
Abstract
The zinc finger-containing transcription factors Egr1 (Krox24) and Egr2 (Krox20) have been implicated in the proliferation and differentiation of many cell types. Egr2 has earlier been shown to play a positive role in adipocyte differentiation, but the function of Egr1 in this context is unknown. We compared the roles of Egr1 and Egr2 in the differentiation of murine 3T3-L1 adipocytes. Egr1 protein was rapidly induced after addition of differentiation cocktail, whereas Egr2 protein initially remained low before increasing on days 1 and 2, concomitant with the disappearance of Egr1. In marked contrast to the effects of Egr2, differentiation was inhibited by ectopic expression of Egr1 and potentiated by knockdown of Egr1. The pro-adipogenic effects of Egr1 knockdown were particularly notable when isobutylmethylxanthine (IBMX) was omitted from the differentiation medium. However, knockdown of Egr1 did not affect CCAAT/enhancer binding protein (C/EBP)β protein expression or phosphorylation of CREB Ser133. Further, Egr1 did not directly affect the activity of promoters for the master adipogenic transcription factors, C/EBPα or peroxisome proliferator-activated receptor-γ2, as assessed in luciferase reporter assays. These data indicate that Egr1 and Egr2 exert opposing influences on adipocyte differentiation and that the careful regulation of both is required for maintaining appropriate levels of adipogenesis. Further, the pro-differentiation effects of IBMX involve suppression of the inhibitory influence of Egr1.
Similar content being viewed by others


Main
Adipose tissue plays a central role in the maintenance of whole-body energy homeostasis. Studies of obese and lipodystrophic humans and rodents show that both increased and decreased adipose tissue mass are associated with insulin resistance and abnormal glucose and lipid metabolism, primary causes of type II diabetes.1, 2 Tight control of adipocyte development, size and insulin sensitivity is therefore of critical importance. Adipose tissue responds to increases in energy flux in part by the differentiation of new adipocyte cells from preadipocyte and stem-cell precursors. The differentiation process requires the carefully orchestrated expression of a cascade of transcription factors.3, 4 It is well established that the core of the transcriptional cascade involves activation of CCAAT/enhancer binding protein (C/EBP) family members C/EBPβ and C/EBPδ followed, in turn, by activation of C/EBPα and the nuclear hormone receptor peroxisome proliferator-activated receptor-γ (PPARγ). Together, PPARγ and C/EBPα control the expression of key genes that confer the differentiated phenotype, including the ability to store lipid in an insulin-sensitive manner. The central adipogenic role of PPARγ is highlighted by the ability of ectopically expressed PPARγ to induce an adipocyte-like phenotype in non-adipose cells.5, 6 The pro-adipogenic transcription factor CREB is also capable of promoting differentiation of preadipocytes in the absence of normal differentiation medium.7, 8
Many other proteins have been identified that impinge on the differentiation process, both positively and negatively. Among these are transcription factors of the Kruppel-like factor (KLF) family, of which KLF5, KLF6 and KLF15 have positive effects, whereas KLF2 and KLF7 have negative effects on adipogenesis.3, 4 Indeed, sequential binding of KLFs to target promoters seems to play a key role in orchestrating differentiation. Other factors that modulate transcription factor activity and promoter accessibility have been shown to be important in fine-tuning the adipogenic process. The co-activator TRAP2209 and histone acetylase CBP/p30010, 11 promote differentiation, whereas the co-repressor ETO/MTG812 and histone deacetylase HDAC113 inhibit differentiation. Studies of the signaling pathways that impact on the adipogenic transcriptional cascade have highlighted the role of secreted proteins such as TGFβ and members of the Wnt and BMP families.3, 4
Early growth response gene 2 (Egr2, also known as Krox20) is a recently described positive regulator of adipogenesis, that apparently exerts its effects through both C/EBPβ-dependent and -independent mechanisms.14 Ectopic expression of Egr2 promoted differentiation in murine 3T3-L1 cells and compensated for the absence of insulin from the differentiation medium.14 Egr2 belongs to a family of Cys2-His2 Zinc-finger transcription factors that also includes Egr1 (also known as NGFI-A, Krox24, tis8 and zif268), Egr3 and Egr4. Egr1 and Egr2 were identified as serum-response genes twenty years ago,15, 16, 17 and are induced by diverse mitogenic stimuli in many different cells. Although Egr1 has been implicated in differentiation of several cell types,18, 19, 20 its role in adipogenesis has not been investigated. This study thus sought to compare the roles of Egr1 and Egr2 in differentiation of 3T3-L1 adipocytes. In contrast to Egr2, Egr1 was found to have an anti-adipogenic action.
Results
Egr1 and Egr2 expression profiles during adipogenesis
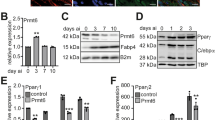
As Egr1 and Egr2 are immediate-early genes induced by a wide variety of stimuli, their expression pattern was assessed in 3T3-L1 cells after addition of differentiation cocktail (MDI: iso-butyl-Methylxanthine, Dexamethasone and Insulin) (Figure 1a). The level of Egr1 protein increased substantially by 1 h and then decreased. In contrast, Egr2 protein did not increase from its initial low level during 4 h after addition of MDI. Levels of Egr1 and Egr2 protein were also determined throughout 6 days of differentiation (Figure 1b). Egr1 protein decreased to near undetectable levels on days 1 and 2, but increased twofold above day 0 levels on day 3 (yet considerably below the peak achieved 1 h after addition of MDI on day 0), which was followed by a return to day 0 levels on days 4 and 5 and a further reduction on day 6. In contrast, Egr2 protein levels increased approximately twofold on days 1 and 2, but decreased to preadipocyte levels thereafter. In general, Egr2 protein expression was highest when Egr1 was lowest, and vise versa, suggesting that they may have distinct roles during differentiation. The exchanges of differentiation medium on days 2, 4 and 5 induced transient increases in expression of Egr1 protein, as on day 0 (data not shown).

