
Abstract
Telomere length is critical for chromosome stability that affects cell proliferation and survival. Telomere elongation by telomerase is inhibited by the telomeric protein, TRF1. Tankyrase-1 (TNKS1) poly(ADP-ribosyl)ates TRF1 and releases TRF1 from telomeres, thereby allowing access of telomerase to the telomeres. TNKS1-mediated poly(ADP-ribosyl)ation also appears to be crucial for regulating the mitotic cell cycle. In searching for proteins that interact with polo-like kinase-1 (Plk1) by using complex proteomics, we identified TNKS1 as a novel Plk1-binding protein. Here, we report that Plk1 forms a complex with TNKS1 in vitro and in vivo, and phosphorylates TNKS1. Phosphorylation of TNKS1 by Plk1 appears to increase TNKS1 stability and telomeric poly(ADP-ribose) polymerase (PARP) activity. By contrast, targeted inhibition of Plk1 or mutation of phosphorylation sites decreased the stability and PARP activity of TNKS1, leading to distort mitotic spindle-pole assembly and telomeric ends. Taken together, our results provide evidence of a novel molecular mechanism in which phosphorylation of TNKS1 by Plk1 may help regulate mitotic spindle assembly and promote telomeric chromatin maintenance.
Similar content being viewed by others

Main
Polo-like kinase-1 (Plk1) is a serine/threonine kinase known to have essential functions in Cdk1–cyclin-B complex activation during the G2-to-M phase transition, centrosome separation and maturation, spindle assembly/formation, chromosome segregation, and cytokinesis.1 A striking feature of Plk1 is its localization to numerous sub-cellular structures during mitosis. This factor associates with the centrosome during prophase, is enriched at kinetochores during prometaphase and metaphase, recruited to the central spindle during anaphase, and then accumulates in the mid-body during telophase.1 The physiologic relevance of these changes in localization is unclear. Plk1 overexpression has been observed in a wide range of tumors and is often associated with a poor prognosis.2 Furthermore, PLK1 mutations may play a part in tumorigenesis.3 A growing body of evidence also indicates that targeted interference of Plk1 function induces prolonged mitotic arrest and subsequent apoptotic cell death.4, 5, 6 Thus, Plk1 is an attractive anticancer target, and deregulation of Plk1 appears to be a considerable causative factor for human diseases such as cancer.
Telomeres are essential for genome stability in all eukaryotes. Changes in telomere functions and associated chromosomal abnormalities have been implicated in numerous human diseases and disorders such as aging and cancer.7, 8, 9, 10 TRF1 is a negative regulator of telomere lengthening by telomerase.9, 10 Overexpression of TRF1 accelerates telomere shortening, whereas a dominant-negative inhibitor of TRF1 leads to telomere elongation.9, 10 TRF2 is required to protect chromosomal ends by stabilizing the terminal t-loop structure, telomere sister chromatid exchange (T-SCE), or recombination with interstitial sites.7, 11
Tankyrase-1 (TNKS1) was identified as a TRF1-binding protein from a yeast-two hybrid screen.12 TNKS1 is a member of the poly(ADP-ribose) polymerase (PARP) family of enzymes. PARPs are cytoplasmic enzymes that use NAD+ to synthesize ADP-ribose polymers on protein acceptors in response to DNA damage.13, 14 Poly(ADP-ribosyl)ation (PARsylation) often dramatically alters protein function,15 and is believed to have a role in the maintenance of genome integrity, although the underlying molecular mechanism is still unclear. TNKS1 PARsylates its binding partner TRF1 and in doing so inhibits TRF1 binding to telomeres, thus allowing access of telomerase to telomeres.8, 9, 10, 12 Overexpression of TNKS1 removes TRF1 from telomeres, resulting in TRF1 ubiquitination and degradation by the proteasome.16, 17 Long-term overexpression of TNKS1 leads to telomere elongation, which is dependent on the catalytic PARP activity of TNKS1 and telomerase,16, 18 whereas long-term inhibition of TNKS1 expression results in telomere shortening.19 Thus, TNKS1 acts as a positive regulator of telomere lengthening by antagonizing TRF1.
Recently, accumulating evidence has suggested that the function of TNKS1 might not be restricted to regulating telomere length. As reviewed by Hsiao and Smith,8 TNKS1 localizes to multiple sub-cellular sites and has many diverse binding partners. Depletion of TNKS1 leads to pre-anaphase arrest.20 This phenotype can be rescued by wild-type (WT) TNKS1 but not a PARP-negative mutant, indicating a requirement for PARsylation in mitotic cell-cycle regulation. In addition, TNKS1-depleted mitotic cells are unable to resolve their telomeres despite separation of sister chromatid arms and centromeres. Recent work has also indicated that poly(ADP-ribose) appears to be a key component of mitotic spindle assembly and structure.8, 21 TNKS1 colocalizes with the nuclear mitotic apparatus (NuMA) protein at the spindle poles during mitosis.22 Localization of TNKS1 at the spindle pole is dependent on the NuMA protein. Interestingly, PARsylation of NuMA by TNKS1 seems to influence the structural integrity of spindle poles and may be essential for protein interactions required for spindle formation.21, 22, 23 Together, these findings indicate that TNKS1 might be an essential enzyme responsible for providing poly(ADP-ribose) to the spindle assembly during mitosis and regulating mitotic cell-cycle progression.
A recent study showed that Plk1 phosphorylates TRF1, which appears to be important for TRF1-telomeric DNA binding.24 A recent report showed that the PARP activity of TNKS1 is regulated by cell-cycle-specific association with the NuMA protein during mitosis. TNKS1 is phosphorylated during mitosis,23, 25 raising the possibility that phosphorylation might influence its PARP activity. However, the mechanisms regulating TNKS1 PARP activity are unclear. In particular, it is unknown whether selective mitotic kinases, such as Aurora kinases, Plk1, and BubR1, are involved in the phosphorylation of TNKS1, or whether mitotic phosphorylation of TNKS1 affects its PARP activity. Here we report that Plk1 directly interacts with and phosphorylates TNKS1, and that Plk1–TNKS1 interaction regulates TNKS1 stability and PARP activity. These findings indicate that Plk1-mediated phosphorylation of TNKS1 is essential for spindle assembly during cell-cycle progression and the regulation of telomere length.
Results
Plk1 forms a complex with TNKS1 in vitro and in vivo
To identify novel Plk1 substrates and/or interacting proteins, we used complex proteomics. Briefly, lysates from HeLa cells stably overexpressing HA-tagged Plk1 were immunoprecipitated using an anti-HA antibody (Figure 1a). The eluted HA–Plk1 immunocomplex was separated by SDS-PAGE. Subsequent matrix-assisted laser desorption ionization–time of flight (MALDI–TOF) analysis led to the identification of novel Plk1-interacting molecules, including TNKS1 polypeptides (Figure 1b).

