
Abstract
SIRT1, the closest mammalian homolog of yeast Sir2, is an NAD+-dependent deacetylase with relevant functions in cancer, aging, and metabolism among other processes. SIRT1 has a diffuse nuclear localization but is recruited to the PML nuclear bodies (PML-NBs) after PML upregulation. However, the functions of SIRT1 in the PML-NBs are unknown. In this study we show that primary mouse embryo fibroblasts lacking SIRT1 contain reduced PML protein levels that are increased after reintroduction of SIRT1. In addition, overexpression of SIRT1 in HEK-293 cells increases the amount of PML protein whereas knockdown of SIRT1 reduces the size and number of PML-NBs and the levels of PML protein in HeLa cells. SIRT1 stimulates PML sumoylation in vitro and in vivo in a deacetylase-independent manner. Importantly, the absence of SIRT1 reduces the apoptotic response of vesicular stomatitis virus-infected cells and favors the extent of this PML-sensitive virus replication. These results show a novel function of SIRT1 in the control of PML and PML-NBs.
Similar content being viewed by others


Main
The tumor suppressor PML is the main and essential component of the nuclear bodies (NBs), the dynamic compartments that participate in a number of cellular processes, including apoptosis, transcriptional regulation, DNA repair, and protection against viral infection.1, 2 PML acts as a tumor suppressor, antagonizing initiation, promotion, and progression of tumors of various histological origins.3 PML is also implicated in the regulation of infection by a variety of RNA viruses, adenoviruses, and human cytomegalovirus.4, 5, 6, 7 Its function is regulated by post-translational modifications such as phosphorylation, sumoylation, ubiquitination, and acetylation. However, only the sumoylation of PML has been shown as essential for the formation of the PML-NBs and a crucial process for PML-dependent apoptosis and transcriptional regulation.8 In addition to PML, PML-NBs contain several other proteins, such as SP100, SUMO-1, pRB, p53, and lately, the NAD+-dependent, type III, histone/protein deacetylase SIRT1.9, 10
SIRT1 is the best-characterized class III histone deacetylase in mammalian cells and the closest homolog to yeast Sir2. However, although most of SIRT1 functions are related to its enzymatic activity, deacetylation-independent activities of SIRT1 have also been proposed.11, 12, 13, 14 SIRT1 is involved in a wide spectrum of biological processes through diverse substrates such as the tumor suppressor p53,9, 15, 16, 17 the transcription factor NF-kB,18 and the FOXO family of transcription factors.12, 14, 19, 20 Although SIRT1 has a diffuse nuclear localization, it is recruited to the PML-NBs after PML upregulation. However, the functional significance of this PML–SIRT1 interaction has not been addressed.
In this report, we show that there is a correlation between the levels of SIRT1 and PML present in both primary and transfected cells. This positive regulation of PML levels by SIRT1 is mediated by an increase in the sumoylation of PML by SIRT1, in a deacetylation-independent manner. Functional significance of this regulation is revealed by the increased replication of the PML-sensitive vesicular stomatitis virus (VSV) in mouse embryo fibroblasts (MEFs) derived from SIRT1−/− mice. These results identify a new function of SIRT1 regulating PML with consequences in the control of virus infection, and that might also be crucial for other PML-mediated activities.
Results
Regulation of PML levels by SIRT1
To study the relationship between SIRT1 and PML, we examined the expression of PML in MEFs derived from genetically modified mice that lack expression of SIRT1 (SIRT1−/−),21 normal wild-type animals (WT), and mice engineered to harbor extra copies of the SIRT1 gene under the control of its own regulatory genomic sequences (SIRT1-tg previously described in Pfluger et al.22). Surprisingly, western blot analysis of cell extracts from SIRT1−/−, WT, or SIRT1-tg MEFs revealed a strong reduction in PML levels in cells lacking SIRT1 in comparison with WT or SIRT1-tg cells (Figure 1a). Similarly, lower PML staining in SIRT1−/− when compared with WT or SIRT1-tg heart tissues was also detected in vivo by immunohistochemical analysis (Figure 1b). PML localizes to specific nuclear subdomains called NBs. To test whether endogenous SIRT1 also regulates PML-NB formation, we analyzed these structures by immunofluorescence using anti-PML antibody and confocal microscopy analysis in SIRT1−/−, WT, and SIRT1-tg MEFs. Cells lacking SIRT1 showed a profound reduction in the number of the PML-NBs relative to WT cells. More than 62% of SIRT1−/− cells showed <15 PML dots per nucleus in comparison with 14% of the WT or SIRT1-tg MEFs (Supplementary Figure 1). To resolve whether SIRT1 absence was inducing PML protein loss through reduction of PML RNA levels, we analyzed PML mRNA from SIRT1−/−, WT, and SIRT1-tg MEFs by quantitative RT-PCR. No differences in the relative PML mRNA levels were found between the different cells (data not shown). To determine whether