
Abstract
Positive α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor modulators include benzamide compounds that allosterically modulate AMPA glutamate receptors. These small molecules that cross the blood–brain barrier have been shown to act as a neuroprotectant by increasing the levels of endogenous brain-derived neurotrophic factor (BDNF). Positive AMPA receptor modulators have also been shown to increase the levels of growth-associated protein-43 (GAP-43). GAP-43 plays a major role in many aspects of neuronal function in vertebrates. The goal of this study was to determine whether GAP-43 was important in mediating the actions of positive AMPA receptor modulator (S18986) and BDNF. Using cortical cultures from GAP-43 knockout and control mice, we show that (1) GAP-43 is upregulated in response to S18986 and BDNF in control cultures; (2) this upregulation of GAP-43 is essential for mediating the neuroprotective effects of S18986 and BDNF; (3) administration of S18986 and BDNF leads to an increase in the expression of the glutamate transporters GLT-1 and GLAST that are key to limiting excitotoxic cell death and this increase in GLT-1 and GLAST expression is completely blocked in the absence of GAP-43. Taken together this study concludes that GAP-43 is an important mediator of the neurotrophic effects of S18986 and BDNF on neuronal survival and plasticity, and is essential for the success of positive AMPA receptor modulator-BDNF-based neurotrophin therapy.
Similar content being viewed by others

Main
Positive α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor modulators allosterically modulate AMPA-type glutamate receptors and facilitate fast excitatory transmission,1, 2 and have been shown to enhance learning and memory.3, 4, 5 Positive AMPA receptor modulators also decrease neuronal loss in excitotoxic neonatal brain lesions.6 Thus, positive AMPA receptor modulators are potential candidates for treatment of diseases characterized by cognitive dysfunction and neuronal loss.7, 8
Some of the effects of positive AMPA receptor modulators are mediated via increased BDNF expression.9, 10 Positive AMPA receptor modulators however are preferred over direct administration of BDNF itself as this class of molecules readily crosses the blood–brain barrier.11
Brain-derived neurotrophic factor is important for appropriate development and connectivity of neurons as well as their survival.12, 13 In adults, activity dependent modulation of BDNF levels is important for synaptic plasticity and remodeling.14, 15 Activation of the extracellular-regulated kinases (ERKs) pathway by BDNF is important for neuronal survival.16, 17 BDNF mediates activity dependent synaptic plasticity by mechanisms that involve receptor regulation and localized gene transcription.18, 19 At present, it is unclear whether the neuroprotection, synaptic remodeling and plasticity effects of BDNF are related to each other through common downstream elements.
Growth-associated protein-43 (GAP-43) is a nervous system specific F-actin regulating phosphoprotein expressed during development that enables differentiating neurons to respond to extracellular signals.20 GAP-43 also plays a role in sprouting responses associated with adult plasticity.21 GAP-43 is expressed along with BDNF in response to neuronal injuries and is upregulated in granule cells following BDNF treatment.22, 23
In the present study, we test whether GAP-43 is an essential downstream effector of positive AMPA receptor modulators and subsequent BDNF-induced responses.
Results
S18986 and BDNF enhance GAP-43 expression
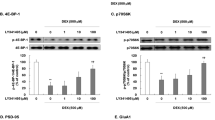
In agreement with previous data,9 there was a threefold increase in BDNF mRNA levels after 3 h of stimulation of cortical cultures with S18986 (CTL (0.024±0.003) vs S18986 (0.087±0.003); P<0.05 Student's t-test). At 3 h post-stimulation of cortical cultures with S18986 or BDNF, there was a significant increase in phosphorylated GAP-43 that could be blocked by the addition of BDNF blocking antibody (Figure 1a and b), although there was no increase in the total amount of GAP-43 protein levels (Figure 1c). At 24 h post–stimulation, there was a significant increase in both total and phosphorylated GAP-43 that could be blocked by the addition of BDNF blocking antibody (Figure 1d-f). Immunocytochemistry confirmed the increase in the intensity of GAP-43 immunoreactivity that could be blocked by the addition of BDNF blocking antibody (Figure 1g).