Plk1 interacts with TNKS1 in vitro and in vivo. (a) Lysates from control AGS (a gastric cancer cell line) cells and ones stably overexpressing HA-tagged Plk1 (AGS-HA-Plk1) were prepared for immunoprecipitation using an anti-HA antibody and subjected to immunoblot analysis. The arrowhead and the asterisk indicate HA-PlK1 and IgG heavy chains, respectively. (b) Lysates from the AGS-HA-Plk1 cells were used for immunoprecipitaion using an anti-HA antibody. The eluted HA-Plk1 immune complexes were analyzed by SDS-PAGE and subsequent Coomassie staining followed by mass spectrometric analysis. TNKS1, HA-Plk1, and TCTP (as a positive control) polypeptides are indicated by arrows. (c) Schematic diagrams of TNKS1. HPS, homopolymeric tracts of histidine, proline, and serine repeats; ANK, ankyrin domain; SAM, sterile alpha motif; PARP, poly(ADP-ribose) polymerase. Purified His-tagged Plk1 were incubated with bead-bound GST or GST-TNKS1 (GST-TNKS1) fusion proteins. The eluted GST and GST fusion proteins were resolved by SDS-PAGE prior to immunoblot analyses. (d) Schematic diagrams of Plk1. GST and GST-Plk1 fusion proteins were incubated with HeLa cell lysates cultured in the presence of nocodazole for 12 h. Bead-bound GST or GST-Plk1s were washed, resolved by SDS-PAGE, and immunoblotted using anti-TNKS1 antibody. (e) HeLa cells were cultured in the absence (Asyn) or presence (Noco) of nocodazole. Lysates were incubated with purified GST or GST-Plk1 fusion protein. Bead-bound GST or GST-Plk1s were washed, resolved by SDS-PAGE, and immunoblotted using anti-TNKS1 antibody. (f) HeLa cells were cultured in the absence (Asyn) or presence (Noco) of nocodazole. Lysates were immunoprecipitaed using normal IgG (negative control) or anti-Plk1 antibody. The immunocomplexes were resolved by SDS-PAGE and immunoblotted using and-Plk1, anti-TNKS1, and anti-actin antibodies. (g) Extracts were prepared from HeLa cells synchronized at mitosis by nocodazole, and fractionated by gel filtration using a Superdex-200 column. Fractions (numbered 7–24) were analyzed by immunoblotting using antibodies against TNKS1 (TNKS1), NuMA, PARP, Plk1, and Aurora-A (Aur-A). Input indicates 4% of the extract
To initially assess the importance of this putative Plk1–TNKS1 interaction, we generated GST-TNKS1 (GST-TNKS1) fusion proteins, which were incubated with purified His-Plk1 (Figure 1c). Pull-down assays revealed that GST-TNKS1 (158–595) and (596–1022), which contained the ankyrin domain (ANK), did in fact bind to His-Plk1. Similarly, we purified GST-Plk1 fusion proteins, which were incubated with cellular extracts from synchronized HeLa cells. As shown in Figure 1d, polypeptides containing the polo-box-binding domain (PBD) of Plk1 formed a complex with TNKS1, unlike the NH2-terminal S/T kinase domain. In addition, we treated asynchronized HeLa cells with the microtubule-inhibiting agent, nocodazole (Figure 1e). Nocodazole clearly induced the hyper-phosphorylation of TNKS1, as reported previously.23, 26 Pull-down assays showed that GST-Plk1 efficiently interacted with endogenous TNKS1, and interaction between these proteins was observed in both synchronized and asynchronized cells. Furthermore, we immunoprecipitated endogenous Plk1 from cells treated with or without nocodazole and recovered Plk1–TNKS1 complexes (Figure 1f).
Gel filtration was used to further evaluate the interaction between Plk1 and TNKS1. Endogenous Plk1, TNKS1, NuMA protein, PARP, and Aurora-A proteins from asynchronized HeLa cells were size-fractionated and constituent proteins were identified by immunoblotting (Figure 1g). We detected both TNKS1 and Plk1 in the higher molecular mass fractions (in particular, fractions 10 and 11); it was determined that the TNKS1-interacting protein NuMA was also isolated. However, TNKS2 was detected in the mid-molecular mass range. PARP was observed in the mid-molecular mass range, which lacked Plk1, but not Aurora-A. These results imply that Plk1 was likely in the same cellular fraction as TNKS1. In summary, our data indicate that Plk1 forms a complex with TNKS1 in vitro and in vivo.
Hyper-phosphorylation of TNKS1 during mitosis may be mediated by Plk1
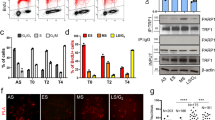
To determine whether the Plk1–TNKS1 interaction leads to TNKS1 phosphorylation, HeLa cells were first synchronized by aphidicolin, released in normal culture media, and lysed at each cell-cycle stage (Figure 2a). The lysates were analyzed by immunoblotting using anti-TNKS1, anti-Plk1, anti-Aurora-A, and anti-cyclin-B1 antibodies (Figure 2b). TNKS1 was gradually hyper-phosphorylated following release from aphidicolin treatment (indicated by arrowheads). This hyper-phosphorylation appeared to be maximized in the mitotic cell population. As expected, the levels of the mitotic kinases Aurora-A, Cdk1, and Plk1 were also increased according to the hyper-phosphorylation of TNKS1. In addition, we comparatively induced TNKS1 hyper-phosphorylation with a separate set of microtubule inhibitors, taxol and nocodazole (Figure 2c). Immunoblotting assays clearly showed marked TNKS1 hyper-phosphorylation in cells treated with microtubule inhibitors. Based on this information, it was important to determine whether hyper-phosphorylation of TNKS1 induced by the microtubule inhibitors was compromised by mitotic kinase inhibitors. Therefore, we treated HeLa cells with nocodazole, ZM447439 (an Aurora kinase inhibitor), TDZD (a GSK3 inhibitor), purvalanol-A (a Cdk1 inhibitor), BI2536 (a Plk1 inhibitor), or purpurogallin (PPG; an inhibitor of polo box domain-dependent recognition). In addition, the cells were also treated with a combination of nocodazole and each of the other five other reagents. As shown in Figure 2d, immunoblotting of the lysates from cells treated with inhibitors that selectively antagonized Aurora kinases or GSK3 showed no changes in TNKS1 phosphorylation. However, treatment with Plk1 (BI2536 or PPG) and its priming kinase, a Cdk1 (purvalanol-A) inhibitor, sharply reduced the hyper-phosphorylation of TNKS1 induced by the microtubule inhibitors.

Plk1 and its priming kinase, Cdk1, are capable of phosphorylating TNKS1. (a and b) HeLa cells were treated with aphidicolin (Aphi; 1.6 μg/ml) for 14 h and then released (Aphi/Rel). Cells were harvested at the indicated time points, stained with propidium iodide (PI), and analyzed by flow cytometry to determine their DNA content. (a) Asyn, Asynchronized cells. (b) Lysates from asynchronized or synchronized HeLa cells were analyzed by immunoblotting using anti-TNKS1 (TNKS1), anti-Plk1, anti-Aurora-A (Aur-A), anti-cyclin-B1 (Cyc-B1), anti-BubR1, anti-Bub3, or anti-actin antibodies. (c) HeLa cells were treated with paclitaxel (Taxol, 33 nM) or nocodazole (Noco, 200 ng/ml) for 16 h. Lysates from the asynchronized or synchronized HeLa cells were analyzed by immunoblotting using the indicated antibodies. Asyn, Asynchronized cells. (d) HeLa cells were treated with the various cell-cycle kinase inhibitors for 12 h as indicated. Lysates were immunoblotted using anti-TNKS1 (TNKS1) and anti-actin antibodies. (e) HeLa cells were treated with (Noco) or without (Asyn) nocodazole (100 ng/ml) for 16 h. Cellular extracts were incubated with or without or lambda phosphatase (PPase) and immunoprecipitated using anti-TNKS1 antibody. The eluted TNKS1 protein was resolved by SDS-PAGE and detected using antibodies against TNKS1, phospho-Ser (P-Ser), phospho-Thr (P-Thr), and actin. (f) HeLa cells were treated with nocodazole and then with purvalanol-A (25 μM) or purpurogallin (PPG, 50 μM). Lysates were immunoprecipitated using an anti-TNKS1 antibody, and the resulting TNKS1 protein was probed using anti-phospho-Ser (P-Ser), anti-phospho-Thr (P-Thr), anti-TNKS1 (TNKS1), and anti-actin antibodies
To further examine whether the slower migrating TNKS1 polypeptide observed during mitosis is the hyper-phosphorylated form, we immunoprecipitated endogenous TNKS1 from cell lysates recovered from both asynchronized and mitotic HeLa cells treated with lambda phosphatase. As shown in Figure 2e, the levels of slower migrating TNKS1 polypeptides appearing in mitotic cells, and recognized by both anti-phospho-Thr and anti-phospho-Ser antibodies, were clearly reduced by lambda phosphatase treatment. However, we were able to exclude the possibility that the appearance of the slower migrating TNKS1 polypeptide corresponding to the hyper-phosphorylated form was not due to other protein modification such as poly(ADP-ribosyl)ation (Supplementary Figure S1). Furthermore, hyper-phosphorylated TNKS1 recognized by anti-phospho-Thr and anti-phospho-Ser was not detected in mitotic HeLa cells cultured in the presence of the selective Plk1 and Cdk1 inhibitors, purvalanol-A and PPG (Figure 2f). Together, these results indicate that hyper-phosphorylation of TNKS1 during mitosis is mediated by Plk1.