there is a causal link between the absence of SIRT1 and the lower levels of PML in human cells, we transiently knocked down SIRT1 by siRNA in HeLa cells. At 24 or 48 h after transfection with siRNA against SIRT1, PML levels were analyzed by western blot. As previously reported, transfection of siRNA against SIRT1 efficiently knocked down the protein,23, 24 leading to almost undetectable levels of SIRT1 protein expression (Figure 1c). In agreement with our observations using genetically modified MEFs, we also detected an important reduction in PML protein levels after knockdown of SIRT1 (approximately 50% of the amount detected in the control cells; Figure 1c). Moreover, immunofluorescence analysis of HeLa cells transfected with siRNA against SIRT1 revealed that those cells showing lower SIRT1 staining showed a profound reduction in the number of the PML-NBs relative to cells containing normal SIRT1 levels (Figure 1d). Finally, to further prove a positive correlation between SIRT1 and PML expression, HEK-293 cells were co-transfected with a plasmid encoding for PML4 together with an empty vector or two different doses of a plasmid encoding for Flag-SIRT1 and a plasmid expressing GFP as a control for transfection efficiency, and the levels of PML were analyzed by western blot. As shown in Figure 1e, transfection of SIRT1 induced a clear increase in the PML protein levels in a dose–response manner. Finally, to further corroborate these results, SIRT1−/− MEFs were transfected with a plasmid encoding for mouse HA-SIRT1 or a GFP expression plasmid using the AMAXA nucleofector system, and the levels of PML were analyzed by western blot. Transfection of MEFs with a vector encoding GFP or a pCDNA empty vector (data not shown) induced a clear increase in the PML levels, as a consequence of the nucleofection-associated stress (Figure 1f). However, and importantly, reintroduction of SIRT1 in SIRT1−/− MEFs resulted in even higher levels of PML protein. All together, these results indicate that SIRT1 controls PML protein levels in both mouse and human cells, and its absence leads to a sharp decline in total PML protein levels as well as to a disorganization of the PML-NBs.

PML levels parallel those of SIRT1. (a) Extracts from SIRT1−/−, WT, or SIRT1-tg MEFs were prepared and analyzed by western blotting using anti-PML antibodies. (b) Heart tissue derived from SIRT1−/−, WT, or SIRT1-tg mice was subjected to immunohistochemistry staining using anti-PML (Sigma, HPA008312) or anti-SIRT1 (Sigma, S5196) antibodies. (c) HeLa cells were transfected with siRNA against SIRT1. At the indicated times after transfection, cells extracts were analyzed by western blotting using anti-PML or anti-SIRT1 antibodies. (d) HeLa cells were transfected with siRNA against SIRT1 and at 48 h were subjected to immunostaining using anti-PML or anti-SIRT1 antibodies followed by confocal microscopy. PML staining in a representative image of cells expressing different SIRT1 levels is shown. Arrows indicate cells in which SIRT1 expression is diminished. (e) HEK-293 cells were transfected with the indicated expression plasmids, and 48 h after transfection cells extracts were analyzed by western blotting using anti-PML or anti-SIRT1 antibodies. Cell extract from untransfected cells (NT) was used as a control. (f) SIRT1−/− MEFs were transfected with the indicated expression plasmids, and 48 h after transfection cells extracts were analyzed by western blotting using anti-PML or anti-SIRT1 antibodies. Cell extract from untransfected cells (NT) was used as a control. The numbers given under the panel show densitometric data after normalization to actin, and compared with control (set as 1)
Control of PML sumoylation by SIRT1
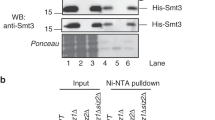
PML is functionally regulated by post-translational modifications such as phosphorylation, sumoylation, ubiquitination, and acetylation. Sumoylation of PML is necessary for the proper formation of PML-NBs and the recruitment of NB-associated proteins, highlighting the importance of this modification in PML function (reviewed in Seeler and Dejean25). To determine whether sumoylation of PML is also required for the PML regulation by SIRT1, MCF-7 cells were transfected with a plasmid encoding for PML4 or a PML sumoylation-defective mutant (PML4-TKO) together with Flag-SIRT1 or an empty vector and the levels of PML protein were analyzed by western blot. Expression of SIRT1 induced an increase in the PML protein but did not alter the PML4-TKO levels (Figure 2a), indicating that SIRT1 regulates sumoylated PML. To examine whether SIRT1 alters PML sumoylation, we carried out in vitro sumoylation assays using PML as a substrate, in the presence or absence of in vitro translated SIRT1. As expected, incubation of PML with SUMO1 in the sumoylation reaction induced the appearance of high-molecular-weight PML forms that correspond to PML–SUMO1 (Figure 2b). The PML–SUMO1 bands were increased when SIRT1 was added to the reaction (Figure 2b), indicating that SIRT1 promoted PML sumoylation in vitro. To determine whether