
Abstract
Phagocytosis serves as one of the key processes involved in development, maintenance of tissue homeostasis, as well as in eliminating pathogens from an organism. Under normal physiological conditions, dying cells (e.g., apoptotic and necrotic cells) and pathogens (e.g., bacteria and fungi) are rapidly detected and removed by professional phagocytes such as macrophages and dendritic cells (DCs). In most cases, specific receptors and opsonins are used by phagocytes to recognize and bind their target cells, which can trigger the intracellular signalling events required for phagocytosis. Depending on the type of target cell, phagocytes may also release both immunomodulatory molecules and growth factors to orchestrate a subsequent immune response and wound healing process. In recent years, evidence is growing that opsonins and receptors involved in the removal of pathogens can also aid the disposal of dying cells at all stages of cell death, in particular plasma membrane-damaged cells such as late apoptotic and necrotic cells. This review provides an overview of the molecular mechanisms and the immunological outcomes of late apoptotic/necrotic cell removal and highlights the striking similarities between late apoptotic/necrotic cell and pathogen clearance.
Similar content being viewed by others
Main
The immune system is constantly under pressure to accurately distinguish foreign materials or pathogens (non-self) from normal healthy tissues (self), and make an appropriate immune response to non-self molecules through a range of effector mechanisms. It is equally important for the immune system to distinguish healthy viable cells (self) from dying cells (altered-self) during the course of tissue remodelling or tissue injury to prevent the release of intracellular molecules that may damage neighbouring cells or stimulate an immunogenic response against self (i.e., an autoimmune response). Professional phagocytes of the innate immune system use a broad range of germline-encoded receptors and opsonins to discriminate viable cells from pathogens and dying cells, and aid the removal of non-self and altered-self through phagocytosis.1, 2 Not surprisingly, impairment of phagocytosis due to a deficiency in key phagocytic components such as C1q, one of the first components of the classical complement cascade, has been implicated in an increased susceptibility to bacterial infection3 as well as in the development of autoimmune diseases such as systemic lupus erythematosus (SLE).4 Therefore, understanding the molecular mechanisms of phagocytosis will provide new insights into numerous physiological and pathological processes.
Under normal physiological conditions, cells of a multicellular organism predominantly die through the well-defined process known as apoptosis or programmed cell death (see Elmore5 for an extensive review). During different stages of apoptotic cell death, a precise set of morphological and biochemical changes occur within the dying cell.5 Dying cells can be characterized as early apoptotic cells, in which the plasma membrane remains intact but exposes phosphatidylserine (PS) on the cell surface to mediate its recognition by phagocytes. Early apoptotic cells can become late apoptotic cells, also known as secondary necrotic cells, when the plasma membrane becomes permeabilized.2 Alternatively, direct exposure of healthy viable cells to trauma, such as extreme temperature, mechanical, and chemical insults, can lead to the generation of membrane-permeabilized necrotic cells (i.e., primary necrotic cells).6 Importantly, depending on the type of dying cells (e.g., early apoptotic versus necrotic cells), recognition and internalization by phagocytes can result in an anti- or a pro-inflammatory response.1, 7 In addition, uptake of apoptotic cells by macrophages can also promote cell growth8 and wound healing9 through the release of vascular endothelial growth factor (VEGF) and transforming growth factor-β (TGF-β), respectively. Thus, the process of phagocytosis in mammals not only serves as an effector mechanism to clear infectious agents, dying cells, or unwanted materials, but also has a fundamental role in determining the subsequent adaptive immune response towards the phagocytosed materials as well as orchestrating the processes of regeneration and angiogenesis at sites of tissue injury. Furthermore, phagocytosis of dying cells, especially early apoptotic cells in an anti-inflammatory context, has a vital role in maintaining immunological tolerance against cell-associated antigens.10
In recent years, significant progress has been made in identifying the molecular and cellular components that are involved in the removal of dying cells. The current review will focus on (1) the complexity of phagocytosis of pathogens and dying cells, (2) the molecular mechanisms of dying cell uptake, in particular the disposal of late apoptotic/necrotic cells, (3) the immunological consequences of late apoptotic/necrotic cell clearance and (4) the striking similarities between pathogen and late apoptotic/necrotic cell removal.
Recognition of Target Cells for Phagocytosis
Although phagocytosis can simply be viewed as the engulfment of large particles (>0.5 μm) by cells through an actin-dependent mechanism, molecular events that govern different phases of phagocytosis are extremely complex11 (Figure 1). To mediate efficient removal of target cells, professional phagocytes such as macrophages, monocytes, microglia, neutrophils and dendritic cells (DCs) are located at high density in specific tissues (e.g., alveoli, spleen and brain), in which there is an increased chance of encountering target cells or being actively attracted to target cells through the so-called ‘come-get-me’ signals.12 For example, generation of C5a molecules after complement activation on microbial surfaces can stimulate chemotaxis of neutrophils, macrophages and monocytes.13, 14 Similarly, early apoptotic cells can release chemotactic factors to attract phagocytes towards sites of apoptotic cell death. The release of apoptotic blebs from human tonsil germinal centre B cells that are dying by apoptosis in culture has been shown to stimulate the migration of human monocytes.15 In addition, the release of soluble molecules such as lysophosphatidylcholine (LPC),16 endothelial monocyte-activating polypeptide II (EMAP II)17 and fractalkine/CX3CL118 by early apoptotic cells has also been suggested to promote the recruitment of macrophages and monocytes towards cells undergoing apoptosis. Interestingly, apoptotic leukocytes (HL-60 cells) generated by heat treatment can secret both S19 ribosomal homodimer and a serine protease to promote and subsequently retard monocyte migration, respectively, during the early and later stages of apoptotic cell death.19 Besides facilitating the chemotaxis of mononuclear phagocytes, induction of apoptosis can also trigger the release of lactoferrin, an anti-inflammatory glycoprotein that selectively inhibits the migration of neutrophils towards early apoptotic cells, possibly to prevent any aberrant inflammation at sites of physiological forms of cell death.20 Although such an elegant phagocyte recruitment system may be necessary in mediating efficient clearance of apoptotic cells at certain tissues, it should be noted that non-professional phagocytes, such as fibroblasts, endothelial and epithelial cells, can also contribute to removing adjacent cells undergoing apoptosis (see Majai et al.21 for an extensive review).21

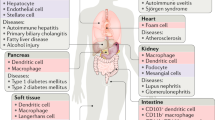
Schematic representation of the phases of phagocytosis and the recognition of target cells. The initial interaction between phagocytes and target cells involves adhesion molecules as well as recognition receptors that can detect, either directly or indirectly through opsonins, various molecules exposed on target cells. Target cells may also release soluble factors that either enhance or inhibit the migration of phagocytes towards the site of homoeostatic cell death, tissue injury or infection. Multiple interactions between a phagocytic cell and a target cell will lead to the formation a ‘phagocytic synapse’, which triggers various intracellular signalling events and induces the reorganization of the cytoskeleton to mediate target cell uptake. After the engulfment process, the internalized target cell is delivered into the phagosome, which will mature into the phagolysosome for target cell killing and degradation. Phagocytic uptake of a target cell can trigger the release of anti- and pro-inflammatory cytokines and growth factors, as well as the presentation of peptides derived from the target cell to T cells through both major histocompatibility complex (MHC) class I and II molecules. Depending on the type of target cell, different levels and types of the so-called ‘don’t-eat-me’ and ‘eat-me’ signals will be exposed. Viable cells expose self-associated molecular patterns (SAMPs) to prevent phagocytosis, whereas pathogens and dying cells (e.g., early and late apoptotic cells) expose pathogen-associated molecular patterns (PAMPs) and apoptotic cell-associated molecular patterns (ACAMPs), respectively, to mediate their uptake by phagocytes. Note that plasma membrane-damaged cells (i.e., late apoptotic and necrotic cells) and pathogens can also release or expose endogenous and exogenous ‘danger’ signals, known as alarmins and PAMPs, respectively, to alert the organism to tissue injury and infection. TCR, T-cell receptor
When a phagocyte comes into close proximity to a target cell, the target cell must expose a sufficient level of ‘eat-me’ signals to trigger phagocytosis. For phagocytes to discriminate pathogens from host cells, Janeway22 proposed that a limited number of germline-encoded pattern recognition molecules (PRMs) are used by the innate immune system to detect conserved molecular structures known as pathogen-associated molecular patterns (PAMPs) exposed on pathogens but absent from healthy host cells (Figure 1). PRMs that are important in detecting PAMPs include the macrophage mannose receptor, macrophage scavenger receptors, C1q, mannose-binding lectin (MBL), C-reactive protein (CRP) and serum amyloid protein (SAP), most of which recognize specific carbohydrate or polyanionic structures exposed on pathogens.23 Similarly, the ability of phagocytes to distinguish dying cells, such as early apoptotic cells from viable cells, also relies on the exposure of the so-called apoptotic-cell-associated molecular patterns (ACAMPs) on apoptotic cells24 (Figure 1). However, because of the complexity of the molecular pathways that initiate cell death as well as the continuous progression through different stages of cell death,5 it is not appropriate to use the term ACAMPs to describe molecular structures exposed by all types of dying cells. To illustrate the complexity of the phagocytic signals exposed by self, altered-self and non-self cells, a number of selected examples will be discussed below and are schematically represented in Figure 2.

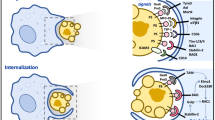
Complexity of phagocytic, immunomodulatory and chemotactic signals exposed on target cells. Phagocytic cells are equipped with a variety of germline-encoded detection mechanisms to distinguish self (viable cells), altered-self (dying cells) and non-self (pathogens) based on the combination of molecular signals exposed on the target cell. For example, viable cells expose a combination of ‘don’t-eat-me’ signals such as CD47, CD31 and specific carbohydrate structures to prevent their uptake by phagocytes through a variety of mechanisms. When cells undergo apoptosis, various ‘eat-me’ signals, such as PS and DNA, are rapidly exposed and several ‘don’t-eat-me’ signals are lost or modified to formulate a specific combination of signals that enhance phagocytosis. When the plasma membrane of apoptotic cells becomes permeabilized (i.e., late apoptotic cells), additional molecules are revealed to shape a new combination of ‘eat-me’ and ‘don’t-eat-me’ signals. Intracellular autoantigens can also be exposed once the plasma membrane is damaged. It is important to acknowledge that the process of cell death is extremely complex and can lead to the exposure of different molecules depending on the pathway of cell death. For example, necrotic cells will not expose molecules that are modified during apoptosis and thus will show a different combination of phagocytic signals compared with late apoptotic cells. In addition, phagocytic cells can recognize unique structures carried by pathogens (e.g., LTA, PCh, flagellin and CpG DNA). A variety of soluble factors are also released by the target cells to attract phagocytes (e.g., LPC) or to directly modulate the immunological outcome of phagocytosis (e.g., TGF-β, HMGB1 and LPS). A, autoantigens; DNA, deoxyribonucleic acid; HMGB1, high-mobility group box 1 protein; LPC; lysophosphatidylcholine; LPS, lipopolysaccharide; LTA, lipoteichoic acid; PCh, phosphocholine; PS, phosphatidylserine; TGF-β, transforming growth factor-β
Besides insufficient levels of ‘eat-me’ signals on healthy host cells, viable cells can actively expose ‘don’t eat-me’ signals (also known as self-associated molecular patterns (SAMPs)) to prevent their uptake by phagocytes25 (Figure 1). Expression of CD47, a ubiquitous member of the immunoglobulin (Ig) superfamily, on viable cells can deliver a negative engulfment signal through the immune inhibitory receptor SIRP-α on macrophages.26 Similarly, another member of the Ig superfamily, CD31 (PECAM-1), can also mediate the detachment of viable leukocytes from macrophages through the homophilic interaction between CD31 expressed on both cell types.27 Furthermore, the inhibition of the complement cascade on viable cells by complement regulators such as CD46 and factor H can prevent the recognition of viable cells by complement receptors (CR) expressed on various phagocytic cells.14 Intriguingly, although the function of putative anti-chemotactic factors in normal healthy tissues is not well understood,28 they may also participate by preventing the interaction between phagocytes and viable cells.