Plk1 phosphorylates TNKS1 in vitro and in vivo
To examine whether Plk1 and its priming kinase, Cdk1, directly phosphorylate TNKS1, we first incubated lysates from asynchronized HeLa cells with purified recombinant Plk1 or Cdk1/cyclin-B and cold ATP in the presence or absence of a selective Cdk or Plk1 inhibitor. As shown in Figure 3a, hyper-phosphorylation of TNKS1 polypeptides was clearly observed in the cell lysates incubated with purified Cdk1/cyclin-B or Plk1, but was abolished in cells additionally treated with a selective Cdk or Plk1 inhibitor, indicating that Cdk1 and Plk1 phosphorylate TNKS1. In addition, we purified His-tagged full-length TNKS1, and then incubated this protein with Cdk1/cyclin-B or Plk1 and [γ32P]ATP (Figure 3b). We observed that Plk1 and its priming kinase Cdk1/cyclin-B efficiently phosphorylated TNKS1. Furthermore, we incubated a series of GST-TNKS1 fusion proteins and GST-TRF1 (as a control) with purified recombinant Plk1 in the presence of [γ32P]ATP (Figure 3c). Interestingly, fragments containing the Plk1-binding region and the PARP activity motif of TNKS1 (residues 596–1022 and 1023–1327) were clearly phosphorylated by Plk1 in vitro.

Plk1 directly phosphorylates TNKS1 in vitro and in vivo. (a) Lysates from asynchronized HeLa cell were incubated with 0.2 mM cold ATP either alone (lane-2) or in combination with purified Cdk1 (1 μg), Cdk1 plus purvalanol-A (25 μM), Plk1 (1 μg), or Plk1 (1 μg) plus BI2536 (300 nM) at 37 °C for 1 h. Samples were separated on 8% SDS-PAGE gel and subsequently immunoblotted using anti-TNKS1 and anti-His antibodies. As control, TNKS1 hyper-phosphorylated was observed in nocodazole-treated (Noco) HeLa cells as indicated by an arrowhead and asterisk. (b) Purified His-TNKS1 was incubated with either recombinant Cdk1 and cyclin-B1 (Cdk1) or Plk1 in the presence of [γ32P]ATP. Protein samples were resolved by SDS-PAGE and visualized by autoradiography. (c) Purified GST-fused TNKS1 mutant proteins (amino acids 1–157, 158–595, 596–1022, and 1023–1327) or the GST-TRF1 fusion protein (as a positive control) were incubated with recombinant His-Plk1 protein in the presence of [γ32P]ATP. GST fusion proteins were resolved by SDS-PAGE and visualized by Coomassie blue staining (left panel) or autoradiography (right panel). (d) Schematic diagrams of TNKS1, including HPS, ANK, SAM, and PARP motifs. Eight putative sites phosphorylated by Plk1 are indicated. (e) Eight putative phosphorylation sites, T643, T785, T839, T930, T938, S978/T982, or T1128, were mutated to alanine (A) using plasmids encoding GST-TNKS1 (596–1022) and GST-TNKS1 (1023–1327). Purified GST-TNKS1 proteins were incubated with recombinant Plk1 in the presence of [γ32P]ATP. Protein samples were resolved by SDS-PAGE and visualized by autoradiography. (f) All five putative phosphorylation sites, T839, T930, S978/T982, and T1128, were mutated in GST-TNKS1 (596–1327) to generate GST-TNKS1 (596–1327)-5A. Purified WT GST-TNKS1 (596–1327) and GST-TNKS1 (596–1327)-5A mutant proteins were incubated with recombinant Plk1 in the presence of [γ32P] ATP and visualized by autoradiography
The amino-acid sequence of TNKS1 includes a number of potential sites phosphorylated by Plk1. To explore their functionality, we generated plasmids encoding glutathione-S-transferase (GST)-fused TNKS1 (GST-TNKS1) mutants, in which each of the eight putative phosphorylation sites was mutated (Figure 3d). Interestingly, replacing T839, T930, S978/T982, or T1128 with alanine (A) significantly reduced phosphorylation by Plk1 in vitro. However, each of those residues failed to completely abolish TNKS1 phosphorylation by Plk1 (Figure 3e). Surprisingly, mutations of all five putative phosphorylation sites (T839, T930, S978/T982, and T1128) sharply prevent the phosphorylation of TNKS1 (596–1327; Figure 3f). Together, these data suggest that Plk1 promotes TNKS1 phosphorylation, and at least five residues of TNKS1 are involved in this process.
Plk1 regulates TNKS1 protein stability
To analyze the effect of Plk1-mediated phosphorylation on TNKS1 function, we transfected shRNA specifically targeting Plk1 into HeLa cells, which were then cultured in the presence or absence of nocodazole (Figure 4a). Interestingly, Plk1 depletion dramatically reduced hyper-the phosphorylation and protein expression of TNKS1, but slightly reduced the levels of TRF1 protein, particularly in cells treated with a microtubule inhibitor. The level of exogenous TRF1 protein was not affected by depletion or overexpression of Plk1 (Supplementary Figure S2). In addition, inhibition of TNKS1 expression did not alter the level of Plk1 protein or its sub-cellular localization (Supplementary Figures S3 and S4).

