Lee JM, Freeman TC, Tiffen PG et al. The genes induced by signal transducer and activators of transcription (STAT)3 and STAT5 in mammary epithelial cells define the roles of these STATs in mammary development. Mol Endocrinol 2006; 20: 675–685.
Gordon KE, Binas B, Chapman RS, Kurian KM, Clarkson RW, Clark AJ et al. A novel cell culture model for studying differentiation and apoptosis in the mouse mammary gland. Breast Cancer Res 2000; 2: 222–235.
Chapman RS, Lourenco PC, Tonner E, Flint DJ, Selbert S, Takeda K et al. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev 1999; 13: 2604–2616.
Khaled WT, Read EK, Nicholson SE, Baxter FO, Brennan AJ, Came PJ et al. The IL-4/IL-13/Stat6 signalling pathway promotes luminal mammary epithelial cell development. Development 2007; 134: 2739–2750.
Kuraishi T, Mizoguchi Y, Sun Y, Aoki F, Imakawa K, Sakai S . The casein mRNA decay changes in parallel with the poly(A) tail length in the mouse mammary gland. Mol Cell Endocrinol 2002; 190: 101–107.
Burdon TG, Maitland KA, Clark AJ, Wallace R, Watson CJ . Regulation of the sheep beta-lactoglobulin gene by lactogenic hormones is mediated by a transcription factor that binds an interferon-gamma activation site-related element. Mol Endocrinol 1994; 8: 1528–1536.
Harris J, Stanford PM, Sutherland K, Oakes SR, Naylor MJ, Robertson FG et al. Socs2 and elf5 mediate prolactin-induced mammary gland development. Mol Endocrinol 2006; 20: 1177–1187.
Hanahan D, Weinberg RA . The hallmarks of cancer. Cell 2000; 100: 57–70.
Wagner KU, Rui H . Jak2/Stat5 signaling in mammogenesis, breast cancer initiation and progression. J Mammary Gland Biol Neoplasia 2008; 13: 93–103.
Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009; 16: 487–497.
Chen E, Beer PA, Godfrey AL, Ortmann CA, Li J, Costa-Pereira AP et al. Distinct clinical phenotypes associated with JAK2V617F reflect differential STAT1 signaling. Cancer Cell 2010; 18: 524–535.
Iavnilovitch E, Groner B, Barash I . Overexpression and forced activation of stat5 in mammary gland of transgenic mice promotes cellular proliferation, enhances differentiation, and delays postlactational apoptosis. Mol Cancer Res 2002; 1: 32–47.
Park DS, Lee H, Frank PG, Razani B, Nguyen AV, Parlow AF et al. Caveolin-1-deficient mice show accelerated mammary gland development during pregnancy, premature lactation, and hyperactivation of the Jak-2/STAT5a signaling cascade. Mol Biol Cell 2002; 13: 3416–3430.
Haffner MC, Petridou B, Peyrat JP, Revillion F, Muller-Holzner E, Daxenbichler G et al. Favorable prognostic value of SOCS2 and IGF-I in breast cancer. BMC Cancer 2007; 7: 136.
Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu H et al. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res 2010; 70: 3119–3127.
Fernandez I, Touraine P, Goffin V . Prolactin and human tumourogenesis. J Neuroendocrinol 2010; 22: 771–777.
Hayashi K, Matsuda S, Machida K, Yamamoto T, Fukuda Y, Nimura Y et al. Invasion activating caveolin-1 mutation in human scirrhous breast cancers. Cancer Res 2001; 61: 2361–2364.
Acknowledgements
This study was supported by a grant from CRUK-Cambridge Cancer Centre (to CJW and ARG). MMC is the recipient of a Fellowship from Fundación Alfonso Martín Escudero and RZ is funded by Ministerio de Ciencia e Innovación, Spain (BFU2010-18253). We thank Katherine Hughes for the analysis of tumour histology, Nigel Miller for technical advice with flow cytometry, Margaret McLeish and Jestina Makomva for tissue histology and all the members of our lab for their continuous support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Deshmukh
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
Caffarel, M., Zaragoza, R., Pensa, S. et al. Constitutive activation of JAK2 in mammary epithelium elevates Stat5 signalling, promotes alveologenesis and resistance to cell death, and contributes to tumourigenesis. Cell Death Differ 19, 511–522 (2012). https://doi.org/10.1038/cdd.2011.122
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.122
Keywords
This article is cited by
-
A randomized, double-blind, phase 2 study of ruxolitinib or placebo in combination with capecitabine in patients with advanced HER2-negative breast cancer and elevated C-reactive protein, a marker of systemic inflammation
Breast Cancer Research and Treatment (2018)
-
C14orf166 overexpression correlates with tumor progression and poor prognosis of breast cancer
Journal of Translational Medicine (2016)
-
STAT5 Reactivation by Catechin Modulates H2O2-Induced Apoptosis Through miR-182/FOXO1 Pathway in SK-N-MC Cells
Cell Biochemistry and Biophysics (2015)
-
Dysregulated STAT1-SOCS1 control of JAK2 promotes mammary luminal progenitor cell survival and drives ERα+ tumorigenesis
Cell Death & Differentiation (2014)