In contrast, when cells undergo apoptosis in response to either extrinsic or intrinsic mediators, a distinct set of morphological (i.e., cell shrinkage and membrane blebbing) and biochemical (e.g., activation of caspases) changes occur.5 These include the release of ‘come-get-me’ signals, the exposure of ‘eat-me’ signals, as well as the loss of ‘don’t eat-me’ signals that collectively facilitate apoptotic cell clearance.12 The loss of phospholipid asymmetry of the plasma membrane during the early stages of apoptosis can lead to the exposure of anionic phospholipids such as PS, which can be recognized by a number of phagocytic receptors and opsonins such as T-cell immunoglobulin mucin (Tim-1) and Tim-4, Stabilin-2, brain-specific angiogenesis inhibitor 1 (BAI1) and scavenger receptors, as well as the opsonins β2-glycoprotein I (β2GPI), milk fat globular-EGF factor 8 protein (MFG-E8) and Protein S.29, 30 It is also important to note that phagocytosis of target cells (e.g., early apoptotic cells) is dependent on both the quantity and quality of positive (i.e., ‘eat-me’) and negative (i.e., don’t eat-me’) phagocytic signals. For example, ‘eat-me’ signals such as PS are not totally absent on viable cells, with a sufficient threshold level of externalized PS being required to initiate phagocytosis.31 In addition, activation of apoptotic pathways can lead to the modification or loss of ‘don’t eat-me’ signals such CD47,26 CD3127 and CD46,32 which can further favour the uptake of apoptotic cells. It is worth noting that the significance of CD47 and CD31 in regulating phagocytosis of early apoptotic cells is only apparent under specific conditions, for example, in the presence or absence of serum opsonins,33, 34 indicating that certain positive or negative phagocytic signals may dominate over other phagocytic signals.
If apoptotic cells persist due to an overload of dying cells and/or an impairment in phagocytosis, early apoptotic cells will progress into late apoptotic cells (i.e., secondary necrotic cells), the states in which the cell membrane becomes more permeable.7 Similarly, generation of (primary) necrotic cells by extreme trauma can also result in permeabilization of the cell membrane.6 Besides physical and chemical insults, necrosis can also be induced by specific signals (e.g., tumour necrosis factor-α (TNF-α) and double-stranded ribonucleic acid (dsRNA)) and controlled by a set of signalling pathways, a process termed necroptosis.35 Importantly, the loss of membrane integrity exposes intracellular molecules that are previously hidden and may exert an effect as additional ‘eat-me’ signals to facilitate the clearance of membrane-damaged cells. Although late apoptotic cells and necrotic cells are often grouped together simply as permeabilized cells (i.e., cells staining positive for PS-binding annexin V and various membrane-impermeable dyes such as Trypan blue, propidium iodine and Hoechst 33258), they are likely to expose different combinations of phagocytic signals because of differences in the molecular events that have occurred before membrane damage (see Figure 2). Although many of the intracellular ‘eat-me’ signals are not well characterized, a growing number of opsonins and receptors have been identified recently that can specifically aid the removal of late apoptotic/necrotic cells (discussed in detail below).
Similar to the ‘immunological synapse’ formed between a T-cell and an antigen-presenting cell, the interaction between a phagocyte and a target cell also requires a combination of both adhesion and recognition molecules to mediate the formation of a ‘phagocytic synapse’,1 which has an important role in initiating various signalling events that trigger cytoskeletal reorganization and the engulfment of the target cell through an actin-based mechanism11 (Figure 1). After the engulfment process, the internalized target cell is delivered to a membrane-limited organelle known as the phagosome, which will eventually mature into a phagolysosome through a series of fusion and fission events with endosomes and lysosomes.1 Most importantly, phagocytic cells such as DCs and macrophages are equipped with the necessary molecular machinery to kill and degrade the internalized target cell, and subsequently process and present antigens derived from the target cell to the adaptive immune system through both major histocompatibility complex (MHC) class I and II molecules1 (Figure 1). Interestingly, besides simply mediating the uptake of dying cells, evidence from Caenorhabditis elegans and mammalian macrophage/microglia studies suggest that phagocytes can also promote cell death during development and tissue remodelling to ensure that certain types of cells are eliminated and cleared appropriately.7
In addition to regulating various biological processes, such as cell proliferation and differentiation, angiogenesis, tissue remodelling and development, the recognition and uptake of target cells can also have an important role in determining the subsequent immunological response at the site in which the phagocytosed material was initially identified. Whether an anti- or a pro-inflammatory response may be initiated depends largely on the type of immunomodulatory signals being released or exposed by the target cell.29 For example, the display of certain PAMPs (also known as exogenous ‘danger’ signals), such as lipopolysaccharide (LPS), lipoteichoic acid (LTA), flagellin and peptidoglycans, on bacteria can activate Toll-like receptors (TLRs) and trigger the release of pro-inflammatory cytokines to alert the host organism to invading pathogens.36, 37 Similarly, the recognition of virus-derived dsRNA and intracellular bacterial-derived peptidoglycans by cytosolic PRMs, such as retinoic acid-inducible gene-I (RIG)-like receptors and nucleotide-binding oligomerization domain (NOD)-like receptors, can also induce the production of pro-inflammatory cytokines.38 In contrast, the uptake of early apoptotic cells by phagocytes can inhibit the secretion of pro-inflammatory cytokines and promote the production of anti-inflammatory cytokines to mediate a nonphlogistic clearance of dying cells. In addition, T cells undergoing apoptotic cell death can also foster an anti-inflammatory response by directly releasing TGF-β and interleukin (IL)-10.10, 29 Interestingly, late apoptotic and necrotic cells can also release endogenous molecules called ‘alarmins’, which function as endogenous ‘danger’ signals and elicit a pro-inflammatory response to indicate tissue injury.6 Well-characterized alarmins include uric acid/monosodium urate (MSU) crystals and high-mobility group box 1 (HMGB1) molecules.39 Both exogenous ‘danger’ signals (i.e., PAMPs) and endogenous ‘danger’ signals (i.e., alarmins) belong to a subgroup of molecules known as danger-associated molecular patterns (DAMPs).6, 40
Therefore, the initial interaction between the phagocyte and the target cell may have a critical role in determining whether an immunogenic or a tolerogenic adaptive immune response will be raised against the internalized material. Although a detailed discussion of how an immunogenic or a tolerogenic immune response is developed and maintained is beyond the scope of this review, it is important to note that the quality and the types of immune response generated towards the phagocytosed material is also influenced by a variety of factors and regulatory mechanisms that are present at different stages of the immune response. These include maturation of DCs and antigen presentation, T- and B-cell activation and clonal expansion as well as control of the immune response by regulatory T cells.1, 10
Molecular Mechanisms of Late Apoptotic/Necrotic Cell Removal
According to the ‘waste disposal’ hypothesis41 and the ‘danger’ hypothesis,42 a failure in apoptotic cell clearance or cell trauma can lead to the generation of plasma membrane-damaged cells (e.g., late apoptotic and necrotic cells) and the exposure of both immunostimulatory molecules and autoantigens to the immune system. Subsequently, phagocytic uptake of late apoptotic/necrotic cells by antigen-presenting cells, such as DCs, can lead to the presentation of autoantigens to autoreactive T cells in a pro-inflammatory context, thus facilitating the onset of an autoimmune response. As apoptotic cell removal is an important ‘check-point’ for autoimmunity, a substantial amount of effort in the last decade has been devoted to identifying the molecular mechanisms of apoptotic cell uptake and is extensively reviewed elsewhere.2, 7, 25, 30 Recently, a number of studies have discovered several novel phagocytic pathways of late apoptotic/necrotic cell recognition and uptake (see Figure 3 and below). A better understanding of the molecular mechanisms underpinning these pathways may shed light on the differences observed in the immunological outcomes after the clearance of early apoptotic cells versus late apoptotic/necrotic cells by phagocytes. Some of the molecular mechanisms involved in late apoptotic/necrotic cell clearance are summarized below.

Molecular mechanisms of late apoptotic/necrotic cell clearance. The interaction between phagocytes and membrane-damaged late apoptotic/necrotic cells involves a variety of receptors, opsonins and cell-associated ligands as indicated. Besides the depicted phagocytic pathways, molecular mechanisms of early apoptotic cell uptake are likely to operate in concert with these pathways to aid late apoptotic and necrotic cell removal. Note that CRP, SAP, PTX3, HRG, Ficolin-2, Ficolin-3, MBL and C1q can all potentially modulate the activation of complement, in addition to IgM, IgG and properdin, to facilitate late apoptotic/necrotic cell clearance. CRP, C-reactive protein; DNA, deoxyribonucleic acid; FcγR, Fcγ receptor; HERDS, heterogeneous ectopic ribonucleoprotein-derived structures; HRG, histidine-rich glycoprotein; HSPG, heparan sulphate proteoglycans; Ig, immunoglobulin; LPC; lysophosphatidylcholine; MBL, mannose-binding lectin; PS, phosphatidylserine; PTX3, pentraxin-3; SAP, serum amyloid protein; TSP-1, thrombospondin-1
Classical complement pathway
The complement system has been known to have a critical role in providing the first line of host defence against intruding pathogens. Complement components can be activated on the surface of pathogens through three different pathways: the classical pathway, the lectin pathway and the alternative pathway. Activation of the complement cascade serves three major roles, namely (1) inducing inflammation through fragments of complement components (e.g., C3a, C4a, C5a), (2) opsonization of pathogens for phagocytosis (e.g., through C3b) and (3) direct killing of pathogens through the assembly of the membrane-attack complex (MAC).13 In addition, recent studies have implicated the complement system in dying cell clearance, in particular the complement component C1q of the classical pathway.
C1q is a member of the collagen family and serves as a key PRM of the classical complement pathway. C1q interacts with pathogen surfaces either through direct interaction with charged and hydrophobic structures or indirectly through pathogen-binding IgG or IgM and subsequently promotes the activation of the serine proteases C1r and C1s.13 Initially, both C1q and factor B were identified as key components in human serum that can enhance the phagocytic uptake of early apoptotic serum-starved human neutrophils and radiation-treated murine thymocytes through CR3 and CR4 on human monocyte-derived macrophages (HMDMs), suggesting that the activation of both the classical and alternative pathways are required to aid early apoptotic cell clearance.43 However, it was subsequently shown that C1q alone can bind directly to membrane blebs on the surface of UV-treated early apoptotic Jurkat T cells,44 possibly through PS,43 cardiolipin45 or deoxyribonucleic acid (DNA) recognition,14 and augment early apoptotic cell removal through calreticulin and CD91 on HMDMs.44 The importance of C1q and the classical pathway in apoptotic cell disposal was further shown by the impairment of dexamethasone-treated early apoptotic murine thymocytes uptake in C1q-deficient mice in vivo as well as UV-treated early apoptotic Jurkat T-cell removal in C1q-deficient patients in vitro.4 Interestingly, complement binding and activation was later found to occur predominantly on heat-killed necrotic peripheral blood lymphocytes (PBLs) and subcellular fragments of radiation-treated late apoptotic PBLs.46 Furthermore, the deposition of C3 and C4 (critical components of the classical complement cascade) on etoposide-treated late apoptotic and heat-killed necrotic Jurkat T cells was also found to be highly dependent on C1q.47 Although phagocytic assays were not performed in these studies to validate the role of complement in late apoptotic and necrotic cell clearance, later studies by Gaipl et al.48 showed that the presence of C1q can enhance the uptake of heat-killed necrotic PBLs by HMDMs.48 Recent studies by Gullstrand et al.49 also showed that an intact classical complement pathway is necessary to aid the phagocytic removal of a mixture of staurosporine-treated early and late apoptotic Jurkat T cells by HMDMs.49 It is worth noting that the classical complement pathway can also be activated indirectly by late apoptotic and necrotic cells through bridging molecules such as IgG, IgM, HRG, CRP and SAP (discussed below).