Plk1 contributes to the regulation of TNKS1 stability. (a) HeLa cells were transfected with shRNA specifically targeting luciferase (shLuc) or Plk1 (shPlk1). Forty-eight hours after transfection, the cells were cultured in the absence (Asyn) or presence (Noco) of nocodazole for another 16 h. Lysates were prepared and analyzed by immunoblotting using the indicated antibodies. (b) HeLa cells were transfected with shLuc or shPlk1, treated with nocodazole, and then released in normal culture media containing the proteasome inhibitor MG132 (10 μm/ml) or DMSO. Cell lysates were analyzed by immunoblotting using anti-TNKS1, anti-BubR1 (a positive control), anti-Plk1, anti-GSK3β (a negative control), and anti-actin antibodies. (c) HeLa cells were treated with the proteasome inhibitor, MG132 (10 μm/ml), for 6 h, and then harvested for immunoblot analysis using antibodies against TNKS1, Plk1, NuMA, TRF1, and actin. (d) HeLa cells were transfected with shLuc or two different shPlk1s, shPlk1 #1 (targeting Plk1 ORF) and shPlk1 #2, (targeting Plk1 3′-UTR), as described under Materials and Methods. Thirty-six hours after transfection, lysates were prepared and subjected to immunoblot analysis using anti-TNKS1, anti-Plk1, anti-TRF1, anti-actin antibodies. (e) HeLa cells were co-transfected with shRNA specific for Plk1 3′-UTR (shPlk1 #2) and expression plasmids encoding shRNA-insensitive (shi), HA-tagged WT Plk1 (HA-Plk1 WT) or a kinase-dead mutant of Plk1 (HA-Plk1 K82R). Thirty-six hours after transfection, lysates were prepared and subjected to immunoblot analysis using the indicated antibodies. (f) U2OS cells were immunostained using an anti-Plk1 (green) or an anti-TNKS1 (TNKS1, red) antibody, or CREST serum (purple). The cells were examined by confocal microscopy. (g) HeLa cells were transfected with shLuc (a negative control) or shPlk1. Thirty-six hours after transfection, the cells were fixed with 4% paraformaldehyde and immunostained using anti-Plk1 (green) or anti-TNKS1 (red) antibodies
Next, we transfected HeLa cells with either shLuc or shPlk1, and then synchronized the cells with nocodazole followed by release in normal culture media (Figure 4b). Immunoblot analyses revealed that Plk1 depletion sharply reduced the hyper-phosphorylation and protein expression of TNKS1. Similar to previous reports,27 both BubR1 and GSK3â were also dysregulated in Plk1-depleted cells. Interestingly, treatment with a proteasome inhibitor markedly increased the levels of TNKS1 but not TRF1 and Plk1, implying that TNKS1 protein stability mediated by Plk1 is regulated by a protein proteasomal degradation pathway (Figure 4c). In addition, we included two different sets of shPlk1, namely shPlk1 #1 and #2, and our immunoblotting results revealed that TNKS1 stability was reduced by specific inhibition of Plk1 expression (Figure 4d). Next, we overexpressed shRNA-insensitive version of HA-tagged WT Plk1 or a kinase-dead mutant (K82R) into HeLa cells in endogenous Plk1 was depleted by shPlk1 transfection (Figure 4e). As expected, overexpression of WT Plk1 significantly increased the protein levels of TNKS1. Importantly, exogenous expression of a kinase-dead Plk1 mutant sharply decreased the stability of TNKS1, thereby demonstrating its dominant-negative effect on TNKS1 stability. Together, these data indicate that depletion of Plk1 inhibits not only the phosphorylation but also the stability of the TNKS1 protein.
To further confirm the role of Plk1 in TNKS1 stability regulation, we immunostained HeLa cells, transfected with either shLuc or shPlk1, with anti-Plk1 and anti-TNKS1 antibodies. The cells were also stained with CREST serum as a marker of kinetochore protein and DAPI to visualize DNA (Figures 4f and g). It has been reported recently that TNKS1 localizes to multiple sub-cellular sites including the spindle poles and the mid-body.22, 26 As shown in Figure 4f, TNKS1 distinctly localized at the spindle poles from interphase to metaphase, and at the mid-body from anaphase to telophase. These patterns of TNKS1 sub-cellular localization were almost identical to those of Plk1. As shown in Figure 4g, depletion of Plk1 clearly reduced the levels of TNKS1 at the spindle poles, indicating that the stability of TNKS1 is regulated by Plk1 in vivo. However, it is still unclear how the sub-cellular localization of TNKS1 to the spindle poles and the mid-body relates to its poly(ADP-ribosyl)ation activity at the telomeres.
Poly(ADP-ribosyl)ation activity of TNKS1 is regulated by Plk1-mediated hyper-phosphorylation
To examine whether Plk1-mediated phosphorylation of TNKS1 affects its poly(ADP-ribosyl)ation activity, lysates from asynchronized and mitotic HeLa cells were first analyzed by immunoblotting. As shown in Figure 5a, TNKS1 was effectively phosphorylated during mitosis and simultaneously enhanced PAR levels, indicating that mitotic phosphorylation of TNKS1 appears to affect intracellular PAR levels. Based on this information, we transfected shPlk1 or shLuc (as a negative control) into HeLa cells that were then cultured in the absence or presence of nocodazole (Figure 5b, left panel). Our immunoblot assay using anti-PAR antibody showed that depletion of Plk1 significantly decreased PAR in both asynchronized and synchronized (at mitosis) cells. By contrast, HeLa cells overexpressing Plk1 had significantly augmented intracellular PAR levels (Figure 5b, right panel).

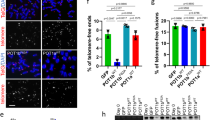
Poly(ADP-ribosyl)ation activity of TNKS1 is regulated by Plk1-mediated hyper-phosphorylation. (a) HeLa cell lysates were prepared in a nuclear extraction buffer from asynchronous (Asyn) or nocodazole-treated (Noco, 200 ng/ml) cells. Lysates were prepared and analyzed by immunoblotting using anti-TNKS1, anti-PAR, or anti-actin antibodies (as a loading control). (b) HeLa cells were transfected with shRNA (shLuc or shPlk1; left panel), pHA, or pHA-Plk1 (right panel). Forty-eight hours after transfection, the cells were cultured in the absence (Asyn) or presence (Noco) of nocodazole for another 16 h. Lysates were separated by electrophoresis and immunoblotted using anti-PAR and anti-actin antibodies. (c) HeLa cells were transfected with shLuc or shPlk1, and cultured in the absence (Asyn) or presence (Noco) of nocodazole for an additional 16 h. Lysates (4 mg of total protein) were used for immunoprecipitation (IP) using anti-TNKS1 antibody (áTNKS1) and analyzed by immunoblotting using anti-Plk1, anti-actin, anti-PAR, or anti-TNKS1 antibodies. (d) Asynchronous (Asyn) or nocodazole-treated (Noco) HeLa cells were further incubated in the absence (DMSO) or presence of a Plk1 inhibitor (50 μM PPG). Lysates were prepared, resolved by SDS-PAGE, and analyzed by immunoblotting using anti-PAR and anti-actin antibodies. (e) Telomeric PNA FISH analysis (FISH using peptide nucleic acid probes) was performed using metaphase spreads of H1299 cells treated with BI2536 (60 nM) or PPG (50 μM) for 12 h, swollen in hypotonic buffer, and fixed in methanol-acetic acid solution. Telomeric repeats were detected by using a Cy3-(CCCTAA)3 PNA probe (red) and DNA was stained with DAPI (blue). (f) Representative images of normal sister telomeres and telomeric fusion in the metaphase spreads of Plk1 inhibitor-treated H1299 cells. Note that in some cases sister telomeric fusions appeared at both ends of the chromosome. (g) The chromosome number distribution of sister telomeric fusions observed in each metaphase was determined and plotted in a graph. More than 100 cells were scored by FISH
To ensure the specificity of TNKS1 PAR, lysates from HeLa cells transfected with shLuc or shPlk1 and subsequently treated with nocodazole were immunoprecipitated using an anti-TNKS1 antibody. The eluted TNKS1 immune complexes were analyzed by immunoblotting (Figure 5c). TNKS1 PARP activity was sharply inhibited by Plk1 depletion. In addition, we treated HeLa cells with the selective Plk1 inhibitor, PPG, and lysates from these cells were immunoblotted using an anti-PAR antibody (Figure 5d). As expected, PAR was clearly perturbed by treatment with PPG. Therefore, these data suggest that targeted inhibition or overexpression of Plk1 leads to dramatic alterations of TNKS1-mediated PARP activity.