Expression of Egr1 and Egr2 during 3T3-L1 differentiation. Two-day post-confluent 3T3-L1 wild-type preadipocytes were differentiated as described in Materials and Methods. Egr1 and Egr2 protein levels were determined in cell lysates by Western blotting with antibodies to Egr1 and Egr2. PI3-kinase p85α subunit was used as loading control. (a) Representative blot showing protein levels immediately after MDI addition. (b) Representative blot showing protein levels throughout differentiation. For comparison with (a) the right-most lane in (b) labeled ‘control’ is a 1 h MDI-treated sample. (c) Quantification of Egr1 and Egr2 protein expression during differentiation. ECL exposures of Western blots were scanned, quantified by densitometry and normalized to PI3-kinase p85α loading control. Error bars represent the S.E.M. of three to four independent experiments
Sustained expression of Egr1 inhibits, whereas Egr2 promotes differentiation
We next compared the effects of ectopically expressing Egr1 and Egr2. Sustained ectopic expression of Egr2 was earlier shown to promote differentiation of 3T3-L1 cells.14 Initial attempts to generate 3T3-L1 preadipocyte cell lines stably overexpressing Egr1 using a retroviral vector were unsuccessful (data not shown), possibly because constitutive expression of Egr1 at high levels is inhibitory to proliferation.21 We therefore employed a strategy for inducible expression of Egrs involving the Cre-inducible retroviral vector pLPNPX1.22 In this system translation of Egr protein from a bi-cistronic mRNA is initiated only after Cre-mediated excision of the upstream intervening sequence encoding the neomycin resistance gene. For flexible introduction of Cre, a recombinant membrane-permeant Cre fusion protein HTNC (His-TAT-NLS-Cre)23 was used. Optimal conditions for incubation of 3T3-L1 cells with HTNC protein so as to maximize the efficiency of gene induction were determined using cells stably expressing an inducible pLPNPX-EGFP construct (Figure 2a). These conditions were then used to induce expression of Egr1 and Egr2 proteins and Western blotting was performed to confirm their induction. No alteration in expression of either Egr1 or Egr2 protein was detectable in the non-induced state. Treatment of confluent pLPNPX-Egr1 or -Egr2 cells with HTNC increased expression of the respective protein, which was maintained for up to 10 days after addition of MDI differentiation cocktail (Figure 2b).

Effects of Cre-mediated induction of Egr1 and Egr2 on adipocyte differentiation. Stably transfected 3T3-L1 cell lines were generated using a pLPNPX-based retrovirus harboring a Cre-inducible cassette for expression of Egr1, Egr2, EGFP or empty vector control (e.v.). Confluent 3T3-L1 cells harboring pLPNPX-EGFP were incubated with or without HTNC protein (8 μM) for 16 h in serum-free medium. Cells were allowed to recover for 56 h after which EGFP expression, as measured by FL1-H channel, was determined by flow cytometry (a). Confluent 3T3-L1 harboring pLPNPX-Egr1, -Egr2 or e.v. control were treated with or without HTNC protein and differentiation was induced 32 h later. Whole-cell lysates were prepared at the indicated times and were subjected to Western blotting with the indicated antibodies. PI3-kinase p85 subunit was used as loading control (b). On day 6, lipid accumulation was visualized by staining with oil red O (c) and quantified by subsequent extraction and absorbance measurement at 515 nm (error bars represent the range of two independent experiments) (d) Relative mRNA levels of biochemical markers of adipogenesis (PPARγ2, C/EBPα, aP2 and GLUT4) determined by semi-quantitative RT-PCR (sqRT-PCR) immediately before addition of MDI (day 0), on days 3 and 6 after addition of MDI (e). Data for each marker are expressed relative to empty vector control on day 3 of differentiation. Open bars, control (e.v.) cells; hatched bars, Egr1-expressing cells; black bars, Egr2-expressing cells. Error bars represent the S.E.M. of three independent experiments, and statistical significance is indicated *P<0.05
To assess the effects of sustained ectopic expression of Egr1 and Egr2 on differentiation, confluent 3T3-L1 cells stably transfected with pLPNPX-Egr1 or -Egr2 were incubated with HTNC protein before addition of MDI cocktail. Lipid accumulation was visualized by oil red O staining after a further 6 days (Figure 2c). Cells expressing Egr1 contained markedly less lipid than controls, whereas cells expressing Egr2 accumulated more lipid. Quantification of oil red O stain showed an approximately twofold decrease in lipid in Egr1-expressing cells and a 2.5-fold increase in Egr2-expressing cells (Figure 2d).
To gain additional insight into the effects of ectopic expression of Egr1 or Egr2 the mRNA levels of biochemical markers of differentiation (C/EBPα, PPARγ2, fatty-acid binding protein 2 (aP2) and glucose transporter 4 (GLUT4)) were determined (Figure 2e). Samples were taken on day 0 to assess effects of overexpression before commencement of differentiation, day 3 as the mid-point of differentiation and day 6 for comparison with oil red O staining. The levels of each marker increased substantially during differentiation of each cell line. On day 0 expression of C/EBPα was already significantly lower in cells expressing Egr1 than in controls, and this difference was maintained at 3 and 6 days of differentiation. In contrast, in cells expressing Egr2 there was a tendency to increased expression of C/EBPα. On day 0 expression of PPARγ2 was already higher in cells expressing Egr2 than in controls and the difference became even greater after 3 and 6 days of differentiation, whereas in Egr1-expressing cells by day 6 expression of PPARγ2 was significantly lower than in controls. The late differentiation markers largely mirrored oil red O staining and expression patterns of the transcription factors C/EBPα and PPARγ2. Compared with controls, levels of both aP2 and GLUT4 were decreased on days 3 and 6 in cells expressing Egr1 and increased in cells expressing Egr2. These data are broadly consistent with earlier studies14 in suggesting that Egr2 is a pro-adipogenic factor, but indicate that Egr1 has an opposite effect and inhibits differentiation when constitutively overexpressed. Further, they indicate that Egr1 exerts its effects in the initial days of differentiation.