IgM and IgG
Antibody has a fundamental role in the adaptive immune response against foreign antigens (1) by forming immune complexes that neutralize and mediate the clearance of pathogen-derived toxins/substances, (2) by opsonizing pathogens to prevent further infection and facilitate their removal by professional phagocytes and (3) by activating the classical complement pathway.50 In the event of autoimmunity, autoantibodies can also be induced against endogenous molecules, such as IgG, in patients with rheumatoid arthritis51 and nuclear components in SLE patients.52 Interestingly, serum IgM antibodies have been shown to have an important role in apoptotic cell removal in vitro and in vivo by facilitating C1q binding and the activation of the classical complement pathway on apoptotic cells.53, 54, 55 Importantly, activation of complement on cells at all stages of cell death is largely dependent on IgM.47, 53, 54, 55, 56 Initially, ‘natural’ IgM antibodies were found to bind specifically to structures exposed on staurosporine-treated late apoptotic and heat-killed necrotic peripheral blood-derived T cells (PBTs),54 possibly by interacting with both LPC54 and oxidized phospholipids.57 More recent studies have also consistently shown that both plasma IgM,47, 56 and poly-reactive ‘natural’ IgM (3B4),58 bind preferentially to late apoptotic etoposide-treated Jurkat T cells47, 56 and dexamethasone-treated murine thymocytes58 and mediate complement activation.47, 56 These studies are in agreement with others that show that complement binding occurs mainly on membrane-damaged late apoptotic and necrotic cells.46 Furthermore, it is worth noting that similar to C1q-deficient mice, mice lacking serum IgM also develop lupus-like autoimmune diseases,59 which seems to be a result of an impairment in dying cell clearance. In addition to a complement-dependent mechanism, IgM can potentially aid plasma membrane-damaged cell clearance through Fc-α/μ receptors, which have been shown to mediate the endocytosis of IgM-coated microbes.60 Besides IgM, both plasma IgG47, 56 and IgG autoantibodies from SLE patients61, 62 were found to bind largely to etoposide-/radiation-treated late apoptotic and heat-killed necrotic cells. However, the precise role of these ‘natural’ and ‘auto’ IgGs in the disposal of late apoptotic/necrotic cells through both complement-dependent and Fcγ receptor (FcγR)-dependent pathways requires further analyses.
Mannose-binding lectin (MBL)
MBL, a member of the collectin family, functions as a PRM that can recognize carbohydrates such as D-mannose and N-acetyl-D-glucosamine (GlcNAc) on microbial surfaces and mediate pathogen removal through the activation of the lectin complement pathway through MBL-associated serine protease (MASP). Besides serving as a circulating innate defence protein against infectious agents, MBL has been shown recently to interact with a variety of endogenous ligands such as dying cells, immunoglobulins, nucleic acids and phospholipids.63 Similar to C1q, MBL was first shown to bind specifically to UV-treated early apoptotic Jurkat T cells and facilitate their removal through calreticulin and CD91 on HMDMs.44 However, later studies showed that MBL binds strongly to radiation-treated late apoptotic and heat-killed necrotic Jurkat T cells, but not early apoptotic cells, through the lectin domain.64 Interestingly, in contrast to the diffuse binding pattern of MBL on certain live cells, such as thymocytes and fibroblasts, MBL was found to cluster on the surface of serum-starved apoptotic thymocytes, suggesting that a clustering of MBL on dying cell surfaces is required for MBL to trigger phagocytosis,65 possibly interacting with fragmented dsDNA that are exposed on cells undergoing apoptosis.66 Although both C1q and MBL compete for the same or adjacent structures on late apoptotic cells,64 C1q may dominate over MBL in enhancing dying cell clearance as the concentration of circulating C1q in human plasma is usually ∼100-fold higher than that of MBL. Indeed, in vivo studies showed that MBL-deficient mice have impaired apoptotic cell clearance but do not develop severe autoimmunity similar to the C1q-deficient mice.65 Nevertheless, as MBL has been suggested to function as an acute-phase protein,67 MBL may have an important role in late apoptotic/necrotic cell removal during the acute-phase response.
Ficolin-2 and Ficolin-3
Similar to MBL, other serum lectins such as Ficolin-2 (L-Ficolin) and Ficolin-3 (H-Ficolin) have also been shown to mediate the clearance of late apoptotic and necrotic Jurkat T cells.68, 69 In humans, three kinds of ficolin have been identified (Ficolin-1, -2 and -3) that show both structural and functional similarities to MBL and C1q.70 Similar to MBL, ficolins can recognize specific carbohydrate structures, such as GlcNAc exposed on the surface of pathogens, and activate the lectin complement pathway through association with MASP.70 Consistent with earlier studies by Kuraya et al.,71 both Ficolin-268 and Ficolin-369 are able to bind to etoposide-treated apoptotic cells, in particular late apoptotic cells as well as heat-killed necrotic Jurkat T cells, and function as opsonins to aid phagocytic uptake by HMDMs. Furthermore, Ficolin-2 was also found to be an important serum factor in activating complement on necrotic cells but not on late apoptotic cells.68 Although it was proposed that Ficolin-2 may exert an effect as an adaptor molecule between exposed DNA on late apoptotic/necrotic cells68, 69 and calreticulin on phagocytes,71 further studies (e.g., using appropriate blocking antibodies) are necessary to elucidate the underlying mechanisms.
Properdin
Besides the components of the classical and the lectin complement pathways, properdin, the only known positive regulator of complement activation, has been shown recently to mediate activation of the alternative complement pathway on cells at all stages of cell death, including necrotic cells.72, 73 Properdin, a single-chained glycoprotein that is present in plasma as cyclic polymers, was proposed to have an important role in the enhancement of complement activation through its interaction with C3b and stabilization of the C3 and C5 convertase complexes.74 Although properdin is only present at the relatively low concentration of ∼5–15 μg/ml in plasma, it can be stored in neutrophil granules and released at sites of inflammation.74 In addition to binding C3b-coated surfaces, recent studies have also shown that properdin can initiate complement activation through direct interaction with microbial surfaces.75 Interestingly, studies by Xu et al.72 showed that properdin binds exclusively to UV-treated late apoptotic and heat-killed necrotic Jurkat T cells independently of C3b, possibly through the recognition of exposed DNA at the later stages of cell death.72 Although conflicting studies have reported that properdin binds predominately to anti-Fas/staurosporin-treated early apoptotic human primary T cells through cell-surface sulphated glycosaminoglycans (GAGs) and aids their uptake by human monocytes-derived dendritic cells (HMDDCs) and HMDMs, either in the presence or absence of complement activation,73 it has been suggested that properdin may also have a critical role in regulating complement activation through the alternative pathway on late apoptotic and necrotic cells.72
Pentraxin family
Pentraxins are a superfamily of proteins characterized by the presence a 200 amino acid pentraxin domain at the C-terminus. Pentraxins can be further subdivided into the classic short pentraxins, such as CRP and SAP, and long pentraxins such as pentraxin-3 (PTX3).76 In humans, CRP is a major acute-phase plasma protein, in which the serum concentration can rapidly increase in response to infection or tissue injury. Being one of the first innate immunity molecules identified, it is well known that CRP binds to phosphocholine (PCh). PCh is a major constituent of many bacterial and fungal polysaccharides and most biological cell membranes, with CRP aiding the removal of pathogens by acting as an adaptor molecule that binds PCh and activates the classical complement pathway through its interaction with C1q. Although the literature regarding the interaction between CRP and various FcγR has been controversial, numerous studies have suggested that CRP can also exert an effect as an opsonin by binding to FcγR.77 In addition to the elimination of pathogens, studies by Gershov et al.78 showed that CRP can also bind to anti-Fas/staurosporin-treated early apoptotic Jurkat T cells in a Ca2+-dependent manner and enhance apoptotic cells uptake by HMDMs when C1q-containing serum is present.78 The researchers also showed that the binding of CRP to early apoptotic Jurkat T cells enhanced the activation of the classical complement pathway but prevented the assembly of the MAC,78 probably by tethering factor H (a complement regulatory protein) to the surface of early apoptotic cells.78, 79 Similarly, CRP has also been proposed to mediate the removal of UV-treated early apoptotic Jurkat T cells through a FcγR-dependent mechanism, based on studies using peritoneal macrophages from Fc receptor common γ-chain-deficient mice.80 Interestingly, recent studies showed that CRP binds exclusively to aged membrane-damaged late apoptotic neutrophils,81 possibly to nuclear structures77 or oxidized phospholipids,82 but had no apparent effect on the phagocytosis of late apoptotic neutrophils by HMDMs in the absence of serum.81 Thus, it is possible that CRP may function predominantly through the classical complement pathway to aid the clearance of dying cells, especially late apoptotic cells, with the role of FcγR in this process still being controversial. Besides binding directly to dying cells, recent studies by Mihlan et al.83 showed that CRP can also be recruited onto heat-killed necrotic cells by another necrotic cell-binding protein, human complement factor H-related protein 4 (CFHR4).83
SAP, another classic short pentraxin, is constitutively present in human serum at ∼30–40 μg/ml and represents a major acute-phase reactant in mice. Similar to other pentraxins, SAP was found to bind complement components, bacteria, various carbohydrate determinants as well as viruses. Interestingly, SAP can also bind chromatin subunits and DNA.76 Similar to CRP, SAP can interact with various FcγR84 and mediate the uptake of UV-treated early apoptotic Jurkat T cells by peritoneal macrophages through a FcγR-dependent mechanism.80 However, later studies showed that SAP binds predominantly to permeabilized radiation-treated late apoptotic Jurkat T cells in a Ca2+-dependent manner and aids the phagocytosis of late apoptotic cells by HMDMs in phagocytic assays containing serum.85 Thus, SAP can potentially mediate the uptake of late apoptotic/necrotic cells through both complement- and FcγR-dependent pathways.
The long pentraxin, PTX3, is a cytokine-inducible innate defence molecule that recognizes microbes and activates the classical complement pathway to enable the removal of pathogens.76 PTX3 binds predominately to UV-treated late apoptotic Jurkat T cells but not to necrotic cells generated by chemical, extreme temperature or mechanical treatments.86 Unlike CRP and SAP, PTX3 binding to UV-treated late apoptotic Jurkat T cells is independent of Ca2+,86 possibly interacting with histones exposed on membrane-damaged cells.76 Paradoxically, PTX3 inhibits the uptake of late apoptotic Jurkat T cells by HMDDCs86 and dampens the ability of HMDDCs to cross-present cell-associated antigens derived from the ingested apoptotic cells, which may prevent the activation of autoreactive T cells.87, 88 Furthermore, PTX3 was found to reduce C1q-enhanced clearance of UV-treated early apoptotic HeLa cells by HMDDCs88 as well as SAP-mediated phagocytosis of aged membrane-permeable late apoptotic neutrophils by HMDMs.89 In contrast, recent studies by Jaillon et al.90 showed that endogenous PTX3 translocates to the surface of aged membrane-damaged late apoptotic neutrophils and aids their removal by HMDMs.90 Therefore, it is of great interest to analyse whether PTX3-deficient animals are more or less prone to autoimmunity and are able to adequately dispose of late apoptotic cells. Interestingly, the ability of PTX3 to bind directly to FcγRIII84 also suggests that PTX3 may regulate the uptake of late apoptotic cells through FcγR.