Because TNKS1 binds to the telomeric protein TRF1 and, like TRF, associates with telomeres in human cells,12 we used fluorescence in situ hybridization (FISH) analysis of metaphase chromosomes to determine whether selective inhibition of Plk1 alters chromosome ends. H1299 cells were treated with two Plk1 inhibitors, BI2536 and PPG, and then cultured in the presence of a microtubule inhibitor (Figures 5e–g). H1299 cells in metaphase were fixed and then hybridized with a Cy3-labeled telomeric PNA probe. As shown in Figure 5e, telomeric repeats were clearly detected at both metaphase chromosome ends in the control cells (left panel). However, inhibition of Plk1 activity profoundly altered the ends of the metaphase chromosomes (middle and right panels), resulting in telomeric fusion (Figure 5f). Most of the control-treated cells (about 80%) had normal telomeric ends, whereas the Plk1 inhibitor-treated cells predominantly contained abnormal telomeric fusions (Figure 5g). These data strongly suggest that Plk1 regulates TNKS1 activity and stability, and contributes to telomeric complex formation.
Absence of Plk1-mediated phosphorylation leads to reduced TNKS1 protein stability and poly(ADP-ribosyl)ation activity
To determine whether mutations of phosphorylation sites compromise TNKS1 function at telomeres during mitosis, we generated plasmids encoding GFP-fused, WT TNKS1 (GFP-TNKS1 WT) or GFP-TNKS1-5A, a Plk1-mediated, phosphorylation-defective mutant in which the five phosphorylation sites were replaced with alanine (Figure 6a). Interestingly, overexpression of GFP-TNKS1-5A led to enlarged cell size and reduced proliferation potential compared with GFP-TNKS1 WT (Figure 6b). These phenoptypes were, in part, similar to that of TNKS1-depleted cells.22 Next, we compared the protein stability of GFP-TNKS1 WT and GFP-TNKS1-5A in asynchronized HeLa cells treated with cycloheximide, a protein translation inhibitor (Figure 6c). As expected, the levels of TNKS1-5A rapidly declined and were almost undetectable 5 h after treatment, whereas those of TNKS1 WT were slightly reduced but still detected 6 h after treatment. However, it is important to note that Plk1-deficient cells harbored less TNKS1 and could be correlated with the reduced PARP activity of TNKS1.

Mutations of TNKS1 Plk1-mediated phosphorylation sites reduce TNKS1 stability and alter spindle-pole localization and telomere separation. (a) Plasmids encoding GFP-fused full-length TNKS1 WT (GFP-TNKS1 WT), TNKS1-5A, or TNKS1 PD were transfected into HeLa cells. Proteins in the lysates from the cells were separated by electrophoresis and immunoblotted using anti-GFP and actin antibodies. (b) HeLa cells transfected with plasmids encoding GFP-TNKS1 WT or GFP-TNKS1-5A were observed by phase-contrast microscopy 60 h after transfection. (c) Asynchronized HeLa cells were transfected with plasmids encoding GFP-TNKS1 WT or GFP-TNKS1-5A and then treated with cycloheximide (CHX). Lysates were analyzed by immunoblotting using anti-GFP and anti-actin antibodies. (d) Transfected HeLa cells described in panel a were immunostained using anti-Aurora-A (red, left panels) or anti-hTRF1 (red, right panels) antibodies and DNA was visualized by DAPI staining. The cells were examined by confocal microscopy. (e) HeLa cells were transfected with GFP-TNKS1 WT, GFP-TNKS1-5A or GFP-TNKS1 PD, and treated then with nocodazole. Cellular extracts were immunoprecipitated using anti-GFP antibody. The eluted GFP-TNKS1 proteins were resolved by SDS-PAGE and probed using antibodies against TNKS1, PAR, and actin. (f and g) HeLa cells were co-transfected with shRNA specifically targeting TNKS1 3′-UTR and expression plasmids encoding shRNA-insensitive (shi), WT GFP-TNKS1 (shi TNKS1 WT) or GFP-TNKS1-5A (shi TNKS1 5A). Telomeric DNA FISH analysis (FISH using peptide nucleic acid probes) was performed on metaphase spreads of transfected HeLa cells swollen in hypotonic buffer and fixed in a methanol-acetic acid solution. Telomeric repeats were detected by using a Cy3-(CCCTAA)3 PNA probe (red) and DNA was stained with DAPI (blue). (f) Representative images of normal sister telomeres (in cells expressing TNKS1 WT) and telomeric fusions (in cells expressing TNKS1-5A mutant) of metaphase spreads. Note that in some cases sister telomeric fusions appeared at both ends of the chromosomes. (g) The chromosome number distribution of the sister telomeric fusions observed in each metaphase was calculated and plotted on a graph. More than 100 cells from each transfectant cell line were scored by FISH
TNKS1 localizes to the mitotic spindle poles and telomeres.22, 26 Therefore, we transfected HeLa cells with TNKS1 WT, TNKS1-5A, or the PARP-dead mutant TNKS1 PD22, 28 to examine whether the inability to be phosphorylated by Plk1 affects TNKS1 sub-cellular localization in spindle poles and telomeres. Immunostaining analyses revealed that mitotic HeLa cells expressing TNKS1-5A had sharply reduced levels of TNKS1 localization at the spindle poles, whereas cells expressing TNKS1 WT or TNKS1 PD showed comparable TNKS1 localization at the spindle poles (Figure 6d, left panel). In addition, both TNKS1 WT and TNKS1 PD localized to the telomeres during interphase, similar to hTRF1. However, we were unable to observe a colocalization of the TNKS1-5A mutant with hTRF1 at the telomeres (Figure 6d, right panel). These results suggest that the absence of Plk1-mediated phosphorylation severely disrupts the stability and the sub-cellular distribution of TNKS1.
To further determine whether abolishing Plk1-mediated phosphorylation affects TNKS1 PARP activity, we expressed GFP-labeled TNKS1 WT, TNKS1-5A, and TNKS1 PD in HeLa cells, and subsequently treated them with nocodazole to enrich the mitotic population. The tagged proteins were immunoprecipitated using an anti-GFP antibody (Figure 6e). Immunoblot analysis using an anti-PAR antibody revealed that mutation of the Plk1-mediated phosphorylation sites significantly abolished TNKS1 PAR similar to the PARP-dead mutant (TNKS1 PD), whereas TNKS1 WT strongly showed PAR. We also performed FISH analysis to determine whether expression of the TNKS1-5A mutant leads to alteration of the chromosome ends. We expressed shRNA-insensitive, TNKS1 WT or the TNKS1-5A mutant in HeLa cells depleted of endogenous TNKS1 by shTNKS1 specifically targeting the TNKS1 3′-UTR (Figures 6f and g). As expected, overexpression of the shRNA-insensitive, WT TNKS1 (shi TNKS1 WT) led to significant recovery from the effects of TNKS1 depletion such as telomeric fusion resulting from altered chromosome ends. However, overexpression of the TNKS1-5A mutant was unable to reduce the number of abnormal telomeric fusions. Taken together, these data strongly indicate that Plk1-mediated phosphorylation of TNKS1 contributes to protein stability and PARP activity.
Discussion
The PARP activity of TNKS1 appears to be subjected to a different type of regulation dictated by its binding partners and post-translational modification.8 In addition, recent studies have found that PAR is enriched in mitotic spindles, and inhibition of TNKS1 PARP activity results in prolonged mitotic arrest along with loss of bipolar spindle assembly. These observations indicate that TNKS1 polymerization of poly(ADP-ribose) is required for mitotic progression and spindle assembly.21, 29 However, little information exists regarding how the PARP activity and post-translational modification of TNKS1 are regulated, and how TNKS1 is phosphorylated during mitosis. In this study, we showed that Plk1 positively regulates the stability and the PARP activity of TNKS1 by phosphorylation, thereby preventing abnormal telomeric fusion. Recently, it has been demonstrated that depletion or inactivation of Plk1 inhibits the complete separation of sister chromatids.30, 31, 32 Similarly, depletion of TNKS1 prevents the separation of sister chromatids, particularly at the telomeres.20 Interestingly, the cells treated with a Plk1-selective inhibitor showed similar abnormal telomeric fusion. Furthermore, overexpression of the TNKS1 mutant, which could not be phosphorylated by Plk1, did not prevent abnormal telomeric fusion induced by TNKS1 depletion, and showed highly reduced stability and spindle pole localization. These results strongly support our hypothesis that Plk1-mediated phosphorylation contributes to TNKS1 stability and PARP activity, leading to telomeric chromatin maintenance and spindle-pole assembly.