Knockdown of Egr1 potentiates, whereas knockdown of Egr2 inhibits, differentiation
To complement the overexpression studies, we tested the effects of RNAi-mediated knockdown of Egr1 and Egr2 on differentiation. Preadipocyte cell lines stably expressing short hairpin RNAs (shRNA) were generated using the pSiren-RetroQ RNAi-Ready retroviral vector. As controls, cells harboring an empty vector or stably expressing an irrelevant shRNA (against the luciferase gene) were used. Similar data were obtained for both controls throughout and data are shown only for one control. Western blotting was used to determine the efficacy of the shRNAs at times, which would otherwise exhibit peak protein expression (Figure 3a and b). Egr1 protein was undetectable in shEgr1 cells 1–2 h after treatment with MDI cocktail, whereas shEgr2 cells exhibited approximately 50% decrease of Egr2 protein on day 1 of differentiation. Therefore, the shEgr1 construct mediates almost complete knockdown of Egr1 protein expression, whereas the shEgr2 construct mediates only partial knockdown of Egr2 protein.

Effects of stable knockdown of Egr1 and Egr2 on differentiation. 3T3-L1 cells stably expressing shRNA targeting Egr1 (shEgr1), Egr2 (shEgr2) or, as a control, the luciferase gene (shLuc) were generated as described in Materials and Methods. After differentiation for the indicated times, protein levels were determined by Western blotting with antibodies to Egr1 (a) and Egr2 (b). PI3-kinase p85α subunit was used as loading control. The black vertical line in (b) indicates where lanes from the same ECL exposure of a single gel were cut and pasted side-by-side. Stable cell lines were differentiated using full MDI cocktail or with omission of individual cocktail components, and lipid accumulation was determined by oil red O staining on day 6 (c). For cells differentiated in the absence of IBMX the relative mRNA levels of biochemical markers of differentiation (PPARγ2, C/EBPα, aP2 and GLUT4) were determined by sqRT-PCR on indicated days (d). Data are normalized to 18S ribosomal RNA and expressed relative to respective mRNA level in shLuc cells on day 3, which was set as one. Open bars, control (shLuc) cells; hatched bars, shEgr1 cells; black bars, shEgr2 cells. Error bars represent the S.E.M. of three independent experiments, and statistical significance is indicated *P<0.05
To address the effects of Egr1 or Egr2 knockdown on 3T3-L1 differentiation, shEgr1, shEgr2 and shLuc cell lines were differentiated and after 6 days lipid accumulation was visualized by oil red O staining (Figure 3c). Consistent with published work,14 shEgr2 cells exhibited a marked decrease in lipid accumulation, whereas shEgr1 cells exhibited a slight increase in lipid accumulation compared with well-differentiated controls. Effects of Egr knockdown were also assessed under sub-optimal differentiation conditions by omission of individual components of the MDI cocktail. Removal of any one of the components greatly impaired differentiation of control cells and no additional inhibitory effects of Egr2 knockdown were visible. Similarly, in the absence of either dexamethasone or insulin shEgr1 cells failed to differentiate. However, in the absence of isobutylmethylxanthine (IBMX), knockdown of Egr1 induced substantial differentiation, with lipid accumulation comparable with that observed in control cells treated with full MDI cocktail. Therefore, knockdown of Egr1 is pro-adipogenic to the extent that lack of Egr1 can effectively compensate for the absence of IBMX.
To gain additional insight into the effects of Egr knockdown, the mRNA levels of biochemical markers of adipogenesis were determined in cells treated with differentiation medium lacking IBMX (Figure 3d). As in the overexpression studies, samples were taken on days 0, 3 and 6 of differentiation. The levels of each of the four markers increased very substantially (30–500-fold) during differentiation of all cell lines. Knockdown of Egr2 had no statistically significant effects on any of the markers at any time point, other than a small inhibition of C/EBPα on day 3. However, knockdown of Egr1 had more widespread effects. On day 0, when all the genes were expressed at relatively low levels, Egr1 knockdown induced a small, but statistically significant increase in only aP2 expression. By day 3 of differentiation all the markers showed higher levels in Egr1 knockdown cells and similar patterns were observed at day 6, when absolute levels of expression had increased still further. Such changes are consistent with the increased lipid deposition in Egr1 knockdown cells. Of the genes studied, C/EBPα was least affected by Egr1 knockdown.
Interrelationships between IBMX and Egr1
The finding that knockdown of Egr1 compensated for the absence of IBMX during differentiation suggested that the roles of Egr1 and IBMX are interrelated, and that IBMX may act to inhibit the activity of Egr1 for differentiation to proceed. However, induction of Egr1 protein by differentiation cocktail on day 0 was the same whether or not IBMX was present (Supplementary Figure 1), and Egr1 protein was localized to the nuclear fraction under both conditions (data not shown).
The transcription factor CREB plays an important role in adipocyte differentiation,7, 24 and IBMX would be expected to activate CREB through inhibition of cAMP phosphodiesterases, elevation of intracellular cAMP and phosphorylation of Ser133 by cAMP-dependent protein kinase. We therefore tested whether knockdown of Egr1 might potentiate this action of IBMX (Figure 4). Basal phosphorylation of CREB Ser133 was very low, but robust phosphorylation was observed within 10 min of addition of full MDI cocktail. Phosphorylation only returned to basal levels between 8 and 24 h. Profiles of CREB phosphorylation were very similar in both shEgr1 cells and controls. Surprisingly, similar levels of CREB phosphorylation were observed after addition of DI cocktail (lacking IBMX), except for a modest decrease at 8 h, but again knockdown of Egr1 did not affect the profile. We conclude that Egr1 does not influence CREB phosphorylation in the early stages of adipocyte differentiation and that the induction of CREB phosphorylation is not substantially dependent on IBMX.

Effect of Egr1 knockdown on phosphorylation of CREB. Post-confluent 3T3-L1 cells stably expressing shLuc control (A) or shEgr1 (B) were treated with FBS medium containing complete differentiation cocktail (MDI) or cocktail lacking IBMX (DI) for the times indicated. Equal amounts of whole-cell protein extracts were subjected to Western blotting with antibody to phosphorylated Ser133 of CREB (P-CREB) and total CREB. Samples taken at 0–8 h and 24 h were loaded on separate gels and the 10 min MDI-stimulated samples were loaded on both gels for comparison of band intensities. Blots representative of three independent experiments are shown. The arrow indicates the phospho-CREB band at 46 kDa
C/EBPβ is an important early transcriptional regulator of adipogenesis and a target of IBMX in preadipocytes.25, 26 In addition to inducing C/EBPβ directly, IBMX is permissive for the induction of C/EBPβ by Egr2/Krox20.14 We therefore investigated whether induction of C/EBPβ was influenced by knockdown of Egr1 or Egr2 (Figure 5). MDI cocktail induced robust and similar increases in C/EBPβ mRNA expression (approx 15-fold at 2 h) in each shRNA-expressing cell line. The omission of IBMX from differentiation cocktail attenuated the induction of C/EBPβ by approximately 40–50% in control and Egr2 knockdown cells, but not in Egr1 knockdown cells. However, removal of insulin from the cocktail did not affect induction of C/EBPβ mRNA compared with full cocktail. In line with these findings full MDI cocktail induced greater expression of C/EBPβ protein in control cells than did cocktail without IBMX, especially at 4 h. However, no difference in C/EBPβ protein expression between Egr1 knockdown and control cells was observed, even in the absence of IBMX. Therefore, the modest difference in expression of C/EBPβ mRNA between control and Egr1 knockdown cells treated with IBMX-depleted medium was not paralleled by a difference in C/EBPβ protein expression.