Histidine-rich glycoprotein (HRG)
HRG, a member of the cystatin supergene family, is an abundant multifunctional protein that is present in the plasma of many vertebrates. HRG has a multidomain structure that allows the molecule to interact with many ligands including heparin, heparan sulphate (HS), plasminogen, fibrinogen, thrombospondin-1 (TSP-1), IgG, FcγR, C1q, haem and Zn2+. The ability of HRG to interact with various ligands simultaneously has suggested that HRG can exert an effect as an adaptor molecule that regulates numerous biological processes such as immune complex and pathogen clearance, angiogenesis, cell adhesion, coagulation and fibrinolysis.91 In addition, studies by Gorgani et al.92 showed that HRG binds more strongly to radiation-treated late apoptotic Jurkat T cells than to viable or early apoptotic cells, possibly by recognizing naked DNA exposed during apoptosis.92 Furthermore, HRG binds to FcγRI on HMDMs and functions as a bridging molecule to enhance the uptake of a mixture of early and late apoptotic cells through a FcγRI-dependent mechanism.92 Similarly, studies by Jones et al.93 also showed that besides cell-surface HS, HRG binds strongly through its N-terminal domains to cytoplasmic ligand(s) exposed on heat-killed necrotic Jurkat T cells and enhances their phagocytic removal by THP-1 monocytic cells.93 Interestingly, HRG has also been shown recently to have a minor role in regulating complement activation on heat-killed necrotic Jurkat T cells, possibly by interacting with complement components such as C1q, factor H and C8.94 As the role of HRG in late apoptotic and necrotic cell clearance in vivo has yet to be examined, further studies using the recently generated HRG-deficient mouse would be of great importance.
Thrombospondin-1 (TSP-1)
TSP-1 is a multifunctional homotrimeric extracellular matrix (ECM) protein that has been implicated in regulating a variety of biological processes, including angiogenesis, chemotaxis, cell adhesion, cell proliferation and apoptosis. TSP-1 has a modular and multidomain structure that allows it to interact with a variety of ligands and receptors such as heparin, Ca2+, integrins, CD36, CD47, fibronectin, latent TGF-β1 and several proteases.95 Interestingly, TSP-1 can also bind directly to peptidoglycans on Gram-positive bacteria and aid their adherence to host cells.96 Although TSP-1 was originally characterized as an α-granule glycoprotein in platelets, it can be synthesized and released by a variety of cell types, including epithelial and mesenchymatous cells,95 as well as macrophages97 and apoptotic monocytes, neutrophils and fibroblasts.98, 99 Importantly, studies by Savill et al.97 and Ren et al.100 showed that TSP-1 has a vital role in the phagocytosis of ageing human neutrophils undergoing apoptosis, possibly by acting as a molecular bridge between late apoptotic cells and CD36 and the αVβ3 integrin (vitronectin receptor) on macrophages.97, 100 Although the apoptotic cell ligand(s) of TSP-1 remains elusive, recent studies suggest that TSP-1 binds predominately to serum-starved late apoptotic monocytes and enhances their uptake by HMDDC.98 Furthermore, TSP-1 has also been shown to aid the clearance of heat-, methanol- and ethanol-killed necrotic PBLs by HMDMs.101
Heparan sulphate proteoglycans (HSPGs)
HSPGs are composed of a protein core and side chains of the complex GAG, HS. HSPGs can be found within the extracellular milieu, including cell surfaces of a variety of cells and tissues, and have been implicated in numerous biological processes, including organization of the ECM as well as cell adhesion, migration, proliferation and differentiation. The ability of HSPGs to regulate various processes often depends on the interaction between the HS component of HSPG and a range of protein ligands such as growth factors, cytokines, chemokines, enzymes and cell-adhesion molecules.102 Furthermore, studies have also shown that HSPGs on epithelial cells can mediate the phagocytosis of HSPG-ligating antibodies-coated latex beads through an actin-dependent mechanism.103 Interestingly, invasive bacteria such as Neisseria gonorrhoeae can hijack the HSPG-dependent phagocytic pathway to facilitate their entry into host epithelial cells through an interaction between HSPG and a pathogen-derived opacity-associated protein.104 Recently, studies by Gebska et al.105 showed that heparin, essentially a more sulphated form of the HS side chains of HSPG, can bind more strongly to camptothecin-treated apoptotic Jurkat T cells than to viable cells, especially to late apoptotic cells. Confocal microscopy studies further indicated that heparin may bind to nuclear materials known as heterogeneous ectopic ribonucleoprotein-derived structures (HERDS). On the basis of the ability of the sulphated polysaccharide pentosan polysulphate to block heparin-binding sites on late apoptotic cells and inhibit their removal by HMDMs, the researchers suggested that HSPGs on macrophages may have an important role in late apoptotic cell clearance.105 However, further studies are necessary to provide direct evidence that HSPGs have a role in mediating late apoptotic/necrotic cell removal.
Molecular mechanisms of early apoptotic cell clearance
As late apoptotic cells originate from uncleared early apoptotic cells, it is not surprising that many of the molecular mechanisms involved in early apoptotic cell clearance are also involved in the phagocytosis of late apoptotic and even necrotic cells. For example, the well-characterized ‘eat-me’ signal PS on early apoptotic cells was shown recently to promote the ability of macrophages to recognize and phagocytosis late apoptotic and necrotic cells.7, 29 Similarly, the glycosylphosphatidyl-inositol-linked plasma-membrane glycoprotein CD14, a PRM that binds the LPS-binding protein (LBP)-LPS complex, was found to be an important receptor on macrophages for phagocytosing dying cells at all stages of cell death and of different cellular origin, possibly by binding to the modified ICAM-3 exposed on dying lymphocytes.106
Immunological consequences of late apoptotic/necrotic cell clearance
The process of phagocytosis not only serves as an effector mechanism to eliminate pathogens and dying cells, but it also has a vital role in orchestrating multiple biological processes, in particular the subsequent adaptive immune response towards the phagocytosed materials. It is often considered that early apoptotic cell uptake is anti-inflammatory and induces a tolerogenic response, whereas late apoptotic and necrotic cell removal is associated with inflammation and promotes immunity/autoimmunity. However, factors that determine the immunological consequences of early apoptotic cell and late apoptotic/necrotic cell clearance are extremely complex and sometimes controversial. The immunological outcome of dying cell removal is influenced by multiple factors such as the type of cell undergoing death, the stages of cell death, the recognition and uptake pathways used by the phagocytes, the types of phagocytes as well as the location and microenvironment of cell death. Owing to this complexity, contrasting results have been reported in the literature,29 possibly due to differences in the experimental models being used. Although the origin of the dying cell (e.g., pancreatic islet cell, lymphocyte or infected cell) may influence the types of cell-associated antigens (e.g., viral proteins in the case of virus infection) being revealed to the immune system, the combination of immunomodulatory signals being exposed by the dying cells and the activation status of the different types of phagocytic cells involved in clearance are key factors in determining the quality and the types of immune response induced.
Membrane integrity of dying cells has been used as one of the key criteria to differentiate different stages of cell death (e.g., early apoptotic versus late apoptotic cells). Functionally, the loss of membrane integrity in late apoptotic and necrotic cells also has a critical role in the release of endogenous ‘danger’ signals (i.e., alarmins) that can induce a pro-inflammatory response through a range of different mechanisms.39 The ‘Danger’ model, originally proposed by Matzinger42, suggests that the immune system has evolved to focus on and respond to materials that are potentially ‘dangerous’, rather than on those that are simply foreign or infectious. Initially, apart from various pathogens, membrane-damaged necrotic cells were also found to directly stimulate an immunogenic response (e.g., by promoting a cytotoxic T-cell response and inducing DC activation) in the absence of exogenous adjuvants.107, 108 Importantly, the immunostimulatory activity of membrane-damaged cells was found to be constitutively present mainly in the cytoplasm but not in the nucleus of cells, and can increase when certain apoptotic pathways are activated.108 Later studies also showed that intracellular components of necrotic human neutrophils generated by freeze/thaw treatment, but not freeze/thaw-treated Jurkat T cells or UV-treated early and late apoptotic neutrophils, could stimulate the production of pro-inflammatory cytokines such as IL-8, TNF-α and IL-10 by HMDMs, possibly mediated through the release of proteolytically active neutrophil elastase.109 As described earlier, a number of endogenous ‘danger’ signals or alarmins have been discovered (see Kono and Rock39 for an extensive review), including uric acid/MSU crystals, HMGB1, dsDNA and spliceosome-associated protein 130 (SAP130).39 For example, HMGB1, an abundant chromatin-binding protein, can be dissociated from the nucleus and released by necrotic cells to trigger a pro-inflammatory and immunogenic response.110, 111 Although the molecular mechanisms underpinning the inflammatory effect of HMGB1 are controversial, it was reported that HMGB1 could form complexes with DNA or DNA-containing immune complexes and stimulate the release of cytokines such as TNF-α and interferon γ (IFN-γ) by murine plasmacytoid DCs through a TLR-9-dependent and receptor for advanced glycation end-products (RAGE)-dependent pathway.112 In contrast, recent studies by Urbonaviciute et al.113 showed that the formation and release of HMGB1–nucleosome complexes by staurosporine-treated late apoptotic Jurkat T cells could enhance the production of pro-inflammatory cytokines such as TNF-α by macrophages through a TLR-2- but not TLR-4-, TLR-9- and RAGE-dependent mechanism.113 It is worth noting that the immunostimulatory activity of HMGB1 could also be regulated by direct oxidation of HMGB1 during UV-induced apoptotic cell death114 or by binding to serum factors such as anti-HMGB1 autoantibodies.115
Besides generating a pro-inflammatory response through the stimulation of professional phagocytes, freeze/thaw-treated or heat-killed necrotic cells of different cellular origin have also been shown to induce inflammation through non-bone-marrow-derived cells using an in vivo model that measures the recruitment of neutrophils to the peritoneal cavity.116, 117 Elegant studies by Chen et al.117 and Eigenbrod et al.116 showed that the influx of neutrophils towards necrotic cell-derived materials is mediated through an IL-1α-, IL-1R- and MyD88-dependent mechanism,116, 117 possibly by inducing IL-1R signalling in mesothelial cells through necrotic cell-derived IL-1α and the release of CXCL1 to promote neutrophil migration.116
In addition to nuclear- and cytosol-derived molecules, the materials originating from the mitochondria of freeze/thaw-treated or heat-killed necrotic HepG2 epithelial cells were found to trigger release of the pro-inflammatory cytokine IL-8 by human monocytes.118 Recent studies by Crouser et al.118 showed that mitochondrial proteins, such as N-formylated peptides and mitochondrial transcription factor A (TFAM, a structural and functional homologue of HMGB1), function synergistically to stimulate the activation of monocytes through a high-affinity formyl peptide receptor (FPR)-dependent mechanism.118 Besides simply revealing intracellular contents to the extracellular environment due to membrane disruption, necrotic but not late apoptotic murine fibrosarcoma cells (L929sAhFas cells) generated by specific treatments (e.g., TNF-α, anti-Fas and dsRNA) can also actively produce the pro-inflammatory cytokine IL-6, possibly mediated through an NF-κB- and p38 MAPK-dependent pathway.119
In addition to late apoptotic and necrotic cells, it is worth noting that early apoptotic cells can also expose various immunostimulatory signals. For example, the chemoattractant LPC, which is released by UV-, staurosprine- and mitomycin C-treated early apoptotic cells,16 can also function as a pro-inflammatory signal to induce the production of pro-inflammatory cytokines such as macrophage inflammatory protein-2 (MIP2)120 and promote the maturation of DCs.121 Similarly, exposure of nucleic acids on the surface of camptothecin- and UV-treated early apoptotic Jurkat T cells32 can potentially deliver a ‘danger’ signal to phagocytes through TLR-9 after apoptotic cell uptake. Thus, the exposure of ‘danger’ signals is not an exclusive event associated with late apoptotic/necrotic cells, which further complicates predicting the immunological consequences of dying cell removal. Furthermore, it is equally important to note that, under certain experimental conditions, recognition of membrane-damaged cells, in particular late apoptotic cells, can promote an anti-inflammatory response that is similar to that induced by early apoptotic cells. For example, the presence of actinomycin D-treated late apoptotic but not heat-killed DO11.10 cells (a murine T-cell hybridoma) was found to inhibit the production of TNF-α and IL-6 by LPS-stimulated J774A.1 macrophages.122 Similarly, freeze/thaw-treated necrotic Jurkat T cells and UV-treated late apoptotic neutrophils, but not freeze/thaw-treated necrotic neutrophils, induced production of the anti-inflammatory cytokine TGF-β by bone marrow-derived macrophages, an effect possibly mediated through membrane components of dying cells.109 Later studies by Hirt et al.123 also showed that both staurosporine-treated early apoptotic and staurosporine-treated ATP-depleted late apoptotic Jurkat T cells could inhibit bacterial-induced production of TNF-α by HMDMs and murine microglia-like BV-2 cells, whereas heat-killed necrotic cells had no significant effect.123 Consistent with this view, both staurosporine-treated early and late apoptotic BUMPT cells (a murine kidney proximal tubule epithelial cell line) but not heat-killed BUMPT cells, induced the same signalling events in murine bone marrow-derived macrophages.124 Collectively, it is clear from the above examples that the type of cell death-inducing stimulus, the stage of cell death and the origin of the dying cells (e.g., Jurkat T cells versus neutrophils) are critical factors in determining the quality and quantity of immunomodulatory signals being exposed by a dying cell.