SIRT1 can also promote PML sumoylation in vivo, and to analyze whether the enzymatic activity of SIRT1 is required, we co-transfected a His-tagged version of PML together with pCDNA-SUMO1 and a Flag-tagged WT SIRT1 or H363Y deacetylase mutant into HEK-293 cells. His-PML was then purified in denaturing conditions using nickel-columns and the purified SUMO1–PML protein was assessed by western blot using anti-SUMO1 antibody. Co-transfection of either SIRT1 WT or SIRT1 H363Y deacetylase mutant resulted in increased accumulation of the SUMO1–PML-bands (Figure 2c). Similar results were also observed after co-transfection of a HA-tagged version of PML and a His-tagged SUMO1 expression plasmid (Supplementary Figure 2). In contrast, the SUMO modification of other PML-NB components did not vary after SIRT1 overexpression. In particular, no changes in the levels of Sp100-SUMO1 in response to SIRT1 expression were detected using a similar approach (Supplementary Figure 3). These results indicate that SIRT1 favors sumoylation of PML in vivo and that the catalytic activity of SIRT1 is dispensable for promoting PML sumoylation. In addition, PML was also purified from lysates of HeLa cells co-transfected with His-tagged PML, pCDNA-SUMO1, and siRNA to SIRT1, and then analyzed for sumoylation with anti-SUMO1 antibody. Silencing of SIRT1 resulted in decreased levels of PML–SUMO1 in the transfected cells (Figure 2d). These results reinforce the idea that SIRT1 favors PML sumoylation.

SIRT1 promotes sumoylation of PML in a deacetylase-independent manner. (a) MCF-7 cells were transfected with the indicated plasmids, and 48 h after transfection cell extracts were analyzed by western blotting using anti-PML or anti-Flag antibodies. (b) Fluorography of dried SDS-PAGE gels after fractionation of in vitro sumoylation assay products using in vitro translated [35S]-PML in the absence or presence of in vitro translated SIRT1. Incubation of the in vitro sumoylation reaction with SUMO1 (+SUMO1) resulted in the appearance of slower migrating bands that correspond to SUMO1–PML. Addition of SIRT1 results in the increase of SUMO1–PML conjugates (black arrowheads). (c) HEK-293 cells were co-transfected with the indicated plasmids and 36 h after transfection, His-PML proteins were purified by nickel affinity chromatography. Purified extracts were analyzed by SDS-PAGE followed by anti-SUMO1 western blot. Cell extracts were also directly analyzed by western blotting using anti-PML and anti-SIRT1 antibodies. (d) HeLa cells were transfected with pCDNA-His-PML4, pCDNA-SUMO1, and siRNA against SIRT1 or control siRNA, and 48 h after transfection whole cell lysates were recovered, and purification of His-PML protein by nickel affinity chromatography was performed. Purified extracts were probed with anti-SUMO1 antibodies as indicated. Input extracts were incubated with anti-SIRT1 and anti-PML antibodies
SIRT1 protects from viral infection in a gene dosage-dependent manner
PML has a role in the control of virus infection, and PML levels are crucial as revealed with the well-characterized VSV.26 Hence, we decided to analyze the effect of different SIRT1 gene dosages on viral infection. For this purpose, MEFs derived from SIRT1−/−, WT, and SIRT1-tg mice were infected with VSV at a multiplicity of infection (MOI) of 5 and virus titers in the supernatants, after all cells died as a result of the infection, were determined. MEFs from SIRT1−/− produced approximately 1 log higher number of infectious virus than WT cells, whereas MEFs from SIRT1-tg produced 5 times less infectious virus than their WT counterparts (Figure 3a), indicating a protective role for SIRT1. This notion was further corroborated by direct inspection of viral protein synthesis after infection of SIRT1−/−, WT, and SIRT1-tg MEFs with VSV. Viral protein synthesis was reduced in a SIRT1 gene dosage-dependent manner when the cells were infected at an MOI of 0.5 (Figure 3b) and an MOI of 5 (Supplementary Figure 4). All together, these results indicate that SIRT1 protects against VSV infection. The implication of PML in antiviral defense was first suggested after the finding that interferons activate PML gene transcription, induce PML expression, and dramatically increase the number and average size of PML NBs.27, 28, 29 To determine whether PML is upregulated in response to VSV infection in the absence of SIRT1, MEFs derived from SIRT1−/− and WT mice were infected with VSV and, at different times after infection, levels of PML protein were analyzed by western blot. As shown in Figure 3c, VSV infection upregulated PML in both SIRT1−/− and WT MEFs. However, the PML protein levels detected at 8 h after VSV infection in SIRT1−/− MEFs were even lower than those observed at zero time in WT MEFs. In addition, only the low-molecular-weight PML form, corresponding to unmodified PML, and not high size PML-containing bands (modified PMLs), were detected in SIRT1−/− cells. To prove the importance of both PML and SIRT1 in the increased VSV replication observed in the cells that do not express SIRT1, MEFs derived