Effect of knockdown of Egr1 and Egr2 on C/EBP β expression. Post-confluent 3T3-L1 preadipocytes stably expressing shEgr1, shEgr2 or shLuc control were treated with complete differentiation cocktail (MDI), cocktail lacking IBMX (DI) or cocktail lacking insulin (MD). (a) Relative levels of C/EBPβ mRNA were determined after 2 h by sqRT-PCR and expressed relative to shLuc cells at time 0. Error bars represent the S.E.M. of four independent experiments (DI and MD) or range of two independent experiments (MDI). P-value of DI-treated shEgr1 versus shLuc=0.06. (b) Whole-cell protein lysates from shEgr1 (A) or shLuc (B) cells treated with IBMX-depleted cocktail were subjected to Western blotting with anti-C/EBPβ, anti-Egr1 or as loading control anti-β-actin antibody. Representative blots from two independent experiments are shown
Effect of Egr1 and Egr2 on PPARγ2 and C/EBPα expression
The finding that C/EBPα and PPARγ2 exhibited altered expression levels in cells ectopically expressing either Egr1 or Egr2, respectively, before addition of MDI cocktail (Figure 3) suggested possible direct effects of Egr proteins on the promoters of those genes. Promoter–reporter experiments, using the Firefly luciferase gene, were used to assess the effects of either Egr1 or Egr2 on expression from either the PPARγ2 or C/EBPα promoter (Figure 6). C/EBPδ is a member of the C/EBP protein family and positive adipogenic regulator, known to induce both PPARγ2 and C/EBPα,27, 28 and was thus used as a positive control. Indeed, expression of C/EBPδ induced three- and fivefold increase in expression of the luciferase reporter from the PPARγ2 and C/EBPα promoters, respectively. However, expression of Egr1 had no effect on luciferase expression from either promoter, whereas, unexpectedly, Egr2 induced a modest inhibition of reporter expression from the C/EBPα promoter. Therefore, the opposing influences of Egr1 and Egr2 on adipogenesis cannot be accounted for by direct transcriptional regulation of the ‘master’ adipogenic transcription factors.

Effect of Egr1 and Egr2 on C/EBP α and PPAR γ 2 expression. 3T3-L1 preadipocytes were transfected with either a C/EBPα promoter- or PPARγ2 promoter-Firefly luciferase construct together with expression vectors for C/EBPδ, Egr1 or Egr2 in the indicated quantities. Data shown are from a single experiment, representative of two
Discussion
The data presented here show, for the first time, that the transcription factor Egr1 (Krox24) acts as a negative regulator of 3T3-L1 differentiation and confirm that the related protein Egr2 (Krox20) acts as a positive regulator. Specifically, stable knockdown of Egr1 enhanced differentiation and largely compensated for the absence of IBMX from differentiation cocktail, whereas induction of ectopic Egr1 expression inhibited differentiation. In contrast, knockdown of Egr2 inhibited differentiation, whereas induction of Egr2 enhanced differentiation.
Our findings in relation to Egr2 are generally in line with the published work of Friedman and colleagues,14 but differ from it in some important respects. Friedman and colleagues documented the expression of Egr2 mRNA, which reached an early peak 1–2 h after addition of differentiation cocktail to confluent 3T3-L1 preadipocytes, declined to a low level by 6 h and then remained essentially undetectable. We confirmed that Egr2 mRNA was rapidly and substantially induced by differentiation medium (data not shown), but found that this was not coupled to an immediate increase in the level of Egr2 protein (Figure 1a). Indeed, Egr2 protein expression increased only between days 1 and 2 of differentiation, and only by a modest amount. It was shown by Friedman and colleagues that ectopic overexpression of Egr2 potentiated the early IBMX- and dexamethasone-dependent increase in C/EBPβ expression.14 More recently it was shown that Egr2 cooperates with the IBMX-regulated transcription factor KLF4 to induce C/EBPβ expression.29 However, the specific window within which this mode of C/EBPβ regulation occurs during the differentiation process remains unclear as Egr2 is expressed only at very low levels in the initial hours of adipogenesis and knockdown of Egr2 has no effect on acute expression of C/EBPβ (Figure 5). It is noteworthy that knockdown of C/EBPβ only partially impaired the pro-adipogenic effect of Egr2,14 indicating that effects of Egr2 are at least partially C/EBPβ-independent. The IBMX dependency of both Egr1 inhibition of adipogenesis (as shown here) and KLF4 expression29 suggests a possible link between the two factors.
Our observation that there is no dramatic early increase in Egr2 protein in differentiating 3T3-L1 cells at the time of initial C/EBPβ induction calls into question the importance of C/EBPβ in mediating pro-adipogenic effects of endogenous Egr2. Indeed, we found that partial knockdown of Egr2 (achieved using the published shRNA sequence14) did not affect the acute induction of C/EBPβ mRNA in response to differentiation cocktail (Figure 5a). The finding that Egr2 binds directly to KLF4 to induce expression of C/EBPβ29 does not conflict with the suggestion that Egr2 also exerts part of its pro-adipogenic effects in parallel with C/EBPβ.