As discussed earlier, professional phagocytes, such as DCs and macrophages, are the key antigen-presenting cells that engulf dying cells and present the cell-associated antigens to the adaptive immune system in either a pro- or anti-inflammatory context.10 To orchestrate an appropriate immune response towards the phagocytosed materials, DCs and macrophages are equipped with receptors that not only aid dying cell uptake, but also sense the pro- and anti-inflammatory signals revealed by cells undergoing different forms and stages of cell death as described above. For example, besides recognizing PAMPs, TLRs are a family of cell surface and endosomal PRRs that has been shown to have an important role in detecting endogenous late apoptotic/necrotic cell-derived ‘danger’ signals such as HMGB1, uric acid/MSU crystals, heat shock proteins (HSPs) and DNA.36, 39 TLRs are differentially expressed on a variety of cell types, including DCs, macrophages and B cells, and have a pivotal role in activating these antigen-presenting cells to initiate an immune response towards both microbial- and host-derived antigens.36, 37 Indeed, TLR-4 and HMGB1 have been implicated in the initiation of an immune response against radiotherapy- and chemotherapy-generated dying tumour cells in vivo, possibly by regulating the ability of DCs to process and present tumour-associated antigens.125 Similarly, TLR-2 and TLR-4 have also been shown to have a minor role in sensing heat-killed necrotic EL4 T cells using a sterile inflammation model in vivo.117 In addition to TLRs, C-type lectins such as Mincle and Clec9A have also been shown recently to function as activating cell-surface receptors on macrophages and DCs that sense late apoptotic and necrotic cells.126, 127 Studies by Yamasaki et al.126 showed that Mincle (also known as Clec4e or Clecsf9), a C-type lectin receptor expressed mainly by macrophages, can detect the exposure of SAP130 (a small nuclear ribonucloprotein) on Fas-ligand-treated late apoptotic 2B4 T cell hybridoma cells, with this interaction aiding the macrophage recognition of these late apoptotic cells through a Fc receptor common γ-chain-dependent mechanism.126 Furthermore, Clec9A (also called DNGR-1), a recently identified C-type lectin expressed predominately on CD8α+ DCs, was found to facilitate the recognition of an unknown cytoplasmic ligand(s) exposed on UV- and anthracyclins-treated or serum-starved late apoptotic cells and freeze/thaw-treated necrotic cells of different cellular origin.127 Interestingly, although neither Mincle nor Clec9A modulated the rate of late apoptotic/necrotic cell uptake,126, 127 Sancho et al.127 showed that Clec9A has a key role in mediating cross-presentation of cell-associated antigens (e.g., ovalbumin) derived from UV-treated late apoptotic mouse embryonic fibroblasts to ovalbumin-specific T-cell receptor (TCR) transgenic CD8+ T cells in vitro, as well as cross-priming of antigens associated with late apoptotic cells in vivo.127 Besides Clec9A and Mincle, triggering receptor expressed on myeloid cells-like 4 (Treml4) was shown recently to bind specifically to radiation-treated late apoptotic and heat-killed necrotic thymocytes. Although the physiological role of Treml4 in detecting late apoptotic/necrotic cells requires further analysis, the high level of Treml4 expression in subsets of macrophages and CD8α+ DCs in the spleen suggests that Treml4, similar to other Trem family members, may have an important role in regulating DC and macrophage activation.128
In addition to non-phagocytic receptors, formation of a ‘phagocytic synapse’ between a phagocyte and a dying cell through various adhesion molecules, phagocytic receptors and opsonins can provide additional signals to shape the subsequent immune response. For example, phagocytosis of necrotic cells through a PS-dependent pathway may favour a non- or an anti-inflammatory response.7, 29 Alternatively, the potential of cross-linking FcγR on DCs and macrophages by IgG-, CRP-, SAP-, PTX3- and HRG-opsonized late apoptotic or necrotic cells is likely to induce the production of pro-inflammatory cytokines as well as regulate antigen presentation.129 It is also important to note that the repertoire of receptors and opsonins used to recognize dying cells depends mainly on the phagocytic cell type as well as their tissue location. It is well known that different CR and FcγR are differentially expressed by various phagocytes (e.g., monocytes, and subsets of macrophages and DCs) and the expression of these receptors may also vary depending on the activation state of the phagocytic cells.129, 130 Similarly, delivery of ‘eat-me’ signals through the CD14-dependent pathway may be limited solely to monocytes and macrophages.106
Collectively, the immune system can use a wide range of receptors and opsonins to sense the presence of late apoptotic/necrotic cells as well as pathogens, a process that could result in the recruitment of additional phagocytes to survey sites of tissue injury or infection and modulate the quality of the resultant immune response. As more recognition mechanisms are being discovered and characterized, it would be of great interest to determine how the downstream signalling pathways induced by these different recognition systems interact and whether certain signalling pathways predominate.
Similarities between pathogen and late apoptotic/necrotic cell recognition and phagocytosis
As more recognition and phagocytic pathways are being identified, it is becoming clear that the same germline-encoded PRMs, such as C1q, MBL, CRP, HRG and TLRs, are used by the innate immune system to detect both pathogens and dying cells, in particular plasma membrane-damaged cells such as late apoptotic and necrotic cells (see above). Although it is obvious that using the same molecular mechanisms may provide an evolutionary advantage for the efficient removal of both ‘non-self’ and ‘altered-self’ materials that could cause harm to the organism, it is not clear which function originated first or whether both functions co-evoluted together. What is even more unclear is how pathogens and membrane-damaged cells can reveal similar or the same ligands to trigger recognition by the same repertoire of PRMs, possibly due to the difficulties in characterizing and validating the endogenous ‘eat-me’ and ‘danger’ signals specifically exposed by late apoptotic and necrotic cells. Nevertheless, these PRMs share a number of striking properties, in particular the ligands they bind (see examples listed in Table 1). Exploring such similarities may help to elucidate why and how the same germline-encoded molecules can recognize both pathogens and late apoptotic/necrotic cells.
One apparently common feature of these PRMs is their ability to interact with a variety of negatively charged molecules, such as DNA, RNA, phospholipids and sulphated GAGs. A variety of pathogens, such as Escherichia coli (O157:H7)131 and Helicobacter pylori,132 have been shown to possess a highly negatively charged surface, whereas membrane-permeabilized cells may also expose an array of negatively charged molecules that are usually hidden intracellularly. Similarly, the ability of antimicrobial peptides, such as neutrophil defensin, to recognize anionic phospholipids exposed on pathogens133 further highlights how the innate immune system has evolved to target fundamental differences in the surface charge density of host cells and microbes. In a similar way, hydrophobic structures have also been proposed to function as a unique molecular pattern expressed by both pathogens and damaged host cells.40 However, as late apoptotic and necrotic cells can potentially expose all kinds of molecules, including positively and negatively charged, hydrophobic, enzyme cleaved and partially processed material, it is difficult to conclude whether any of these are the common molecular pattern shared by both pathogens and membrane-damaged host cells.
Alternatively, PRMs may recognize identical molecules that are constitutively expressed by pathogens and exposed in membrane-damaged host cells. As mitochondria are thought to have originated from bacterial ancestors, and permeabilization of the plasma membrane reveals intracellular organelles, it is not surprising that mitochondrial-derived molecules are potential late apoptotic/necrotic cell ligands for PRMs. For example, cardiolipin, an anionic phospholipid found on the inner membrane of mitochondria as well as on the outer cytoplasmic membrane of certain bacteria, has been suggested to bind C1q and aid the activation of the classical complement pathway.45 Similarly, the release of N-formylated peptides from bacteria or disrupted mitochondria can also stimulate chemotaxis and immune activation through FPR and FPR-like receptors.118, 134 Interestingly, studies by Ohta et al.135 also suggested that HRG could directly interact with ATP synthase, a well-conserved enzyme found in the plasma membrane of bacteria and in the inner membrane of mitochondria.135 Furthermore, recognition of the universal genetic material DNA and RNA by TLR-3, TLR-7, TLR-8 and TLR-9,36, 37 SAP,76 Ficolin-2,68, 69 C1q,14 MBL,66 HRG92 and properdin72 further highlights the ability of these PRMs to sense pathogen-derived nucleic acids and similar nucleic acid structures revealed in late apoptotic/necrotic host cells. It is also possible for these PRMs to detect modified structures on dying cells that are similar to pathogen-derived molecules. For example, oxidation of phospholipids and low-density lipoproteins during apoptotic cell death can generate the PCh moiety that resembles the PCh headgroup of bacterial and fungal polysaccharides, and that could be recognized by CRP and certain natural antibodies.57, 82 Similarly, Kuraya et al.71 also suggested that the glycosylation of cytoplasmic and nuclear proteins with O-linked GlcNAc during signal transduction may facilitate the binding of Ficolin-2 to dying cells,71 possibly when the cell membrane is disrupted.68 Thus, lectins such as MBL and Ficolins may sense dying cells through the same molecular mechanism that is used for recognizing GlcNAc exposed on microbial surfaces. In addition to glycosylation, other post-translational modifications, such as phosphorylation, are also essential for a variety of cellular processes. Interestingly, the ability of CRP to bind to pathogens, such as Leishmania donovani, has been shown to be mediated through the recognition of phosphorylated carbohydrates.136 As phosphorylated molecules, such as signalling proteins and phosphoinositides, are often hidden from the extracellular environment of mammalian cells unless the plasma membrane is damaged, it is tempting to speculate that intracellular phosphorylated molecules exposed in late apoptotic/necrotic cells may provide novel molecular patterns that can be recognized by the innate immune system. Indeed, MBL137 and HRG (IKH Poon et al., unpublished observations) can interact specifically with specific intracellular phosphorylated phosphatidylinositol molecules. Similarly, SAP binding to certain carbohydrates is also dependent on phosphorylation.138
To date, the molecular mechanisms underpinning how PRMs can interact with both pathogens and late apoptotic/necrotic host cells remains poorly understood. First, few studies provide direct evidence that the proposed PRM ligands are indeed the endogenous ligands exposed by the host cells, possibly because of the technical difficulties in selectively blocking the ligand using antibodies, in removing the ligand enzymatically or in generating a ligand-deficient cell line. Furthermore, as many of the PRMs, especially the opsonins, are multifunctional and have a modular domain structure, they could potentially interact with many different types of ligands, including proteins, lipids and carbohydrates. Therefore, mapping the recognition sites on PRMs could help to define the nature of the endogenous ligands and may provide clues as to how the PRMs interact with late apoptotic/necrotic cells. In addition, cross-blocking studies with a wide range of endogenous and exogenous PRM ligands (Table 1) may provide useful information as to whether similar or unique molecular patterns are recognized by the different PRMs on late apoptotic/necrotic cells.