from SIRT1−/− mice were transfected with GFP, a plasmid encoding HA-SIRT1 or PML, and 36 h later, cells were infected with VSV at an MOI of 10. Viral titers in the supernatants of transfected cells were calculated 24 h after infection. As shown in Table 1, the titers in the supernatant of the PML- or SIRT1-transfected cells were reduced around 2 times compared with GFP-transfected cells (Table 1). It has been well established that PML is a proapoptotic factor required for the induction of apoptosis by different stimuli, including type I and II interferons.30 To determine whether the differences in PML protein levels observed in the cells with different SIRT1 gene dosages could result in a different degree of apoptosis induction in response to VSV infection, MEFs derived from SIRT1−/−, WT, and SIRT1-tg mice were infected with VSV at an MOI of 5, and 16 h after infection, cells were processed to measure apoptosis induction. The analysis of cell death revealed that MEFs derived from SIRT1−/− mice showed a clear reduction in apoptosis induction in comparison with the levels of apoptosis detected in infected WT cells (Figure 3d). Even more, a higher number of apoptotic cells were observed after VSV infection of MEFs derived from SIRT1-tg mice compared with WT MEFs (Figure 3d). PML expression contributes to apoptosis, among other mechanisms, by enhancing p53-mediated apoptotic activity and, in particular, it increases p53 transcriptional activity on the promoter of the proapoptotic PIG3 gene.31 For this reason, we transiently transfected the PIG3 promoter fused to the luciferase gene (PIG3-luc) in MEFs with different SIRT1 gene dosages, and the levels of transactivation in response to VSV infection were measured. PIG3-luc reporter was clearly transactivated by VSV infection in both WT and in SIRT1-tg cells, but not in SIRT1−/− cells, which showed a slight repression (Figure 3e). As a negative control, a mutant version of the promoter that lacks p53-binding sites was used (Figure 3e). Altogether, these results showed that the levels of PML positively regulated by SIRT1 correlated with the apoptosis levels triggered by VSV infection.

SIRT1-deficient MEFs are less protected from viral infection, accumulate less PML in response to VSV infection, and are more susceptible to VSV-induced apoptosis. (a) MEFs derived from SIRT1−/−, WT, or SIRT1-tg mice were infected in triplicate with VSV at an MOI of 5, and quantification of the virus yield after total destruction of the monolayer was assessed. The same results were obtained in at least three different experiments and using MEFs derived from three different WT or transgenic embryos. Data represent means±S.E. for one experiment. *P<0.05 compared with WT cells, Student's t-test. (b) SIRT1−/−, WT, or SIRT1-tg MEFs were infected with VSV at an MOI of 0.5, and at the indicated times western blotting using antibodies against both the M and N protein from VSV was performed. (c) SIRT1−/− or WT MEFs were infected with VSV at an MOI of 5, and at the indicated times after infection cells extracts were analyzed by western blotting using anti-PML or anti-SIRT1 antibodies. *Nonspecific band. (d) MEFs derived from SIRT1−/−, WT, or SIRT1-tg mice were infected with VSV at an MOI of 5, and 16 h after infection propidium iodide staining and flow cytometry analysis was carried out. The apoptotic response was measured by calculating the cell fraction with less than G1 DNA content. Results shown represent the average of at least three independent experiments±S.E. *P<0.05, **P<0.005, compared with WT cells, Student's t-test. (e) Activation of the proapoptotic promoter PIG3 in response to VSV infection is impaired in SIRT1−/− MEFs. SIRT1−/−, WT, or SIRT1-tg MEFs were transfected in triplicate with the indicated reporter plasmids. At 24 h after transfection, cells were infected with VSV at an MOI of 5, and at 15 h after infection luciferase activity was measured. Results shown represent means±S.E. for one experiment. Similar results were observed in at least three independent experiments. *P<0.05, compared with WT cells, Student's t-test
Discussion
In this study, we have provided evidence for the regulation of PML protein levels as well as of the PML-NBs by SIRT1. Analysis of PML in cells with different levels of expression of SIRT1 gene dosage reveals that absence of SIRT1 expression caused a reduction in the number of the PML-NBs that was accompanied by a decrease in PML protein levels. In contrast, SIRT1 expression induced an increase in the PML protein levels and the accumulation of SUMO-modified PML forms. Consistent with these data, siRNA knockdown of SIRT1 also induced a decrease in PML and PML–SUMO1 protein levels. Altogether, these data show that SIRT1 has an important role in PML sumoylation and PML-NB formation and that the loss of SIRT1 leads to a reduction in the sumoylation and steady-state accumulation of PML. As this effect was also observed after overexpression of a catalytically dead SIRT1, these results indicate that this SIRT1 function is independent of its deacetylase activity.