A mechanistic link between C/EBPβ and the anti-differentiative effects of Egr1 was suggested by the observation that knockdown of Egr1 rescued the defect in C/EBPβ mRNA induction mediated by the removal of IBMX from differentiation medium, although there was no comparable rescue of the attenuation of C/EBPβ protein expression (Figure 5). The reasons for this discrepancy are unclear. Further investigation is clearly required to determine whether Egr1 influences the expression or activity of C/EBPβ at later stages of differentiation. Nevertheless, the finding that knockdown of Egr1 enhanced differentiation in medium lacking IBMX, but not dexamethasone or insulin, suggests that under normal differentiation conditions IBMX may act in part by inhibiting Egr1 action. However, we could not show any effect of IBMX on the expression (Supplementary Figure 1) or nuclear localization (data not shown) of Egr1 protein, nor was there any effect of Egr1 knockdown on the phosphorylation of CREB (Figure 4), a known target of IBMX and important regulator of adipogenesis.24
It seems paradoxical that a protein with the capacity to inhibit differentiation should exhibit a rapid and substantial increase in expression as an early response to addition of MDI cocktail. However, the critical time window within which Egr1 exerts its inhibitory effect on the overall differentiation program is unknown. Clearly, the transient MDI-induced induction of Egr1 expression is dispensable for differentiation (Figure 3) and it is likely that the later decrease in Egr1 expression, between days 1 and 2, is more important for differentiation to occur. Unfortunately, currently available techniques do not allow rapid and reversible knockdown or overexpression of proteins within narrow time windows so as to determine the critical period of Egr1 action.
Important outstanding questions are whether the opposing effects of Egr1 and Egr2 on adipogenesis are exerted at the same or distinct phases of the differentiation program and whether they reflect antagonistic actions on shared or distinct targets. There is a degree of reciprocity in the fluctuating levels of Egr1 and Egr2 proteins, in that Egr1 is near undetectable by day 2 of differentiation, concomitant with maximum expression of Egr2, whereas Egr1 reappears on day 3 as Egr2 decreases (Figure 1). Sequential binding of Egr1 and Egr2 to target promoters may be necessary to orchestrate the differentiation process. Egr1 and Egr2 share almost identical consensus binding sites,30 and consensus sites within certain promoters have been shown to be capable of interacting with either Egr1 or Egr2. However, other binding sites, usually dependent on promoter sequence context, can interact with one of these Egrs, but not the other.31, 32 Such differential modulation of gene expression is attributed to the ability of different Egr proteins to interact with different secondary factors through their divergent activation domains. Although we found that cells overexpressing Egr1 or Egr2 had altered levels of both PPARγ2 and C/EBPα mRNA, there was no evidence in promoter–reporter assays that Egr1 or Egr2 could directly modulate the expression of either gene. Whether Egr proteins affect C/EBP-dependent expression from these promoters requires further investigation. Egr1 can induce expression of other genes, such as Pref-1 and PAI-1, which are known to inhibit adipocyte differentiation.33, 34
In conclusion, our data add to the growing body of evidence that Egr proteins play important, but complex, roles in the differentiation of a number of cell types. However, the effects of Egr1 and Egr2 are cell- and context-dependent. Similar to the situation described here Egr1 and Egr2 displayed opposing expression patterns and function during Schwann cell differentiation35 and T-cell activation.36 Indeed, the latter study provided evidence that Egr2 can suppress Egr1 expression mechanistically. In contrast, Egr proteins were found to play similar and redundant roles in both the suppression of neutrophil differentiation, in favor of macrophage differentiation, and the promotion of thymocyte differentiation.37, 38 Further work is clearly required to understand the mechanisms by which the differentiation of different cell types is regulated by the tonic balance of Egr1 and Egr2.
Materials and Methods
Reagents
All laboratory chemicals were from Sigma Aldrich (Gillingham, UK), except where stated.
Cell culture and differentiation
3T3-L1 preadipocytes (ATCC) were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% neonatal calf serum plus 2 mM L-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin (NCS medium), in a humidified incubator under 5% C02/95% air. Two days after confluence (differentiation day 0) medium containing 0.5 mM IBMX, 1 μM dexamethasone and 5 μg/ml insulin (MDI cocktail) and 10% fetal calf serum (FCS) was added. After two days this was replaced with medium containing 5 μg/ml insulin and FCS. After four days cells were maintained in medium containing FCS alone, with fresh medium added daily. For submaximal differentiation of 3T3-L1 cells, the relevant components of the MDI cocktail, as indicated in figure legends, were omitted for the entire period that they would normally be used. The retroviral packaging cell line HEK293-BOSC was maintained in DMEM containing 10% FCS plus 2 mM L-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin, in a humidified incubator under 10% C02/90% air.
Plasmids and retroviral infection
Stable knockdown of Egr1 and Egr2 was achieved using a retroviral-based shRNA expression system. An siRNA against Egr1 was designed using a web-based tool (Dharmacon, CO, USA) and modified to include a loop sequence (see below) to make it a functional shRNA. The shRNA targeting Egr2 was as described earlier.14 The relevant pairs of oligonucleotides were annealed and introduced into the BamHI/EcoRI sites of pSiren-RetroQ RNAi-Ready (Clontech, CA, USA). A shRNA targeting the luciferase gene (Clontech) and circularized empty vector were included as controls. Retroviruses were generated by transfection of the HEK293-BOSC packaging cell line with the relevant plasmids using Fugene 6 (Roche, Lewes, UK). Viral medium supplemented with 2 μg/ml polybrene was added to 3T3-L1 cells 48 h later. 3T3-L1 cells were selected using 4 μg/ml puromycin and maintained in 2 μg/ml puromycin. Sequences for the oligonucleotides cloned into pSiren-RetroQ were as follows (siRNA sense and antisense sequences are underlined, loop sequences are in bold): shEgr1-sense, 5′-GATCCATGCGTAACTTCAGTCGTA AGAGAACTT TACGACTGAAGTTACGCATTTTTTTCTCGAGG-3′; shEgr1-antisense, 5′-AATTCCTCGAGAAAAAAATGCGTAACTTCAGTCGTA AAGTTCTCT TACGACTGAAGTTACGCATG-3′; shEgr2-sense, 5′-GATCCGGCTCTGGCTGACACACCAG TTCAAGAGA CTGGTGTGTCAGCCAGAGCCTTTTTTCTCGAGG-3′; shEgr2-antisense, 5′-AATTCCTCGAGAAAAAAGGCTCTGGCTGACACACCAG TCTCTTGAA CTGGTGTGTCAGCCAGAGCCG-3′. The Gateway system (Invitrogen, Paisley, UK) was employed for cloning Egr1 and Egr2 cDNAs into N-terminal HA-tagging vector pDEST-475 (Invitrogen) and Cre-inducible pLPNPX plasmid.22 Egr1 cDNA was amplified by PCR from IMAGE clone #2631117 (Geneservice, Cambridge, UK). Egr2 cDNA was obtained by reverse transcription (RT)-PCR from an insulin-induced 3T3-L1 adipocyte sample. PCR products were introduced to the Gateway pENTR vector and from there these were transferred to pDEST-475 and pLPNPX destination vectors according to the manufacturer's instructions. Sequences for the primers used to amplify Egr1 and Egr2 are given in Table 1 (Supplementary Information).