Besides interacting with multiple cell-associated endogenous and exogenous ligands, another apparent feature shown in Table 1 is that most of the opsonins listed can also bind to one another. As mentioned earlier, CRP, SAP, immunoglobulin, PTX3 and HRG can all interact with C1q.13, 76, 91 HRG and MBL can also bind immunoglobulin,63, 91 whereas TSP-1 can form a complex with HRG.91 In addition, Ficolin-2 has been shown recently to bind directly to both PTX3 and CRP.139, 140 Thus, it is of particular interest to analyse whether these different PRMs can form multimers and function as soluble recognition complexes that possess multiple pattern recognition sites and receptor binding sites. Indeed, immunoprecipitation studies by Manderson et al.94 have shown that HRG can form complexes with C1q, C4b-binding protein (C4BP) and factor H in normal human serum and in synovial fluid collected from rheumatoid arthritis patients.94 Similarly, CRP and Ficolin-2 can also form complexes in human serum and function cooperatively to aid the clearance of Pseudomonas aeruginosa through a complement-dependent mechanism.140
As discussed above, there are numerous examples showing that the same recognition systems are used to detect both pathogen-derived and late apoptotic/necrotic cell-derived molecules. However, the question remains whether the immune system can differentiate between pathogens and membrane-damaged self cells (Figure 4). Recently, elegant studies by Chen et al.141 showed that endogenous ‘danger’ signals, such as HMGB1, HSP70 and HSP90, can negatively regulate their ability to activate the NF-κB pathway and induce pro-inflammatory cytokines production by bone-marrow derived DCs, by interacting with CD24, which provides an inhibitory signal by associating with immunoreceptor tyrosine-based inhibitor motifs (ITIM)-containing Siglec-G. In contrast, exogenous ‘danger’ signals, such as LPS and poly(I:C) (a synthetic dsRNA), were unable to negatively regulate their immunostimulatory effects through CD24- and the Siglec-G-dependent pathway.141 Therefore, although both endogenous and exogenous ‘danger’ signals can activate the immune system through PRMs, such as the TLRs,37, 39 endogenous molecules may be able to selectively finetune the immune response such that the presence of pathogens can be distinguished from tissue damage. Similarly, the extent of complement activation on a target cell may allow the immune system to differentiate between endogenous and exogenous cells, with there being multiple mechanisms that can limit complement activation on self cells, whether they are early apoptotic, late apoptotic or necrotic.14 Indeed, many of the opsonins that can bind selectively to certain late apoptotic/necrotic cells (see above and Table 1), such as CRP, PTX3, HRG and TSP-1, have all been shown to bind to the complement inhibitory factor H,79, 94, 142, 143 with CRP, SAP and HRG also interacting with C4BP, another negative regulator of complement activation.14, 94 In addition, C4BP can also bind directly to heat-, ethanol- and freeze/thaw-treated necrotic Jurkat T cells, possibly through DNA recognition, and inhibit complement activation.144 Besides using a common recognition mechanism to sense both pathogens and late apoptotic/necrotic cells, the immune system can also detect endogenous and exogenous substances through unique recognition pathways, such as sensing the exposure of PS on late apoptotic cells through Tim-4 and possibly other PS-binding opsonins/receptors,30 as well as detecting bacterial flagellin through TLR-5.37 Thus, it is likely that the immune system uses common recognition pathways to efficiently sense invading pathogens and tissue injury, whereas unique recognition pathways are also needed to gather additional information to determine whether or not an immune response is induced and, if so, the quality and quantity of the ensuring immune response (Figure 4).

Sensing pathogens and late apoptotic/necrotic cells through common and unique recognition pathways. The innate immune system uses a repertoire of common recognition mechanisms to detect both pathogens and late apoptotic/necrotic cells. These common recognition pathways (e.g., detecting N-formylated peptides and PCh headgroup by FPR and CRP, respectively) are essential for sensing and responding to infection or tissue injury efficiently but may not be able to discriminate between whether the ‘danger’ signal is endogenous or exogenous. Thus, unique pathways (e.g., recognizing flagellin and PS by TLR-5 and CD24, respectively) are needed to gather more information regarding the source of ‘danger’. CRP, C-reactive protein; DNA, deoxyribonucleic acid; FPR, formyl peptide receptor; HMGB1, high-mobility group box 1 protein; HRG, histidine-rich glycoprotein; PCh, phosphocholine; PS, phosphatidylserine; TIM, T-cell immunoglobulin mucin; TLR, Toll-like receptor
Concluding Remarks
The uptake of dying cells has been shown to have a critical role in development, tissue homeostasis and wound healing, as well as in providing a source of cell-associated antigens and immunomodulatory signals for immune activation, the maintenance of immunological tolerance or inducing signals that regulate the quality of any resultant immune response. As late apoptotic and necrotic cells are generally considered as activators of the immune system, elucidating the molecular mechanisms underpinning the clearance of such cells have important implications for our understanding of the initiation and maintenance of autoimmune diseases such as SLE. Conversely, such molecular mechanisms may be harnessed to develop cancer immunotherapies by enhancing the immunogenicity of dying cancer cells after their uptake by phagocytes, such as DCs. Despite the recent advances in defining the mechanisms and consequences of late apoptotic/necrotic cell clearance, much more still needs to be done before we fully understand this important and complex process, especially its striking similarities with pathogen recognition.
Abbreviations
- ACAMP:
-
apoptotic cell-associated molecular pattern
- β2GPI:
-
β2-glycoprotein I
- BAI1:
-
brain-specific angiogenesis inhibitor 1
- C4BP:
-
C4b-binding protein
- CFHR4:
-
human complement factor H-related protein 4
- CR:
-
complement receptors
- CRP:
-
C-reactive protein
- DAMP:
-
danger-associated molecular pattern
- DC:
-
dendritic cell
- DNA:
-
deoxyribonucleic acid
- ds:
-
double-stranded
- EMAP II:
-
endothelial monocyte-activating polypeptide II
- FcγR:
-
Fcγ receptor
- FPR:
-
formyl peptide receptor
- GAG:
-
glycosaminoglycan
- GlcNAc:
-
N-acetyl-D-glucosamine
- HERDS:
-
heterogeneous ectopic ribonucleoprotein-derived structures
- HMDDC:
-
human monocyte-derived dendritic cell
- HMDM:
-
human monocyte-derived macrophage
- HMGB1:
-
high mobility group box 1 protein
- HRG:
-
histidine-rich glycoprotein
- HS:
-
heparan sulphate
- HSP:
-
heat shock protein
- HSPG:
-
heparan sulphate proteoglycans
- IFN-γ:
-
interferon-γ
- Ig:
-
immunoglobulin
- IL:
-
interleukin
- ITIM:
-
immunoreceptor tyrosine-based inhibitor motif
- LBP:
-
LPS-binding protein
- LPC:
-
lysophosphatidylcholine
- LPS:
-
lipopolysaccharide
- LTA:
-
lipoteichoic acid
- MAC:
-
membrane-attack complex
- MASP:
-
MBL-associated serine protease
- MBL:
-
mannose-binding lectin
- MFG-E8:
-
milk fat globular-EGF factor 8 protein
- MIP2:
-
macrophage-inflammatory protein-2
- MHC:
-
major histocompatibility complex
- MSU:
-
monosodium urate
- NOD-like:
-
nucleotide-binding oligomerization domain
- PAMP:
-
pathogen-associated molecular pattern
- PBL:
-
peripheral blood lymphocyte
- PCh:
-
phosphocholine
- PRM:
-
pattern recognition molecule
- PS:
-
phosphatidylserine
- PTX3:
-
pentraxin-3
- RAGE:
-
receptor for advanced glycation end-products
- RIG-like:
-
retinoic acid-inducible gene-I-like
- RNA:
-
ribonucleic acid
- SAMP:
-
self-associated molecular pattern
- SAP:
-
serum amyloid protein
- SAP130:
-
spliceosome-associated protein 130
- SLE:
-
systemic lupus erythematosus
- TCR:
-
T-cell receptor
- TFAM:
-
mitochondrial transcription factor A
- TGF-β:
-
transforming growth factor-β
- TIM:
-
T-cell immunoglobulin mucin
- TLR:
-
Toll-like receptor
- TNF-α:
-
tumour necrosis factor-α
- Trem:
-
triggering receptor expressed on myeloid cells
- TSP-1:
-
thrombospondin-1
- VEGF:
-
vascular endothelial growth factor
References
Stuart LM, Ezekowitz RA . Phagocytosis: elegant complexity. Immunity 2005; 22: 539–550.
Gregory CD, Devitt A . The macrophage and the apoptotic cell: an innate immune interaction viewed simplistically? Immunology 2004; 113: 1–14.
Yuste J, Ali S, Sriskandan S, Hyams C, Botto M, Brown JS . Roles of the alternative complement pathway and C1q during innate immunity to Streptococcus pyogenes. J Immunol 2006; 176: 6112–6120.
Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med 2000; 192: 359–366.
Elmore S . Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35: 495–516.
Bianchi ME . DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 2007; 81: 1–5.
Krysko DV, D′Herde K, Vandenabeele P . Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 2006; 11: 1709–1726.
Golpon HA, Fadok VA, Taraseviciene-Stewart L, Scerbavicius R, Sauer C, Welte T et al. Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J 2004; 18: 1716–1718.
Sindrilaru A, Peters T, Schymeinsky J, Oreshkova T, Wang H, Gompf A et al. Wound healing defect of Vav3−/− mice due to impaired {beta}2-integrin-dependent macrophage phagocytosis of apoptotic neutrophils. Blood 2009; 113: 5266–5276.
Green DR, Ferguson T, Zitvogel L, Kroemer G . Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009; 9: 353–363.
Aderem A, Underhill DM . Mechanisms of phagocytosis in macrophages. Annu Rev Immunol 1999; 17: 593–623.
Grimsley C, Ravichandran KS . Cues for apoptotic cell engulfment: eat-me, don’t eat-me and come-get-me signals. Trends Cell Biol 2003; 13: 648–656.
Sim RB, Tsiftsoglou SA . Proteases of the complement system. Biochem Soc Trans 2004; 32: 21–27.
Sjoberg AP, Trouw LA, Blom AM . Complement activation and inhibition: a delicate balance. Trends Immunol 2009; 30: 83–90.
Segundo C, Medina F, Rodriguez C, Martinez-Palencia R, Leyva-Cobian F, Brieva JA . Surface molecule loss and bleb formation by human germinal center B cells undergoing apoptosis: role of apoptotic blebs in monocyte chemotaxis. Blood 1999; 94: 1012–1020.
Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003; 113: 717–730.
Knies UE, Behrensdorf HA, Mitchell CA, Deutsch U, Risau W, Drexler HC et al. Regulation of endothelial monocyte-activating polypeptide II release by apoptosis. Proc Natl Acad Sci U S A 1998; 95: 12322–12327.
Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 2008; 112: 5026–5036.
Horino K, Nishiura H, Ohsako T, Shibuya Y, Hiraoka T, Kitamura N et al. A monocyte chemotactic factor, S19 ribosomal protein dimer, in phagocytic clearance of apoptotic cells. Lab Invest 1998; 78: 603–617.
Bournazou I, Pound JD, Duffin R, Bournazos S, Melville LA, Brown SB et al. Apoptotic human cells inhibit migration of granulocytes via release of lactoferrin. J Clin Invest 2009; 119: 20–32.