A regulation of PML by SIRT1 was already presented as a hypothesis to explain the functional significance of the interaction between PML and SIRT1.9 This interaction seems to be independent of SIRT1 enzymatic activity as it can also be verified using a catalytically dead SIRT1 (Supplementary Figure 5). It is not clear exactly how SIRT1 positively regulates the sumoylation levels of PML. One potential mechanism could be that SIRT1 prevents the desumoylation of PML. As SIRT1 is also a substrate for SUMO modification (Supplementary Figure 6 and Yang et al.32) and the sumoylation of both proteins, PML and SIRT1, is regulated by the SUMO-specific protease SENP1,32, 33, 34 SIRT1 could potentially compete with PML for the interaction with this desumoylase. However, we could not detect a reduction in SENP1-mediated desumoylation of PML in the presence of SIRT1 (Supplementary Figure 7), pointing to alternative explanations. A second explanation might be that the binding of SIRT1 to PML acts as a scaffold for the interaction of PML with components of the sumoylation system, thanks to a putative zinc-binding domain that has been annotated in SIRT1,35 or through the putative SUMO interaction motifs (SIMs)36, 37 that can be found in SIRT1. Other possibilities may be that, being a transcriptional corepressor, SIRT1 could potentially affect expression of the genes encoding the sumoylation machinery such as SUMO1. However, we detected no variations in the relative SUMO1 mRNA levels (data not shown) or in the amount of non-conjugated SUMO1 protein (Supplementary Figure 8) in cells expressing different levels of SIRT1. Interestingly, it has been described that in testis derived from SIRT1−/− mice there is an overrepresentation of genes involved in sumoylation, a finding that has been proposed to represent a putative compensatory mechanism to promote sumoylation of specific proteins.38
Regulation of PML by other HDACs has been reported. Thus, class I HDACs interact with PML and this interaction is required for the transcriptional repression function of PML.39, 40 In addition, class IIa HDACs – HDAC4, HDAC5, and HDAC7 – increase PML sumoylation in an acetylation-independent manner,41 and although it is not clear exactly how HDAC4 and related members positively regulate the sumoylation levels of PML, a putative SUMO E3 ligase activity has been suggested.42
PML has an inhibitory effect on virus infections in vitro and in vivo.5 Thus, overexpression of PML confers resistance against VSV, influenza virus, human foamy virus (HFV), and lymphocytic choriomeningitis virus (LMCV).26, 43 Our results show that SIRT1 can control VSV infection in a dosage-dependent manner. Although we cannot discard that SIRT1 controls virus infection by additional mechanisms, the correlation between the levels of SIRT1 and PML showed here suggests that PML may have a role as a mediator of the antiviral activity exerted by SIRT1. In addition, our results show that SIRT1 augments the apoptosis in response to VSV infection. As PML is required for interferon-induced apoptosis,30 these results point to PML as a putative downstream mediator of the proapoptotic activity observed for SIRT1.
In conclusion, in this study we show that SIRT1 is capable of stimulating PML sumoylation in both a reconstituted extracellular system and in mammalian cells, and that it is required for a proper PML–SUMO conjugation, and consequently, proper PML-NB formation. To our knowledge, this is the first time that SIRT1 has been shown to stimulate PML sumoylation, a new function of SIRT1 that is independent of its deacetylase activity, which might explain some of the contradictory results obtained while investigating the role of SIRT1 in cancer and aging.
Materials and Methods
Mice, cell culture, virus, and transfections
SIRT1-tg and SIRT1−/− mice have been previously described.21, 22 MEFs were isolated and cultured as described previously.44 All MEFs were used before spontaneous immortalization. HeLa, MCF-7, and green African monkey BSC-40 cells were cultured following a standard procedure. Infections were carried out using VSV of Indiana strain and virus yields were measured by plaque assays in BSC-40 cells. Transfection of MCF-7 was performed using FuGene (Roche, Barcelona, Spain) following the manufacturer's instructions. siRNAs were transfected into HeLa cells using Lipofectamine 2000 (Invitrogen, Barcelona, Spain). MEFs were transfected using an Amaxa nucleofector apparatus (program A-023) and Amaxa MEF nucleofector Kit 1 according to the manufacturer's instructions (Amaxa, GmbH, Cologne, Germany).
Plasmids and reagents
siRNA against SIRT1 (SiGenome Smartpool M-003540-01) was purchased from Dharmacon (Thermo Scientific, Epson, Surrey, UK). pcDNA-SIRT1 was kindly supplied by Tony Kouzarides (Gurdon Institute, University of Cambridge). Flag-SIRT1 (1791Addgene) and Flag-SIRT1 H363Y (1792Addgene)19 were kindly supplied by Dr. Michael Greenberg (Children's Hospital Center for Life Sciences). HA-SIRT1 (10962 Addgene) was kindly supplied by Dr. Toren Finkel (National Heart Lung and Blood Institute). pcDNA3-His-PML4 plasmid was provided by Dr. Kun-Sang Chang (MD Anderson Cancer Center, Houston) and the pcDNA3-His-PML4-TKO has been previously described.45 pcDNA-SUMO1 plasmid was kindly supplied by Dr. Keith D Robertson (Shands Cancer Center, University of Florida).