Real time PCR
Total RNA was islolated from 3T3-L1 cells using RNeasy Mini Kit (Qiagen, Crawley, UK). 300 ng total RNA was used for each RT reaction. Primer Express software (version 1.0, Perkin-Elmer Applied Biosystems, Warrington, UK) was used to design the primers and probes for semi-quantitative real time PCR to determine murine mRNA expression levels, except for C/EBPα for which a pre-designed primer/probe set was used (Applied Biosystems). SYBR-Green-based semi-quantitative real time PCR analysis was used to determine mRNA expression levels for Egr1, Egr2 and Glut4. TaqMan analysis was used to determine expression levels for PPARγ2, C/EBPα and 18S ribosomal RNA. Sequences for primers and probes used are given in Table 1 (Supplementary Information). 18S rRNA was used as internal control in which replicate experiments were not run on individual real time PCR plates. Data in Figure 2e were obtained from three experiments in which RTs and real time PCR analysis for all samples were prepared in parallel without internal normalization.
Western blotting
Whole-cell lysates were extracted into lysis buffer (20 mM HEPES, 50 mM NaF, 30 mM C3H7Na2O6P, 5 mM EGTA, 5 mM EDTA, 10 mM NA4P2O7, 1% (v/v) Triton X100, 4 mM Na3VO4 and protease inhibitor cocktail (1 : 200; Sigma)) and diluted 1 : 5 (v/v) in Laemmli loading buffer (containing 100 mM DTT final concentration). Equivalent amounts of lysates were subjected to SDS-PAGE using Mini-Protean II apparatus (Bio-Rad, Paisley, UK), and transferred to PVDF membrane (Millipore, Watford, UK) using semi-dry Western blotting apparatus. Membranes were probed with primary antibodies: Egr1 (sc-189, Santa Cruz, CA, USA), Egr2 (CRP Inc., PA, USA), phospho-CREB and CREB (cat. #9196 and #9197, Cell Signalling, MA, USA), C/EBPβ (sc-150, Santa Cruz), β-actin (AbCam, Cambridge, UK) and PI3-kinase-p85 subunit (generated in-house). Membranes were then incubated in HRP-conjugated secondary antibodies (DAKO Ltd, Ely, UK) and emission detected using enhanced chemiluminescence, Hyper-ECL film (Amersham, Little Chalfont, UK) and X-OMAT (Fujifilm, Bedfordshire, UK). For quantification of Western blots relevant ECL exposures were scanned and densitometry analysis performed using AIDA software (Raytest Isotopenmessgeraete, Germany). Egr1 and Egr2 expression values were normalized to PI3-kinase p85α expression.
Transduction of cells with HTNC protein
Cell-permeant Cre recombinase fusion protein containing a nuclear localization signal (HTNC) was generated in E coli using the pTriEx1.1 construct, as described.23 Bacteria were grown and induced with 0.5 mM isopropyl β-D-thiogalactoside (IPTG). After centrifugation the bacterial pellet was resuspended in 10 ml phosphate Tris buffer (PTB; 50 mM NaH2PO4, 5 mM Tris-HCl, pH 7.8) per liter of culture and incubated at room temperature first with lysosyme (1 mg/ml, 20 min) and then benzonase (1 : 1000, 15 min, Novagen, Nottingham, UK). After sonication 1 ml of cold tartaric salt buffer (TSB: PTB containing 2 M L-tartaric acid, 20 mM imidazole) was added per ml of suspension, mixed and incubated on ice for 5 min. The solution was centrifuged for 30 min at 17 000 rpm (SS 34, Kendro) at 4°C and 2 ml of washed 50% Ni-NTA slurry (Qiagen) per liter culture was added to the supernatant and shaken gently for 1 h at 4°C The suspension was applied onto a column (Bio-Rad), washed twice with 5 resin volumes of wash buffer (1 × PTB, 500 mM NaCl, 20 mM imidazole) and protein eluted (1 × PTB, 500 mM NaCl, 250 mM imidazole) with 2–3 resin volumes. Eluant was filtered (0.22 μm) and dialysed firstly against high salt buffer (600 mM NaCl, 20 mM HEPES, pH 7.4) then against 50% (v/v) glycerol (containing 500 mM NaCl, 20 mM HEPES, pH 7.4), both overnight at 4°C. Purified HTNC protein was stored at −80°C. Optimal conditions for incubation of HTNC protein with 3T3-L1 preadipocytes were determined using inducible pLPNPX-EGFP cells. On the day of confluency fresh NCS medium was added to 3T3-L1 cells 4–6 h before addition of HTNC. Cells were washed twice with serum-free medium and HTNC protein was incubated at 8 μM for 16 h in serum-free medium. Cells were rinsed and allowed to recover in NCS-medium for 32 h, after which differentiation was induced.
Flow cytometry
3T3-L1 preadipocytes were washed twice with phosphate buffered saline (PBS) and detached (trypsin 1 : 5 in 5 mM EDTA/PBS) from plate surface. Cells were pelleted at 300 g at 4°C resuspended in 0.5% BSA/5 mM EDTA/PBS, pelleted again and resuspended in fixation medium (2% paraformaldehyde/PBS) and stored at 4°C. Analysis was performed using a FACSCalibur instrument and CELLQUEST software (BD Biosciences, CA, USA). EGFP fluorescence was measured using the FL1-H channel.