Majai G, Petrovski G, Fesus L . Inflammation and the apopto-phagocytic system. Immunol Lett 2006; 104: 94–101.
Janeway Jr CA . Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 1989; 54 (Part 1): 1–13.
Janeway Jr CA, Medzhitov R . Innate immune recognition. Annu Rev Immunol 2002; 20: 197–216.
Franc NC, White K, Ezekowitz RA . Phagocytosis and development: back to the future. Curr Opin Immunol 1999; 11: 47–52.
Elward K, Gasque P . ‘Eat me’ and ‘don’t eat me’ signals govern the innate immune response and tissue repair in the CNS: emphasis on the critical role of the complement system. Mol Immunol 2003; 40: 85–94.
Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005; 123: 321–334.
Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J . Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature 2002; 418: 200–203.
Grutkoski PS, Graeber CT, Lim YP, Ayala A, Simms HH . Alpha-defensin 1 (human neutrophil protein 1) as an antichemotactic agent for human polymorphonuclear leukocytes. Antimicrob Agents Chemother 2003; 47: 2666–2668.
Krysko DV, Vandenabeele P . From regulation of dying cell engulfment to development of anti-cancer therapy. Cell Death Differ 2008; 15: 29–38.
Bratton DL, Henson PM . Apoptotic cell recognition: will the real phosphatidylserine receptor(s) please stand up? Curr Biol 2008; 18: R76–R79.
Borisenko GG, Matsura T, Liu SX, Tyurin VA, Jianfei J, Serinkan FB et al. Macrophage recognition of externalized phosphatidylserine and phagocytosis of apoptotic Jurkat cells--existence of a threshold. Arch Biochem Biophys 2003; 413: 41–52.
Elward K, Griffiths M, Mizuno M, Harris CL, Neal JW, Morgan BP et al. CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. J Biol Chem 2005; 280: 36342–36354.
Potter PK, Larbi KY, Nourshargh S, Botto M . Efficient clearance of opsonised apoptotic cells in the absence of PECAM-1. Mol Immunol 2007; 44: 1135–1140.
Nilsson A, Oldenborg PA . CD47 promotes both phosphatidylserine-independent and phosphatidylserine-dependent phagocytosis of apoptotic murine thymocytes by non-activated macrophages. Biochem Biophys Res Commun 2009; 387: 58–63.
Festjens N, Vanden Berghe T, Vandenabeele P . Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta 2006; 1757: 1371–1387.
Marshak-Rothstein A . Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol 2006; 6: 823–835.
Iwasaki A, Medzhitov R . Toll-like receptor control of the adaptive immune responses. Nat Immunol 2004; 5: 987–995.
Kumar H, Kawai T, Akira S . Pathogen recognition in the innate immune response. Biochem J 2009; 420: 1–16.
Kono H, Rock KL . How dying cells alert the immune system to danger. Nat Rev Immunol 2008; 8: 279–289.
Seong SY, Matzinger P . Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 2004; 4: 469–478.
Walport MJ . Complement. Second of two parts. N Engl J Med 2001; 344: 1140–1144.
Matzinger P . Tolerance, danger, and the extended family. Annu Rev Immunol 1994; 12: 991–1045.
Mevorach D, Mascarenhas JO, Gershov D, Elkon KB . Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med 1998; 188: 2313–2320.
Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med 2001; 194: 781–795.
Peitsch MC, Tschopp J, Kress A, Isliker H . Antibody-independent activation of the complement system by mitochondria is mediated by cardiolipin. Biochem J 1988; 249: 495–500.
Gaipl US, Kuenkele S, Voll RE, Beyer TD, Kolowos W, Heyder P et al. Complement binding is an early feature of necrotic and a rather late event during apoptotic cell death. Cell Death Differ 2001; 8: 327–334.
Ciurana CL, Zwart B, van Mierlo G, Hack CE . Complement activation by necrotic cells in normal plasma environment compares to that by late apoptotic cells and involves predominantly IgM. Eur J Immunol 2004; 34: 2609–2619.
Gaipl US, Beyer TD, Heyder P, Kuenkele S, Bottcher A, Voll RE et al. Cooperation between C1q and DNase I in the clearance of necrotic cell-derived chromatin. Arthritis Rheum 2004; 50: 640–649.
Gullstrand B, Martensson U, Sturfelt G, Bengtsson AA, Truedsson L . Complement classical pathway components are all important in clearance of apoptotic and secondary necrotic cells. Clin Exp Immunol 2009; 156: 303–311.
Mix E, Goertsches R, Zett UK . Immunoglobulins--basic considerations. J Neurol 2006; 253 (Suppl 5): V9–17.
Dorner T, Egerer K, Feist E, Burmester GR . Rheumatoid factor revisited. Curr Opin Rheumatol 2004; 16: 246–253.
Gaipl US, Voll RE, Sheriff A, Franz S, Kalden JR, Herrmann M . Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun Rev 2005; 4: 189–194.
Ogden CA, Kowalewski R, Peng Y, Montenegro V, Elkon KB . IGM is required for efficient complement mediated phagocytosis of apoptotic cells in vivo. Autoimmunity 2005; 38: 259–264.
Kim SJ, Gershov D, Ma X, Brot N, Elkon KB . I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med 2002; 196: 655–665.
Quartier P, Potter PK, Ehrenstein MR, Walport MJ, Botto M . Predominant role of IgM-dependent activation of the classical pathway in the clearance of dying cells by murine bone marrow-derived macrophages in vitro. Eur J Immunol 2005; 35: 252–260.
Zwart B, Ciurana C, Rensink I, Manoe R, Hack CE, Aarden LA . Complement activation by apoptotic cells occurs predominantly via IgM and is limited to late apoptotic (secondary necrotic) cells. Autoimmunity 2004; 37: 95–102.
Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest 2009; 119: 1335–1349.
Fu M, Fan PS, Li W, Li CX, Xing Y, An JG et al. Identification of poly-reactive natural IgM antibody that recognizes late apoptotic cells and promotes phagocytosis of the cells. Apoptosis 2007; 12: 355–362.
Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J . Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci USA 2000; 97: 1184–1189.
Shibuya A, Sakamoto N, Shimizu Y, Shibuya K, Osawa M, Hiroyama T et al. Fc alpha/mu receptor mediates endocytosis of IgM-coated microbes. Nat Immunol 2000; 1: 441–446.
Grossmayer GE, Munoz LE, Weber CK, Franz S, Voll RE, Kern PM et al. IgG autoantibodies bound to surfaces of necrotic cells and complement C4 comprise the phagocytosis promoting activity for necrotic cells of systemic lupus erythaematosus sera. Ann Rheum Dis 2008; 67: 1626–1632.
Reefman E, Horst G, Nijk MT, Limburg PC, Kallenberg CG, Bijl M . Opsonization of late apoptotic cells by systemic lupus erythematosus autoantibodies inhibits their uptake via an Fcgamma receptor-dependent mechanism. Arthritis Rheum 2007; 56: 3399–3411.
Takahashi K, Ip WE, Michelow IC, Ezekowitz RA . The mannose-binding lectin: a prototypic pattern recognition molecule. Curr Opin Immunol 2006; 18: 16–23.
Nauta AJ, Raaschou-Jensen N, Roos A, Daha MR, Madsen HO, Borrias-Essers MC et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol 2003; 33: 2853–2863.
Stuart LM, Takahashi K, Shi L, Savill J, Ezekowitz RA . Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J Immunol 2005; 174: 3220–3226.
Nakamura N, Nonaka M, Ma BY, Matsumoto S, Kawasaki N, Asano S et al. Characterization of the interaction between serum mannan-binding protein and nucleic acid ligands. J Leukoc Biol 2009; 86: 737–748.
Ezekowitz RA, Day LE, Herman GA . A human mannose-binding protein is an acute-phase reactant that shares sequence homology with other vertebrate lectins. J Exp Med 1988; 167: 1034–1046.
Jensen ML, Honore C, Hummelshoj T, Hansen BE, Madsen HO, Garred P . Ficolin-2 recognizes DNA and participates in the clearance of dying host cells. Mol Immunol 2007; 44: 856–865.
Honore C, Hummelshoj T, Hansen BE, Madsen HO, Eggleton P, Garred P . The innate immune component ficolin 3 (Hakata antigen) mediates the clearance of late apoptotic cells. Arthritis Rheum 2007; 56: 1598–1607.
Endo Y, Matsushita M, Fujita T . Role of ficolin in innate immunity and its molecular basis. Immunobiology 2007; 212: 371–379.
Kuraya M, Ming Z, Liu X, Matsushita M, Fujita T . Specific binding of L-ficolin and H-ficolin to apoptotic cells leads to complement activation. Immunobiology 2005; 209: 689–697.
Xu W, Berger SP, Trouw LA, de Boer HC, Schlagwein N, Mutsaers C et al. Properdin binds to late apoptotic and necrotic cells independently of c3b and regulates alternative pathway complement activation. J Immunol 2008; 180: 7613–7621.
Kemper C, Mitchell LM, Zhang L, Hourcade DE . The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci USA 2008; 105: 9023–9028.
Schwaeble WJ, Reid KB . Does properdin crosslink the cellular and the humoral immune response? Immunol Today 1999; 20: 17–21.
Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE . Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol 2007; 179: 2600–2608.
Garlanda C, Bottazzi B, Bastone A, Mantovani A . Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu Rev Immunol 2005; 23: 337–366.
Marnell L, Mold C, Du Clos TW . C-reactive protein: ligands, receptors and role in inflammation. Clin Immunol 2005; 117: 104–111.
Gershov D, Kim S, Brot N, Elkon KB . C-Reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med 2000; 192: 1353–1364.
Mihlan M, Stippa S, Jozsi M, Zipfel PF . Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting factor H 2009; 16: 1630–1640.
Mold C, Baca R, Du Clos TW . Serum amyloid P component and C-reactive protein opsonize apoptotic cells for phagocytosis through Fcgamma receptors. J Autoimmun 2002; 19: 147–154.
Hart SP, Alexander KM, MacCall SM, Dransfield I . C-reactive protein does not opsonize early apoptotic human neutrophils, but binds only membrane-permeable late apoptotic cells and has no effect on their phagocytosis by macrophages. J Inflamm (Lond) 2005; 2: 5.
Chang MK, Binder CJ, Torzewski M, Witztum JL . C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: Phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci USA 2002; 99: 13043–13048.
Mihlan M, Hebecker M, Dahse HM, Halbich S, Huber-Lang M, Dahse R et al. Human complement factor H-related protein 4 binds and recruits native pentameric C-reactive protein to necrotic cells. Mol Immunol 2009; 46: 335–344.
Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD . Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature 2008; 456: 989–992.
Bijl M, Horst G, Bijzet J, Bootsma H, Limburg PC, Kallenberg CG . Serum amyloid P component binds to late apoptotic cells and mediates their uptake by monocyte-derived macrophages. Arthritis Rheum 2003; 48: 248–254.
Rovere P, Peri G, Fazzini F, Bottazzi B, Doni A, Bondanza A et al. The long pentraxin PTX3 binds to apoptotic cells and regulates their clearance by antigen-presenting dendritic cells. Blood 2000; 96: 4300–4306.
Baruah P, Propato A, Dumitriu IE, Rovere-Querini P, Russo V, Fontana R et al. The pattern recognition receptor PTX3 is recruited at the synapse between dying and dendritic cells, and edits the cross-presentation of self, viral, and tumor antigens. Blood 2006; 107: 151–158.
Baruah P, Dumitriu IE, Peri G, Russo V, Mantovani A, Manfredi AA et al. The tissue pentraxin PTX3 limits C1q-mediated complement activation and phagocytosis of apoptotic cells by dendritic cells. J Leukoc Biol 2006; 80: 87–95.
van Rossum AP, Fazzini F, Limburg PC, Manfredi AA, Rovere-Querini P, Mantovani A et al. The prototypic tissue pentraxin PTX3, in contrast to the short pentraxin serum amyloid P, inhibits phagocytosis of late apoptotic neutrophils by macrophages. Arthritis Rheum 2004; 50: 2667–2674.