Western blot analysis and antibodies
Cells were washed in PBS, scraped in SDS-gel loading buffer, and boiled for 5 min. Proteins of total extracts were separated by SDS-PAGE, transferred to nitrocellulose, and incubated with the corresponding antibodies. The following antibodies were used: anti-SIRT1 (Abcam, Cambridge, UK, AB12193), anti-VSV, anti-human PML (Chemicon, Millipore, Madrid, Spain, AB-1370), anti-mouse PML (Upstate, Millipore, Madrid, Spain, 05-718), anti-Flag (Sigma, M2), anti-SUMO1 (Abcam, AB11672), anti-actin (MP Biomedicals, Illkirch, France), and anti-α-tubulin (Serotec, Kidlington, Oxford, UK).
Immunofluorescence and confocal microscopy
Cells were seeded onto glass coverslips, fixed, and stained as described previously.46 Antibodies against PML (Chemicon, AB-1370 or Upstate, 05-718), or anti-SIRT1 (Abcam, AB12193) were used, followed by Alexa 488-conjugated anti-rabbit and Alexa 594-conjugated anti-mouse immunoglobulins (Molecular Probes, Leiden, The Netherlands). Analysis of the samples was carried out on a Leica TCS SP5 confocal laser microscope (Leica Microsystems, Heidelberg, Mannheim, Germany) using simultaneous scans to avoid shift between the optical channels. Images were exported using Adobe Photoshop (Adobe Systems Inc, San Jose, CA, USA).
Immunohistochemistry
Heart tissue derived from PML−/−, SIRT1−/−, WT, or SIRT1-tg mice was subjected to immunohistochemistry staining using anti-PML (Sigma, HPA008312) or anti-SIRT1 (Sigma, S5196) antibodies.
In vitro SUMO1 conjugation assay
In vitro transcribed/translated, [35S]methionine-labelled, PML proteins were incubated with E1 in a 10 μl reaction including an ATP regenerating system (50 mM Tris pH 7.6, 5 mM MgCl2, 2 mM ATP, 10 mM creatine phosphate, 3.5 U/ml of creatine kinase, and 0.6 U/ml of inorganic pyrophosphatase), 10 μg SUMO1, and 600 ng Ubc9. Reactions were incubated at 30°C for 45 min. After terminating the reactions with SDS sample buffer containing β-mercaptoethanol, reaction products were fractionated by SDS-PAGE (8%) and detected by fluorography.
In vitro expression of proteins
In vitro transcription/translation of proteins was performed using 1 μg of pCDNA-SIRT1 or pCDNA3-His-PML4 plasmid DNA and rabbit reticulocyte coupled transcription/translation system according to the instructions provided by the manufacturer (TNT-coupled reticulocyte lysate system; Promega, Madrid, Spain). [35S]methionine was used in some reactions to generate radiolabelled proteins.
Reporter assay
MEFs derived from SIRT1−/−, WT, or SIRT1-tg mice were transfected in 24-well plates with the PIG3-luc or PIG3-luc mutant plasmids.47 pCMV-β-gal plasmid was co-transfected to determine transfection efficiencies. At 24 h after transfection, cells were infected with VSV at an MOI of 5, and 15 h after infection, cells were harvested and luciferase activity was measured using the Luciferase Reporter Gene Assay (Roche) according to the manufacturer's instructions. Triplicate measurements were performed for all experiments and each experiment was repeated at least three times.
Purification of His-tagged PML conjugates
HEK-293 cells were transfected with pCDNA3-His-PML4 plasmid and pCDNA-SUMO1 in combination with the plasmids Flag-SIRT1, Flag-SIRT1 H363Y, or empty vector. At 36 h after transfection, cells were lysed in 4 ml of 6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl pH 8.0 plus 5 mM imidazole, and 10 mM β-mercaptoethanol per 75-cm2 flask. Lysates were then mixed with 50 μl of Ni2+-NTA-agarose beads prewashed with lysis buffer and incubated for 2 h at room temperature. The beads were successively washed with the following: 6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl pH 8.0 plus 10 mM β-mercaptoethanol; 8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl pH 8.0, 10 mM β-mercaptoethanol; 8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-HCl pH 6.3, 10 mM β-mercaptoethanol (buffer A) plus 0.2% Triton X-100; buffer A; and then buffer A plus 0.1% Triton X-100. After the last wash with buffer A the beads were eluted with 200 mM imidazole in 5% SDS, 0.15 M Tris-HCl pH 6.7, 30% glycerol, and 0.72 M β-mercaptoethanol. The eluates were subjected to SDS-PAGE (8%), and western blot was performed as indicated above.