Several recent studies have shown that TNKS1 expression is upregulated in many human cancers such as breast and colorectal cancer, and non-Hodgkin's lymphomas.33, 34 These findings suggesting that there could be an association between aberrant regulation of TNKS1 expression and human cancers. Importantly, a number of studies have also reported that Plk1 is highly upregulated in a wide range of human cancers and is often associated with poor prognosis.2 In addition, TNKS1 shows almost identical sub-cellular localization to that of Plk1 during mitosis. TNKS1 colocalizes with Plk1 at the spindle poles and centrosomes from prophase to metaphase, and at the mid-body during telophase. Together, these findings strongly support our proposed mechanism that the physiologic activity of TNKS1 is associated with a Plk1-dependent signaling pathway. Indeed, our results showed that the Plk1-mediated phosphorylation of TNKS1 is associated with its stability and PARsylation activity, whereas these are dramatically decreased by targeted inhibition of Plk1. Therefore, it may be possible that aberrant Plk1 expression directly influences the PARP activity of TNKS1 and the PARsylation of its protein-binding partner. Our data partially support the notion that aberrant expression of Plk1 is highly associated with dysregulated TNKS1 expression in human cancer cells (Supplementary Figure S5).
A recent approach to identifying Plk1-interacting proteins by using a yeast two-hybrid system demonstrated that Plk1 is associated with TRF1, and that Plk1-mediated phosphorylation of TRF1 is important for TRF1 binding to telomeres.24 These observations indicate that Plk1 may to have a bona fide role in telomeric protein phosphorylation and regulation. Our in vitro binding assay showed that the ANK repeat of TNKS1 may be responsible for interaction with Plk1, raising the possibility that Plk1 might compete with TRF1 for binding to the ANK repeat. However, interaction of TNKS1 with TRF1 was independent of TNKS1 Plk1-mediated phosphorylation (Supplementary Figure S6). Moreover, a previous report indicated that phosphorylation of TRF1 by Plk1 increases the telomeric DNA binding of TRF1.24 This seems to contradict the physiologic role of Plk1-mediated TNKS1 phosphorylation in terms of regulating telomere length. Further studies will be required to better understand the physiologic mechanisms underlying the Plk1-mediated phosphorylation of TRF1 and TNKS1. In particular, it is important to understand whether TRF1 and TNKS1 phosphorylation events are interrelated or independent. However, based on the data shown in Supplementary Figure S6, we can exclude the possibility that the phosphorylation of TNKS1 by Plk1 does not prevent interaction with TRF1 or vice versa. In summary, our study suggests that Plk1 regulates TNKS1 activity and stability, thereby contributing to the formation of telomeric complexes and mitotic cell-cycle progression. Thus, Plk1–TNKS1 signaling may be important for regulating mitotic progression and telomere length, and could be an attractive target for anticancer therapy.
Materials and Methods
Plasmid construction and antibodies
The full-length cDNA sequence of the human Plk1 gene has been published previously.35 cDNA fragments of the Plk1 gene were PCR-amplified and inserted into the pCMV-HA vector to generate pCMV-HA-Plk1 WT. A Plk1 kinase-dead mutant, K82R, was generated by site-directed mutagenesis using pCMV-HA-Plk1 WT as the template. An HA-tagged Plk1 construct was sub-cloned into a pIRESpuro3 vector using pcDNA3-HA-Plk1 to create stable cell lines. His-tagged Plk1 fusion constructs were cloned into a pFastBac vector (Invitrogen, Carlsbad, CA, USA), and used to generate a recombinant baculovirus in Sf9 insect cells by using the Bacto-Bac baculovirus system (Invitrogen). GST fusion constructs for expression in Escherichia coli were generated by in-frame insertion of PCR fragments encoding Plk1 amino-acid residues 1–603, 1–308, and 309–603 into a pGEX-KG vector (Amersham Pharmacia, GE Healthcare, Little Chalfont, UK).
The full-length cDNA sequence of the human Tankyrase-1 gene has been described previously.12 cDNA fragments of the Tankyrase-1 gene were PCR-amplified and inserted into the pGEX-KG vector to generate pGEX-KG-TNKS1 amino-acid residues 1–158, 158–595, 596–1022, 1023–1327. pGEX-6T-TRF1 has been described previously.24, 36 TNKS1 mutants were produced by site-directed mutagenesis. GST-tagged or GFP-tagged human TNKS1-5A (T643A, T785A, T839A, T930A, T938A, S978/T982A, and T1128A) constructs were sub-cloned into pGEX-KG or peGFP-C1 (Clontech, Mountain View, CA, USA) vectors. All TNKS1 mutations were verified by sequencing. WT GFP TNKS1 (WT) and the PARP-dead (PD) mutant were a gift from Dr. Paul Chang.22 All GFP-tagged TNKS1 (WT, PD, and 5A) constructs contained exogenous nuclear localization sequences (NLS). The small interfering RNA (siRNA) expression plasmid was created by inserting oligonucleotides encoding shRNA against human TNKS1 (shTNKS1: 5′-aacaattcaccgtcgtcctct-3′; 3′-UTR shTNKS1: 5′-ccagatcagatttcaacct-3′), human Plk1 (shPlk1#1: 5′-ggtccattgggtgtatcatgt-3′; shPlk1#2 (3′-UTR shPlk1): 5′-gtgtgggttctacagcctt-3′), human Aurora-A (5′-acaagccggttcagaatca-3′), or luciferase (5′-cgtacgcggaatacttcga-3′) as a control, and synthesized (Genomine, Pohang, South Korea) into the pSuper-puro vector containing an H1 promoter and T5 terminal sequences (Oligoengine, Seattle, WA, USA). TRF1 depletion was performed by using 10 nM of an siRNA duplex (5′-ggaacaugacaaacuucauga-3′/5′-ucaugaaguuugucauguucc-3′) specific for nucleotides 489–509 of the coding region relative to the first nucleotide of the start codon as described previously.24
The following antibodies were purchased for subsequent studies: anti-TNKS1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-His (BD Biosciences PharMingen, San Diego, CA, USA), anti-PARP (Chemicon, Millipore, Billerica, MA, USA), anti-Plk1 (Santa Cruz Biotechnology), anti-cyclin-B1 (Santa Cruz Biotechnology), anti-NuMA (BD Biosciences PharMingen), anti-BubR1 (BD Biosciences PharMingen), anti-Aurora-A (BD Biosciences PharMingen), anti-GSK3β (Epitomics, Burlingame, CA, USA), anti-TRF1 (Abcam, Cambridge, UK), anti-PAR (Trevigen, Gaithersburg, MD, USA), anti-actin (Sigma, St. Louis, MO, USA), anti-HA (Roche, Basel, Switzerland), anti-phospho-Ser (Zymed, Invitrogen, Carlsbad, CA, USA), anti-phospho-Thr (Zymed), anti-α-tubulin (Cell Signaling Technology, Danvers, MA, USA), anti-MPM2 (Abcam), anti-phospho-histone-H3 at Ser10 (P-H3; Upstate Biotechnology, Millipore, Bellerica, MA, USA), anti-GFP (Santa Cruz Biotechnology) and human CREST autoimmune serum (Immunovision, Springdale, AR, USA).