Jaillon S, Jeannin P, Hamon Y, Fremaux I, Doni A, Bottazzi B et al. Endogenous PTX3 translocates at the membrane of late apoptotic human neutrophils and is involved in their engulfment by macrophages. Cell Death Differ 2009; 16: 465–474.
Jones AL, Hulett MD, Parish CR . Histidine-rich glycoprotein: a novel adaptor protein in plasma that modulates the immune, vascular and coagulation systems. Immunol Cell Biol 2005; 83: 106–118.
Gorgani NN, Smith BA, Kono DH, Theofilopoulos AN . Histidine-rich glycoprotein binds to DNA and Fc gamma RI and potentiates the ingestion of apoptotic cells by macrophages. J Immunol 2002; 169: 4745–4751.
Jones AL, Poon IK, Hulett MD, Parish CR . Histidine-rich glycoprotein specifically binds to necrotic cells via its amino-terminal domain and facilitates necrotic cell phagocytosis. J Biol Chem 2005; 280: 35733–35741.
Manderson GA, Martin M, Onnerfjord P, Saxne T, Schmidtchen A, Mollnes TE et al. Interactions of histidine-rich glycoprotein with immunoglobulins and proteins of the complement system. Mol Immunol 2009; 46: 3388–3398.
Sid B, Sartelet H, Bellon G, El Btaouri H, Rath G, Delorme N et al. Thrombospondin 1: a multifunctional protein implicated in the regulation of tumor growth. Crit Rev Oncol Hematol 2004; 49: 245–258.
Rennemeier C, Hammerschmidt S, Niemann S, Inamura S, Zahringer U, Kehrel BE . Thrombospondin-1 promotes cellular adherence of gram-positive pathogens via recognition of peptidoglycan. FASEB J 2007; 21: 3118–3132.
Savill J, Hogg N, Ren Y, Haslett C . Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J Clin Invest 1992; 90: 1513–1522.
Krispin A, Bledi Y, Atallah M, Trahtemberg U, Verbovetski I, Nahari E et al. Apoptotic cell thrombospondin-1 and heparin-binding domain lead to dendritic-cell phagocytic and tolerizing states. Blood 2006; 108: 3580–3589.
Moodley Y, Rigby P, Bundell C, Bunt S, Hayashi H, Misso N et al. Macrophage recognition and phagocytosis of apoptotic fibroblasts is critically dependent on fibroblast-derived thrombospondin 1 and CD36. Am J Pathol 2003; 162: 771–779.
Ren Y, Stuart L, Lindberg FP, Rosenkranz AR, Chen Y, Mayadas TN et al. Nonphlogistic clearance of late apoptotic neutrophils by macrophages: efficient phagocytosis independent of beta 2 integrins. J Immunol 2001; 166: 4743–4750.
Bottcher A, Gaipl US, Furnrohr BG, Herrmann M, Girkontaite I, Kalden JR et al. Involvement of phosphatidylserine, alphavbeta3, CD14, CD36, and complement C1q in the phagocytosis of primary necrotic lymphocytes by macrophages. Arthritis Rheum 2006; 54: 927–938.
Tumova S, Woods A, Couchman JR . Heparan sulfate proteoglycans on the cell surface: versatile coordinators of cellular functions. Int J Biochem Cell Biol 2000; 32: 269–288.
Dehio C, Freissler E, Lanz C, Gomez-Duarte OG, David G, Meyer TF . Ligation of cell surface heparan sulfate proteoglycans by antibody-coated beads stimulates phagocytic uptake into epithelial cells: a model for cellular invasion by Neisseria gonorrhoeae. Exp Cell Res 1998; 242: 528–539.
Naumann M, Rudel T, Meyer TF . Host cell interactions and signalling with Neisseria gonorrhoeae. Curr Opin Microbiol 1999; 2: 62–70.
Gebska MA, Titley I, Paterson HF, Morilla RM, Davies DC, Gruszka-Westwood AM et al. High-affinity binding sites for heparin generated on leukocytes during apoptosis arise from nuclear structures segregated during cell death. Blood 2002; 99: 2221–2227.
Gregory CD . CD14-dependent clearance of apoptotic cells: relevance to the immune system. Curr Opin Immunol 2000; 12: 27–34.
Gallucci S, Lolkema M, Matzinger P . Natural adjuvants: endogenous activators of dendritic cells. Nat Med 1999; 5: 1249–1255.
Shi Y, Zheng W, Rock KL . Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc Natl Acad Sci USA 2000; 97: 14590–14595.
Fadok VA, Bratton DL, Guthrie L, Henson PM . Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: role of proteases. J Immunol 2001; 166: 6847–6854.
Scaffidi P, Misteli T, Bianchi ME . Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418: 191–195.
Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep 2004; 5: 825–830.
Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 2007; 8: 487–496.
Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med 2008; 205: 3007–3018.
Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA . Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008; 29: 21–32.
Urbonaviciute V, Furnrohr BG, Weber C, Haslbeck M, Wilhelm S, Herrmann M et al. Factors masking HMGB1 in human serum and plasma. J Leukoc Biol 2007; 81: 67–74.
Eigenbrod T, Park JH, Harder J, Iwakura Y, Nunez G . Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J Immunol 2008; 181: 8194–8198.
Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL . Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 2007; 13: 851–856.
Crouser ED, Shao G, Julian MW, Macre JE, Shadel GS, Tridandapani S et al. Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors. Crit Care Med 2009; 37: 2000–2009.
Vanden Berghe T, Kalai M, Denecker G, Meeus A, Saelens X, Vandenabeele P . Necrosis is associated with IL-6 production but apoptosis is not. Cell Signal 2006; 18: 328–335.
Olofsson KE, Andersson L, Nilsson J, Bjorkbacka H . Nanomolar concentrations of lysophosphatidylcholine recruit monocytes and induce pro-inflammatory cytokine production in macrophages. Biochem Biophys Res Commun 2008; 370: 348–352.
Coutant F, Perrin-Cocon L, Agaugue S, Delair T, Andre P, Lotteau V . Mature dendritic cell generation promoted by lysophosphatidylcholine. J Immunol 2002; 169: 1688–1695.
Cocco RE, Ucker DS . Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol Biol Cell 2001; 12: 919–930.
Hirt UA, Leist M . Rapid, noninflammatory and PS-dependent phagocytic clearance of necrotic cells. Cell Death Differ 2003; 10: 1156–1164.
Patel VA, Longacre A, Hsiao K, Fan H, Meng F, Mitchell JE et al. Apoptotic cells, at all stages of the death process, trigger characteristic signaling events that are divergent from and dominant over those triggered by necrotic cells: implications for the delayed clearance model of autoimmunity. J Biol Chem 2006; 281: 4663–4670.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007; 13: 1050–1059.
Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T . Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol 2008; 9: 1179–1188.
Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, Hernanz-Falcon P et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009; 458: 899–903.
Hemmi H, Idoyaga J, Suda K, Suda N, Kennedy K, Noda M et al. A new triggering receptor expressed on myeloid cells (Trem) family member, Trem-like 4, binds to dead cells and is a DNAX activation protein 12-linked marker for subsets of mouse macrophages and dendritic cells. J Immunol 2009; 182: 1278–1286.
Nimmerjahn F, Ravetch JV . Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 2008; 8: 34–47.
Bajtay Z, Csomor E, Sandor N, Erdei A . Expression and role of Fc- and complement-receptors on human dendritic cells. Immunol Lett 2006; 104: 46–52.
Sherman P, Soni R, Petric M, Karmali M . Surface properties of the Vero cytotoxin-producing Escherichia coli O157:H7. Infect Immun 1987; 55: 1824–1829.
Smith JI, Drumm B, Neumann AW, Policova Z, Sherman PM . In vitro surface properties of the newly recognized gastric pathogen Helicobacter pylori. Infect Immun 1990; 58: 3056–3060.
Hristova K, Selsted ME, White SH . Critical role of lipid composition in membrane permeabilization by rabbit neutrophil defensins. J Biol Chem 1997; 272: 24224–24233.
Rabiet MJ, Huet E, Boulay F . The N-formyl peptide receptors and the anaphylatoxin C5a receptors: an overview. Biochimie 2007; 89: 1089–1106.
Ohta T, Ikemoto Y, Usami A, Koide T, Wakabayashi S . High affinity interaction between histidine-rich glycoprotein and the cell surface type ATP synthase on T cells. Biochim Biophys Acta 2009; 1788: 1099–1107.
Culley FJ, Harris RA, Kaye PM, McAdam KP, Raynes JG . C-reactive protein binds to a novel ligand on Leishmania donovani and increases uptake into human macrophages. J Immunol 1996; 156: 4691–4696.
Kuroki Y, Honma T, Chiba H, Sano H, Saitoh M, Ogasawara Y et al. A novel type of binding specificity to phospholipids for rat mannose-binding proteins isolated from serum and liver. FEBS Lett 1997; 414: 387–392.
Loveless RW, Floyd-O′Sullivan G, Raynes JG, Yuen CT, Feizi T . Human serum amyloid P is a multispecific adhesive protein whose ligands include 6-phosphorylated mannose and the 3-sulphated saccharides galactose, N-acetylgalactosamine and glucuronic acid. EMBOJ 1992; 11: 813–819.
Ma YJ, Doni A, Hummelshoj T, Honore C, Bastone A, Mantovani A et al. Synergy between ficolin-2 and pentraxin 3 boosts innate immune recognition and complement deposition. J Biol Chem 2009; 284: 28263–28275.
Zhang J, Koh J, Lu J, Thiel S, Leong BS, Sethi S et al. Local inflammation induces complement crosstalk which amplifies the antimicrobial response. PLoS Pathog 2009; 5: e1000282.
Chen GY, Tang J, Zheng P, Liu Y . CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 2009; 323: 1722–1725.
Deban L, Jarva H, Lehtinen MJ, Bottazzi B, Bastone A, Doni A et al. Binding of the long pentraxin PTX3 to factor H: interacting domains and function in the regulation of complement activation. J Immunol 2008; 181: 8433–8440.
Vaziri-Sani F, Hellwage J, Zipfel PF, Sjoholm AG, Iancu R, Karpman D . Factor H binds to washed human platelets. J Thromb Haemost 2005; 3: 154–162.
Trouw LA, Nilsson SC, Goncalves I, Landberg G, Blom AM . C4b-binding protein binds to necrotic cells and DNA, limiting DNA release and inhibiting complement activation. J Exp Med 2005; 201: 1937–1948.
Acknowledgements
We are grateful for the project and program grant support provided by the Australian National Health and Medical Research Council (NHMRC). Owing to space considerations we apologize for not citing many important contributions to this field. The supplemental data contain a list of these important works.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by P Vandenabeele
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Poon, I., Hulett, M. & Parish, C. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ 17, 381–397 (2010). https://doi.org/10.1038/cdd.2009.195
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.195
Keywords
This article is cited by
-
Immunosuppressive effects of the mycotoxin patulin in macrophages
Archives of Microbiology (2024)
-
Unbiased proteomics and multivariable regularized regression techniques identify SMOC1, NOG, APCS, and NTN1 in an Alzheimer’s disease brain proteomic signature
npj Aging (2023)
-
Endophytic Fungus of Achyrocline satureioides: Molecular Identification, Chemical Characterization, and Cytotoxic Evaluation of its Metabolites in Human Melanoma cell line
Applied Biochemistry and Biotechnology (2023)
-
Implantable micro-scale LED device guided photodynamic therapy to potentiate antitumor immunity with mild visible light
Biomaterials Research (2022)
-
The interrelationships between neuronal viability, synaptic integrity, microglial responses, and amyloid-beta formation in an in vitro neurotrauma model
Scientific Reports (2022)