Abbreviations
- PML-NB:
-
PML nuclear body
- NB:
-
nuclear body
- VSV:
-
vesicular stomatitis virus
- SIM:
-
SUMO interaction motif
- HFV:
-
human foamy virus
- LMCV:
-
lymphocytic choriomeningitis virus
- MEF:
-
mouse embryo fibroblast
- WT:
-
wild-type
- MOI:
-
multiplicity of infection
References
Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T et al. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol 1999; 147: 221–234.
Zhong S, Salomoni P, Pandolfi PP . The transcriptional role of PML and the nuclear body. Nat Cell Biol 2000; 2: E85–E90.
Wang ZG, Delva L, Gaboli M, Rivi R, Giorgio M, Cordon-Cardo C et al. Role of PML in cell growth and the retinoic acid pathway. Science 1998; 279: 1547–1551.
Ahn JH, Hayward GS . Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology 2000; 274: 39–55.
Bonilla WV, Pinschewer DD, Klenerman P, Rousson V, Gaboli M, Pandolfi PP et al. Effects of promyelocytic leukemia protein on virus-host balance. J Virol 2002; 76: 3810–3818.
Doucas V, Ishov AM, Romo A, Juguilon H, Weitzman MD, Evans RM et al. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev 1996; 10: 196–207.
Regad T, Chelbi-Alix MK . Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 2001; 20: 7274–7286.
Zhong S, Muller S, Ronchetti S, Freemont PS, Dejean A, Pandolfi PP . Role of SUMO-1-modified PML in nuclear body formation. Blood 2000; 95: 2748–2752.
Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J 2002; 21: 2383–2396.
Maul GG, Negorev D, Bell P, Ishov AM . Review: properties and assembly mechanisms of ND10, PML bodies, or PODs. J Struct Biol 2000; 129: 278–287.
Ghosh HS, Spencer JV, Ng B, McBurney MW, Robbins PD . Sirt1 interacts with transducin-like enhancer of split-1 to inhibit nuclear factor kappaB-mediated transcription. Biochem J 2007; 408: 105–111.
Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W et al. Mammalian SIRT1 represses forkhead transcription factors. Cell 2004; 116: 551–563.
Vaitiekunaite R, Butkiewicz D, Krzesniak M, Przybylek M, Gryc A, Snietura M et al. Expression and localization of Werner syndrome protein is modulated by SIRT1 and PML. Mech Ageing Dev 2007; 128: 650–661.
van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM . FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J Biol Chem 2004; 279: 28873–28879.
Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001; 107: 137–148.
Smith J . Human Sir2 and the ‘silencing’ of p53 activity. Trends Cell Biol 2002; 12: 404–406.
Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001; 107: 149–159.
Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 2004; 23: 2369–2380.
Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004; 303: 2011–2015.
Daitoku H, Hatta M, Matsuzaki H, Aratani S, Ohshima T, Miyagishi M et al. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci USA 2004; 101: 10042–10047.
Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA 2003; 100: 10794–10799.
Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH . Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci USA 2008; 105: 9793–9798.
Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA 2008; 105: 3374–3379.
Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W . Negative regulation of the deacetylase SIRT1 by DBC1. Nature 2008; 451: 587–590.
Seeler JS, Dejean A . SUMO: of branched proteins and nuclear bodies. Oncogene 2001; 20: 7243–7249.
Chelbi-Alix MK, Quignon F, Pelicano L, Koken MH, de The H . Resistance to virus infection conferred by the interferon-induced promyelocytic leukemia protein. J Virol 1998; 72: 1043–1051.
Chelbi-Alix MK, Pelicano L, Quignon F, Koken MH, Venturini L, Stadler M et al. Induction of the PML protein by interferons in normal and APL cells. Leukemia 1995; 9: 2027–2033.
Lavau C, Marchio A, Fagioli M, Jansen J, Falini B, Lebon P et al. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene 1995; 11: 871–876.
Stadler M, Chelbi-Alix MK, Koken MH, Venturini L, Lee C, Saib A et al. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene 1995; 11: 2565–2573.
Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R et al. PML is essential for multiple apoptotic pathways. Nat Genet 1998; 20: 266–272.
Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K et al. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J 2000; 19: 6185–6195.
Yang Y, Fu W, Chen J, Olashaw N, Zhang X, Nicosia SV et al. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat Cell Biol 2007; 9: 1253–1262.
Gong L, Millas S, Maul GG, Yeh ET . Differential regulation of sentrinized proteins by a novel sentrin-specific protease. J Biol Chem 2000; 275: 3355–3359.