Oil red O staining
Cells were washed with PBS and then fixed with 0.5% glutaraldehyde (in PBS) for 5 min. After washing twice with PBS, followed by rinsing with 60% isopropanol, cells were stained in oil red O solution (3 : 2 isopropanol, 0.25% oil red O/H2O) for 30 min, and finally rinsed with 60% isopropanol and washed with PBS. Digital images of stained plates were taken using Nikon Digital Sight DS-U1 camera. Quantification of lipid accumulation was achieved by extraction of oil red O stain from cells by incubation with 100% ethanol for 10 min and absorbance at 515 nm measured using an Anthos HT II (Labtech, Ringmer, UK) plate reader.
Luciferase reporter assays
3T3-L1 preadipocytes were transfected with 50 ng of either pGL3-C/EBPα promoter–reporter construct (containing C/EBPα nucleotides −1450 to +125)39 or pGL3-PPARγ2 promoter–reporter construct (containing PPARγ nucleotides −609 to +25)28 in the presence of pcDNA3.1-C/EBPδ (50 ng), pDEST-475-HA-Egr1 (50 or 200 ng) or pDEST-475-HA-Egr2 (50 or 200 ng) as indicated with Lipofectamine 2000 (Invitrogen). Approximately equal amounts of Egr1 and Egr2 protein expression from pDEST-475 was confirmed by transient transfection of HEK293 cells and detection by anti-HA antibody (data not shown). Empty vector pcDNA3.1 was used to equalize DNA quantity per transfection. Luciferase expression levels were assayed 24 h after transfection by using a dual luciferase reporter assay system (Promega, Southampton, UK). Values were normalized to pRL-CMV constitutive Renilla luciferase reporter vector.
Abbreviations
- aP2:
-
fatty-acid binding protein 2
- C/EBP:
-
CCAAT/enhancer binding protein
- DMEM:
-
Dulbecco's modified Eagle's medium
- GLUT4:
-
glucose transporter 4
- HTNC:
-
His-TAT-NLS-Cre recombinase fusion protein
- IBMX:
-
isobutylmethylxanthine
- IPTG:
-
isopropyl β-D-thiogalactoside
- KLF:
-
Kruppel-like factor
- MDI:
-
IBMX-dexamethasone and insulin
- PBS:
-
phosphate buffered saline
- PTB:
-
phosphate Tris buffer
- PPAR:
-
peroxisome proliferator-activated receptor
- SDS-PAGE:
-
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- RT-PCR:
-
reverse transcriptase polymerase chain reaction
- shRNA:
-
short hairpin ribonucleic acid
References
Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B et al. Life without white fat: a transgenic mouse. Genes Dev 1998; 12: 3168–3181.
Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 2002; 109: 1345–1350.
Farmer SR . Transcriptional control of adipocyte formation. Cell Metab 2006; 4: 263–273.
Rosen ED, MacDougald OA . Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 2006; 7: 885–896.
El-Jack AK, Hamm JK, Pilch PF, Farmer SR . Reconstitution of insulin-sensitive glucose transport in fibroblasts requires expression of both PPARgamma and C/EBPalpha. J Biol Chem 1999; 274: 7946–7951.
Wu Z, Rosen ED, Brun R, Hauser S, Adelmant G, Troy AE et al. Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol Cell 1999; 3: 151–158.
Fox KE, Fankell DM, Erickson PF, Majka SM, Crossno Jr JT, Klemm DJ . Depletion of cAMP-response element-binding protein/ATF1 inhibits adipogenic conversion of 3T3-L1 cells ectopically expressing CCAAT/enhancer-binding protein (C/EBP) alpha, C/EBP beta, or PPAR gamma 2. J Biol Chem 2006; 281: 40341–40353.
Klemm DJ, Leitner JW, Watson P, Nesterova A, Reusch JE, Goalstone ML et al. Insulin-induced adipocyte differentiation. Activation of CREB rescues adipogenesis from the arrest caused by inhibition of prenylation. J Biol Chem 2001; 276: 28430–28435.
Ge K, Guermah M, Yuan CX, Ito M, Wallberg AE, Spiegelman BM et al. Transcription coactivator TRAP220 is required for PPAR gamma 2-stimulated adipogenesis. Nature 2002; 417: 563–567.
Takahashi N, Kawada T, Yamamoto T, Goto T, Taimatsu A, Aoki N et al. Overexpression and ribozyme-mediated targeting of transcriptional coactivators CREB-binding protein and p300 revealed their indispensable roles in adipocyte differentiation through the regulation of peroxisome proliferator-activated receptor gamma. J Biol Chem 2002; 277: 16906–16912.
Yamauchi T, Oike Y, Kamon J, Waki H, Komeda K, Tsuchida A et al. Increased insulin sensitivity despite lipodystrophy in Crebbp heterozygous mice. Nat Genet 2002; 30: 221–226.
Rochford JJ, Semple RK, Laudes M, Boyle KB, Christodoulides C, Mulligan C et al. ETO/MTG8 is an inhibitor of C/EBPbeta activity and a regulator of early adipogenesis. Mol Cell Biol 2004; 24: 9863–9872.
Wiper-Bergeron N, Wu D, Pope L, Schild-Poulter C, Hache RJ . Stimulation of preadipocyte differentiation by steroid through targeting of an HDAC1 complex. EMBO J 2003; 22: 2135–2145.
Chen Z, Torrens JI, Anand A, Spiegelman BM, Friedman JM . Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell Metab 2005; 1: 93–106.
Joseph LJ, Le Beau MM, Jamieson Jr GA, Acharya S, Shows TB, Rowley JD et al. Molecular cloning, sequencing, and mapping of EGR2, a human early growth response gene encoding a protein with ‘zinc-binding finger’ structure. Proc Natl Acad Sci USA 1988; 85: 7164–7168.
Milbrandt J . A nerve growth factor-induced gene encodes a possible transcriptional regulatory factor. Science 1987; 238: 797–799.
Lau LF, Nathans D . Expression of a set of growth-related immediate early genes in BALB/c 3T3 cells: coordinate regulation with c-fos or c-myc. Proc Natl Acad Sci USA 1987; 84: 1182–1186.
Dinkel A, Warnatz K, Ledermann B, Rolink A, Zipfel PF, Burki K et al. The transcription factor early growth response 1 (Egr-1) advances differentiation of pre-B and immature B cells. J Exp Med 1998; 188: 2215–2224.