Stable cell lines
A cell line stably overexpressing Plk1 was generated by transfecting pIRES-puro-HAPlk1 or the empty pIRES-puro vector (as a control) into human gastric epithelial (AGS) cells. Colonies showing resistance to puromycin (10 μg/ml) were isolated and analyzed by immunoblotting and immunofluorescence assays.
In vitro binding and immunoprecipitation assays
For the GST pull-down assays, the GST fusion proteins were absorbed onto glutathione–protein-A/G–Sepharose beads (Amersham Biosciences, GE Healthcare, Little Chalfont, UK) and incubated with whole-cell extracts from asynchronized or nocodazole-treated HeLa cells. The bound proteins were separated by SDS-PAGE and TNKS1 and Plk1 were detected by immunoblotting using the corresponding antibodies. For immunoprecipitation, asynchronized or nocodazole-treated HeLa cells were lysed in immunoprecipitation buffer (50 mmol/l Tris-HCl (pH 7.5), 150 mmol/l NaCl, 1% NP40, 1 mmol/l EDTA, 1 mmol/l phenylmethylsulfonyl fluoride (PMSF), and 1 mmol/l DTT) containing a protease inhibitor cocktail (Sigma). Each cell extract was incubated with antibodies against TNKS1, Plk1, GFP, or normal human IgG (control) for 2 h at 4 °C followed by incubation with protein-A/G–Sepharose beads for an additional 2 h. The beads were pelleted, washed four times with immunoprecipitation buffer, and analyzed by immunoblotting.
In vitro and in vivo kinase assays
For in vivo kinase assays, asynchronized and nocodazole-treated HeLa cells were harvested by shaking or scraping. The cells then washed twice in cold PBS and lysed in kinase buffer (100 mmol/l Tris-HCl (pH 7.5), 2 mmol/l EDTA (pH 8.0), 20 mmol/l MgCl2, 10 mmol/l MnCl2, 1 mmol/l DTT, 1 mmol/l PMSF, and protease inhibitor cocktail). Lysates from asynchronized cells (DMSO-treated) were incubated with 0.2 mM ATP alone (negative control) or in combination with purified His-Cdk1 (1 μg), His-Cdk1 with 25 μM purvalanol-A, His-Plk1 (1 μg), or His-Plk1 (1 μg) with 300 nM BI2536 at 37 °C for 1 h. Samples were separated on a 8% SDS-PAGE gel and immunoblotted. Beads with purified GST or GST-TNKS1 were washed twice with a kinase buffer and incubated with 4 μg of purified GST or GST-conjugated TNKS1 deletion mutants (residues 1–157, 158–595, 596–1022, and 1023–1327), TNKS1 point mutants (T643A, T785A, T839A, T930A, T938A, S978/T982A, and T1128A), or His-TNKS1 (WT) using purified baculovirus-expressed His-Plk1 and Cdk1/cyclin-B (Invitrogen) in the presence of 10 μCi of [γ-32P]ATP at 37 °C for 30 min.
Immunoblot and immunofluorescence assays
For immunoblot analysis, asynchronized and synchronized cells were harvested, washed twice in cold PBS, and lysed in lysis buffer (50 mmol/l Tris-HCl (pH 7.5), 150 mmol/l NaCl, 1% NP40, 1 mmol/l PMSF, and 1 mmol/l DTT) containing a protease inhibitor cocktail (Sigma). Equal amounts of protein (quantified by a Bio-Rad Protein Assay) from each sample were separated by SDS-PAGE, transferred to a nitrocellulose membrane, blocked, and probed with anti-TNKS1, anti-Plk1, anti-cyclin-B1 (Santa Cruz Biotechnology), anti-NuMA, anti-BubR1, anti-Aurora-A, anti-Bub3 (BD Biosciences PharMingen), anti-GSK3β (Epitomics Inc.), anti-TRF1 (Abcam), anti-PAR (Trevigen), anti-actin (Sigma), and anti-HA antibodies (Roche). HeLa cells were treated with the following cell-cycle-related kinase inhibitors for 12 h: 1 μM ZM447439 (an Aurora kinase inhibitor; Biomol, Hamburg, Germany), 2 μM TDZD-8 (a GSK3 inhibitor; A.G. Scientific Inc., San Diego, CA, USA), 25 μM purvalanol-A (a Cdk inhibitor; A.G. Scientific Inc.), 60 nM BI2536 (a Plk1 inhibitor; Axon Medchem, Groningen, The Netherlands), and 50 μM PPG (an inhibitor of polo box domain-dependent recognition; Sigma).
For immunofluorescence studies, the cells were fixed in 4% paraformaldehyde for 5 min. The fixed cells were washed, permeabilized in PBS containing 0.1% Triton X-100, and incubated with the appropriate primary antibodies at room temperature for 2 h. The cells were then washed and further incubated for 1 h with goat anti-mouse IgG conjugated with either FITC or Cy5. The cells were washed, stained with Hoechst dye to visualize DNA, and viewed under a confocal microscope (Zeiss 510 Meta; Carl Zeiss, Oberkochen, Germany).
Analytic gel filtration
Gel filtration was performed on a Superdex-200 10/300 GL column (Amersham Pharmacia Biotech, GE Healthcare, Little Chalfont, UK). HeLa cells were lysed in TNE buffer (10 mmol/l Tris-HCl (pH 7.4), 200 mmol/l NaCl, 1 mmol/l EDTA, 1 mmol/l PMSF, and 1 mmol/l DTT containing a protease inhibitor cocktail (Sigma)) and cleared by ultracentrifugation. The sample (500 μl, 20 μg/μl) was loaded onto a Superdex-200 10/300 GL column (10 mm i.d. × 300 mm) equilibrated with TNE buffer. Proteins were fractionated with TNE buffer at a linear flow rate of 0.4 ml/min, and 1-ml fractions were collected.
Telomeric PNA FISH on metaphase chromosome spreading
H1299 cells were treated with the Plk1 inhibitors, BI2536 (60 nM), and PPG (50 μM), and then cultured in the presence of nocodazole (660 nM/ml) for 12 h. HeLa cells were co-transfected with shRNA specifically targeting the TNKS1 3′-UTR and expression plasmids encoding shRNA-insensitive, WT GFP-TNKS1 or GFP-TNKS1-5A mutant. After washing and swelling in a hypotonic (0.075 M KCl) solution for 10 min at 37 °C, the cells were fixed and stored in a 3 : 1 (v/v) methanol/acetic acid solution. The cells were fixed to slides by spinning small volumes (10–100 μl) of cells in 2 ml of 50% acetic acid. The slides were dried overnight in air and immersed in PBS for 5 min prior to fixation in 4% formaldehyde in PBS for 2 min. The slides were then washed in PBS (three times × 5 min) and treated with pepsin (Sigma) at 1 mg/ml for 10 min at 3 °C in 10 mM glycine (pH 2.0). After a brief rinse in PBS, formaldehyde fixation and washes were repeated. The slides were then dehydrated in ethanol (70, 95, and 100%) and air dried. A 10-μl volume of a hybridization mixture containing 70% formamide, 0.3 μg/ml Cy3-(CCCTAA)3 PNA probe (Panagene, Daejeon, South Korea), and 1% (w/v) blocking reagent (Boehringer-Mannheim, Ingelheim am Rhein, Germany) in 10 mM Tris-HCl (pH 7.2) was added to the slide, a coverslip (180 mm) was added, and the DNA was denatured.
After hybridization for 2 h at room temperature, the slides were washed at room temperature with 70% formamide/10 mM Tris-HCl (pH 7.2; 2 times for 15 min), 0.05 M Tris-HCl (pH 7.2), and 0.15 M NaCl (pH 7.5) containing 0.05% Tween-20 (three times for 5 min). The slides were then dehydrated with increasing concentrations of ethanol (70, 95, and 100%), air-dried, and covered with 5–10 μl of an anti-fade solution (Sigma) containing 0.1 μg/ml DAPI. The slides were finally examined under a confocal microscope (Zeiss 510 Meta).