Meinecke I, Cinski A, Baier A, Peters MA, Dankbar B, Wille A et al. Modification of nuclear PML protein by SUMO-1 regulates Fas-induced apoptosis in rheumatoid arthritis synovial fibroblasts. Proc Natl Acad Sci USA 2007; 104: 5073–5078.
Sherman JM, Stone EM, Freeman-Cook LL, Brachmann CB, Boeke JD, Pillus L . The conserved core of a human SIR2 homologue functions in yeast silencing. Mol Biol Cell 1999; 10: 3045–3059.
Hecker CM, Rabiller M, Haglund K, Bayer P, Dikic I . Specification of SUMO1- and SUMO2-interacting motifs. J Biol Chem 2006; 281: 16117–16127.
Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y . Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc Natl Acad Sci USA 2004; 101: 14373–14378.
Coussens M, Maresh JG, Yanagimachi R, Maeda G, Allsopp R . Sirt1 deficiency attenuates spermatogenesis and germ cell function. PLoS ONE 2008; 3: e1571.
Khan MM, Nomura T, Kim H, Kaul SC, Wadhwa R, Zhong S et al. PML-RARalpha alleviates the transcriptional repression mediated by tumor suppressor Rb. J Biol Chem 2001; 276: 43491–43494.
Wu WS, Vallian S, Seto E, Yang WM, Edmondson D, Roth S et al. The growth suppressor PML represses transcription by functionally and physically interacting with histone deacetylases. Mol Cell Biol 2001; 21: 2259–2268.
Gao C, Ho CC, Reineke E, Lam M, Cheng X, Stanya KJ et al. Histone deacetylase 7 promotes PML sumoylation and is essential for PML nuclear body formation. Mol Cell Biol 2008; 28: 5658–5667.
Zhao X, Sternsdorf T, Bolger TA, Evans RM, Yao TP . Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol Cell Biol 2005; 25: 8456–8464.
Djavani M, Rodas J, Lukashevich IS, Horejsh D, Pandolfi PP, Borden KL et al. Role of the promyelocytic leukemia protein PML in the interferon sensitivity of lymphocytic choriomeningitis virus. J Virol 2001; 75: 6204–6208.
Palmero I, Serrano M . Induction of senescence by oncogenic Ras. Methods Enzymol 2001; 333: 247–256.
Marcos-Villar L, Lopitz-Otsoa F, Gallego P, Muñoz-Fontela C, González-Santamaría J, Campagna M et al. KSHV protein LANA2 disrupts PML oncogenic domains and inhibits PML-mediated transcriptional repression of Survivin gene. J Virol 2009; 83: 8849–8858.
Gil J, Garcia MA, Gomez-Puertas P, Guerra S, Rullas J, Nakano H et al. TRAF family proteins link PKR with NF-kappa B activation. Mol Cell Biol 2004; 24: 4502–4512.
Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B . A model for p53-induced apoptosis. Nature 1997; 389: 300–305.
Acknowledgements
We acknowledge the technical assistance of Victoria Jiménez and Maribel Muñoz. We are indebted to Dr. Michael Greenberg (Children's Hospital Center for Life Sciences), Dr. Kun-Sang Chang (MD Anderson Cancer Center, Houston), Dr. Tony Kouzarides (Gurdon Institute, University of Cambridge), Dr. Toren Finkel (National Heart Lung and Blood Institute), and Dr. Keith D Robertson (Shands Cancer Center, University of Florida), for kindly providing reagents. Funding at the laboratory of CR is provided by BFU-2008-03784 and CSIC. ME is supported by SAF2008-02036 and Foundation Botín. M Campagna is supported by Fundación Botín and Juan de la Cierva Programme. D Herranz is supported by a predoctoral fellowship from the Spanish Ministry of Health and from the ‘Francisco Cobos’ Foundation. M Collado is an investigator of the Ramón y Cajal Programme. The funders had no role in the study design, data collection and analysis, and decision to publish, or in the preparation of the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by P Salomoni
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Campagna, M., Herranz, D., Garcia, M. et al. SIRT1 stabilizes PML promoting its sumoylation. Cell Death Differ 18, 72–79 (2011). https://doi.org/10.1038/cdd.2010.77
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2010.77
Keywords
This article is cited by
-
SUMOylation modulates eIF5A activities in both yeast and pancreatic ductal adenocarcinoma cells
Cellular & Molecular Biology Letters (2024)
-
Synergistic roles of CBX4 chromo and SIM domains in regulating senescence of primary human osteoarthritic chondrocytes
Arthritis Research & Therapy (2023)
-
PML: Regulation and multifaceted function beyond tumor suppression
Cell & Bioscience (2018)
-
Cervical cancer is addicted to SIRT1 disarming the AIM2 antiviral defense
Oncogene (2018)
-
Phosphorylable tyrosine residue 162 in the double-stranded RNA-dependent kinase PKR modulates its interaction with SUMO
Scientific Reports (2017)