Nguyen HQ, Hoffman-Liebermann B, Liebermann DA . The zinc finger transcription factor Egr-1 is essential for and restricts differentiation along the macrophage lineage. Cell 1993; 72: 197–209.
Pignatelli M, Cortes-Canteli M, Santos A, Perez-Castillo A . Involvement of the NGFI-A gene in the differentiation of neuroblastoma cells. FEBS Lett 1999; 461: 37–42.
Lucerna M, Pomyje J, Mechtcheriakova D, Kadl A, Gruber F, Bilban M et al. Sustained expression of early growth response protein-1 blocks angiogenesis and tumor growth. Cancer Res 2006; 66: 6708–6713.
Lawrence JT, Birnbaum MJ . ADP-ribosylation factor 6 delineates separate pathways used by endothelin 1 and insulin for stimulating glucose uptake in 3T3-L1 adipocytes. Mol Cell Biol 2001; 21: 5276–5285.
Peitz M, Pfannkuche K, Rajewsky K, Edenhofer F . Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: a tool for efficient genetic engineering of mammalian genomes. Proc Natl Acad Sci USA 2002; 99: 4489–4494.
Reusch JE, Colton LA, Klemm DJ . CREB activation induces adipogenesis in 3T3-L1 cells. Mol Cell Biol 2000; 20: 1008–1020.
Cao Z, Umek RM, McKnight SL . Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev 1991; 5: 1538–1552.
Yeh WC, Cao Z, Classon M, McKnight SL . Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev 1995; 9: 168–181.
Christy RJ, Kaestner KH, Geiman DE, Lane MD . CCAAT/enhancer binding protein gene promoter: binding of nuclear factors during differentiation of 3T3-L1 preadipocytes. Proc Natl Acad Sci USA 1991; 88: 2593–2597.
Clarke SL, Robinson CE, Gimble JM . CAAT/enhancer binding proteins directly modulate transcription from the peroxisome proliferator-activated receptor gamma 2 promoter. Biochem Biophys Res Commun 1997; 240: 99–103.
Birsoy K, Chen Z, Friedman J . Transcriptional regulation of adipogenesis by KLF4. Cell Metab 2008; 7: 339–347.
Swirnoff AH, Milbrandt J . DNA-binding specificity of NGFI-A and related zinc finger transcription factors. Mol Cell Biol 1995; 15: 2275–2287.
Decker EL, Nehmann N, Kampen E, Eibel H, Zipfel PF, Skerka C . Early growth response proteins (EGR) and nuclear factors of activated T cells (NFAT) form heterodimers and regulate proinflammatory cytokine gene expression. Nucleic Acids Res 2003; 31: 911–921.
Dzialo-Hatton R, Milbrandt J, Hockett Jr RD, Weaver CT . Differential expression of Fas ligand in Th1 and Th2 cells is regulated by early growth response gene and NF-AT family members. J Immunol 2001; 166: 4534–4542.
Takemori H, Doi J, Katoh Y, Halder SK, Lin XZ, Horike N et al. Characterization of a proximal element in the rat preadipocyte factor-1 (Pref-1) gene promoter. Eur J Biochem 2001; 268: 205–217.
Liao H, Hyman MC, Lawrence DA, Pinsky DJ . Molecular regulation of the PAI-1 gene by hypoxia: contributions of Egr-1, HIF-1{alpha}, and C/EBP{alpha}. FASEB J 2007; 21: 935–949.
Topilko P, Levi G, Merlo G, Mantero S, Desmarquet C, Mancardi G et al. Differential regulation of the zinc finger genes Krox-20 and Krox-24 (Egr-1) suggests antagonistic roles in Schwann cells. J Neurosci Res 1997; 50: 702–712.
Collins S, Lutz MA, Zarek PE, Anders RA, Kersh GJ, Powell JD . Opposing regulation of T cell function by Egr-1/NAB2 and Egr-2/Egr-3. Eur J Immunol 2008; 38: 528–536.
Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee HJ, Sciammas R et al. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell 2006; 126: 755–766.
Carter JH, Lefebvre JM, Wiest DL, Tourtellotte WG . Redundant role for early growth response transcriptional regulators in thymocyte differentiation and survival. J Immunol 2007; 178: 6796–6805.
Tang QQ, Jiang MS, Lane MD . Repression of transcription mediated by dual elements in the CCAAT/enhancer binding protein alpha gene. Proc Natl Acad Sci USA 1997; 94: 13571–13575.
Acknowledgements
We are very grateful to the Wellcome Trust for financial support for the Biological Atlas of Insulin Resistance (BAIR) program (GR066786), and to all members of the BAIR consortium for helpful discussion. We also acknowledge support from the BBSRC (project grant C20109 for SHR), MRC (studentships for SV and WPC) and European Union COST Action BM0602. We thank Dr. F Edenhofer (Institute of Reconstructive Neurobiology, University of Bonn, Germany) for the pTriEx1.1 plasmid and advice on the production of HTNC protein, Dr. MJ Birnbaum (Howard Hughes Medical Institute, PA, USA) for pLPNPX1 plasmid and Dr. Justin Rochford (University of Cambridge) for helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Federici
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Boyle, K., Hadaschik, D., Virtue, S. et al. The transcription factors Egr1 and Egr2 have opposing influences on adipocyte differentiation. Cell Death Differ 16, 782–789 (2009). https://doi.org/10.1038/cdd.2009.11
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.11
Keywords
This article is cited by
-
Transcriptomic profiling of the telomerase transformed Mesenchymal stromal cells derived adipocytes in response to rosiglitazone
BMC Genomic Data (2022)
-
EEF1A1 transcription cofactor gene polymorphism is associated with muscle gene expression and residual feed intake in Nelore cattle
Mammalian Genome (2022)
-
Whole transcriptome RNA-seq reveals key regulatory factors involved in type 2 diabetes pathology in peripheral fat of Asian Indians
Scientific Reports (2021)
-
Co-expression network analysis predicts a key role of microRNAs in the adaptation of the porcine skeletal muscle to nutrient supply
Journal of Animal Science and Biotechnology (2020)
-
Adipogenesis and metabolic health
Nature Reviews Molecular Cell Biology (2019)