Abbreviations
- Plk1:
-
Polo-like kinase-1
- TNKS1:
-
Tankyrase-1
- T-SCE:
-
telomere sister chromatid exchange
- PARP:
-
poly(ADP-ribose) polymerase
- MALDI–TOF:
-
matrix-assisted laser desorption ionization–time of flight
- PBD:
-
polo-box-binding domain
- FISH:
-
fluorescence in situ hybridization
- PD:
-
PARP-dead mutant
References
Petronczki M, Lenart P, Peters JM . Polo on the rise-from mitotic entry to cytokinesis with Plk1. Dev Cell 2008; 14: 646–659.
Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I . Polo-like kinases (Plks) and cancer. Oncogene 2005; 24: 287–291.
Simizu S, Osada H . Mutations in the Plk gene lead to instability of Plk protein in human tumour cell lines. Nat Cell Biol 2000; 2: 852–854.
Lapenna S, Giordano A . Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov 2009; 8: 547–566.
Lowery DM, Clauser KR, Hjerrild M, Lim D, Alexander J, Kishi K et al. Proteomic screen defines the Polo-box domain interactome and identifies Rock2 as a Plk1 substrate. EMBO J 2007; 26: 2262–2273.
Barr FA, Sillje HH, Nigg EA . Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 2004; 5: 429–440.
de Lange T . Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005; 19: 2100–2110.
Hsiao SJ, Smith S . Tankyrase function at telomeres, spindle poles, and beyond. Biochimie 2008; 90: 83–92.
van Steensel B, de Lange T . Control of telomere length by the human telomeric protein TRF1. Nature 1997; 385: 740–743.
Ancelin K, Brunori M, Bauwens S, Koering CE, Brun C, Ricoul M et al. Targeting assay to study the cis functions of human telomeric proteins: evidence for inhibition of telomerase by TRF1 and for activation of telomere degradation by TRF2. Mol Cell Biol 2002; 22: 3474–3487.
Palm W, de Lange T . How shelterin protects mammalian telomeres. Annu Rev Genet 2008; 42: 301–334.
Smith S, Giriat I, Schmitt A, de Lange T . Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 1998; 282: 1484–1487.
Jeggo PA . DNA repair: PARP – another guardian angel? Curr Biol 1998; 8: R49–R51.
Schreiber V, Dantzer F, Ame JC, de Murcia G . Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 2006; 7: 517–528.
D'Amours D, Desnoyers S, D'Silva I, Poirier GG . Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 1999; 342 (Pt 2): 249–268.
Cook BD, Dynek JN, Chang W, Shostak G, Smith S . Role for the related poly(ADP-ribose) polymerases tankyrase 1 and 2 at human telomeres. Mol Cell Biol 2002; 22: 332–342.
Chang W, Dynek JN, Smith S . TRF1 is degraded by ubiquitin-mediated proteolysis after release from telomeres. Genes Dev 2003; 17: 1328–1333.
Smith S, de Lange T . Tankyrase promotes telomere elongation in human cells. Curr Biol 2000; 10: 1299–1302.
Donigian JR, de Lange T . The role of the poly(ADP-ribose) polymerase tankyrase1 in telomere length control by the TRF1 component of the shelterin complex. J Biol Chem 2007; 282: 22662–22667.
Dynek JN, Smith S . Resolution of sister telomere association is required for progression through mitosis. Science 2004; 304: 97–100.
Chang P, Coughlin M, Mitchison TJ . Interaction between poly(ADP-ribose) and NuMA contributes to mitotic spindle pole assembly. Mol Biol Cell 2009; 20: 4575–4585.
Chang P, Coughlin M, Mitchison TJ . Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nat Cell Biol 2005; 7: 1133–1139.
Chang W, Dynek JN, Smith S . NuMA is a major acceptor of poly(ADPribosyl) ation by tankyrase 1 in mitosis. Biochem J 2005; 391: 177–184.
Wu ZQ, Yang X, Weber G, Liu X . Plk1 phosphorylation of TRF1 is essential for its binding to telomeres. J Biol Chem 2008; 283: 25503–25513.
Taylor S, Peters JM . Polo and Aurora kinases: lessons derived from chemical biology. Curr Opin Cell Biol 2008; 20: 77–84.
Smith S, de Lange T . Cell cycle dependent localization of the telomeric PARP, tankyrase, to nuclear pore complexes and centrosomes. J Cell Sci 1999; 112 (Pt 21): 3649–3656.
Elowe S, Hummer S, Uldschmid A, Li X, Nigg EA . Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev 2007; 21: 2205–2219.
Ye JZ, de Lange T . TIN2 is a tankyrase 1 PARP modulator in the TRF1 telomere length control complex. Nat Genet 2004; 36: 618–623.
Compton DA . Regulation of mitosis by poly(ADP-ribosyl)ation. Biochem J 2005; 391: e5–e6.
Losada A, Hirano M, Hirano T . Cohesin release is required for sister chromatid resolution, but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev 2002; 16: 3004–3016.
Gimenez-Abian JF, Sumara I, Hirota T, Hauf S, Gerlich D, de la Torre C et al. Regulation of sister chromatid cohesion between chromosome arms. Curr Biol 2004; 14: 1187–1193.
Hauf S, Roitinger E, Koch B, Dittrich CM, Mechtler K, Peters JM . Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol 2005; 3: e69.
Gelmini S, Poggesi M, Distante V, Bianchi S, Simi L, Luconi M et al. Tankyrase, a positive regulator of telomere elongation, is overexpressed in human breast cancer. Cancer Lett 2004; 216: 81–87.
MacNamara B, Wang W, Chen Z, Hou M, Mazur J, Gruber A et al. Telomerase activity in relation to pro- and anti-apoptotic protein expression in high grade non-Hodgkin's lymphomas. Haematologica 2001; 86: 386–393.
Golsteyn RM, Schultz SJ, Bartek J, Ziemiecki A, Ried T, Nigg EA . Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J Cell Sci 1994; 107 (Pt 6): 1509–1517.
Seimiya H, Muramatsu Y, Ohishi T, Tsuruo T . Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell 2005; 7: 25–37.
Acknowledgements
We thank Dr. Susan Smith (New York University School of Medicine) for the baculovirus-expressed human TNKS1 protein and a TNKS1 expression plasmid, and Dr. Kwan-Hyuck Baek (Sungkyunkwan University School of Medicine) for helpful discussions and comments. This work was supported by research grants from the National Research and Development Program for Cancer Control (0920290 and 1120220) and the Korea Healthcare Technology Research and Development Project (A08-4965-AA2023-08N1-00010A), Ministry for Health, Welfare and Family Affairs, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by B Zhivotovsky
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Ha, GH., Kim, HS., Go, H. et al. Tankyrase-1 function at telomeres and during mitosis is regulated by Polo-like kinase-1-mediated phosphorylation. Cell Death Differ 19, 321–332 (2012). https://doi.org/10.1038/cdd.2011.101
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.101
Keywords
This article is cited by
-
Tankyrase: a promising therapeutic target with pleiotropic action
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)
-
Tankyrase inhibitors: emerging and promising therapeutics for cancer treatment
Medicinal Chemistry Research (2021)
-
Oncogenic potential of BEX4 is conferred by Polo-like kinase 1-mediated phosphorylation
Experimental & Molecular Medicine (2018)
-
A systematic analysis of the PARP protein family identifies new functions critical for cell physiology
Nature Communications (2013)
-
Tankyrase-targeted therapeutics: expanding opportunities in the PARP family
Nature Reviews Drug Discovery (2012)