GAP-43 expression in cortical cultures. (a) Western blot probed with phospho-specific 2G12 antibody, total GAP-43 and total tubulin after 3 h of stimulation. (b) Quantitation of phospho-GAP-43 normalized to total GAP-43 after 3 h of stimulation. (c) Quantitation of total GAP-43 normalized to tubulin after 3 h of stimulation. (d) Western blot probed with phospho-specific 2G12 antibody, total GAP-43 and total tubulin after 24 h of stimulation. (e) Quantitation of phospho-GAP-43 normalized to total GAP-43 after 24 h of stimulation. (f) Quantitation of total GAP-43 normalized to tubulin after 24 h of stimulation. (g) Immunocytochemistry for GAP-43 (green) and βIII tubulin (red). Images were captured at a constant exposure time to enable comparison between different conditions. Scale bar=8 μm. Western blots shown are representative blots. Bars represent normalized relative optical densities (ODs) plotted as mean±S.E.M calculated from six independent experiments for 3-h stimulation with BDNF and three independent experiments for 24-h BDNF stimulation experiments. Asterisks indicate statistically significant difference from controls (CTL) (*P<0.05, ***P<0.001 in one-way ANOVA with Bonferroni's test). Lane 1=control, lane 2=S18986, lane 3=BDNF and lane 4=S18986+BDNF blocking antibody (a-BDNF)
S18986 and BDNF rescues cortical cell death through ERK pathway
Addition of S18986 and BDNF to cortical cultures resulted in a significant decrease in the percentage of neuronal cell death as compared to the control condition when measured after 24 h (Figure 2A). Addition of BDNF blocking antibodies along with S18986 or BDNF resulted in an increase in cell death as compared to S18986 or BDNF alone (Figure 2A).

S18986 and BDNF enhance neuronal survival. (A) Percentage cell death calculated as TUJ1- and TUNEL-positive cells divided by the total number of cells. Experimental conditions are labeled on the x axis. Bars represent means±S.E.M of three independent experiments. Asterisks indicate statistically significant difference from cultures treated with S18986 or BDNF alone (**P<0.01, ***P<0.001 in one-way ANOVA with Bonferroni's test). (B) Representative western blot (n=4) from cultured neurons. (C) Fold induction was calculated by normalizing relative optical density values to that of the control (CTL) value that was taken as 1 (white bar). Black bars and gray represent cultures treated with S18986 and BDNF, respectively. (***P<0.001 in one-way ANOVA with Dunnett's test compared to CTL). (D) Graph of the number of βIII tubulin and TUNEL double-positive cells divided by the total number of cells. White bar=GAP-43(−/−) and black bar=GAP-43(+/+). Bars represent means±S.E.M of five independent experiments. Asterisks indicate statistically significant difference from GAP-43(+/+) control cultures for GAP-43(+/+) cultures and GAP-43(−/−) control cultures. Treatment conditions in the GAP-43(−/−) conditions were compared to the GAP-43(−/−) control condition (***P<0.001 in two-way ANOVA genotype × treatment with post hoc t-test). In GAP-43(−/−) cultures, S18986 and BDNF had no significant effect on cell death when compared to controls (KO-CTL). (E) Immunocytochemistry of cortical cultures. βIII tubulin (green), TUNEL (red) and DAPI (blue). Scale bar=25 μm. Wortman, wortmannin; Akt inh, Akt inhibitor; PD, PD98059; a-BDNF, anti-BDNF blocking antibody
Addition of PD98059 (ERK inhibitor) along with S18986 or BDNF resulted in an increase in cell death as compared to S18986 or BDNF alone (Figure 2A), suggesting that ERK pathway is important for the increase in neuronal survival observed in culture. Interestingly, inhibitors of PI3K (wortmannin) or its downstream target Akt (1L6-hydroxymethyl-chiro-inositol-2-(R)-2-O-methyl-3-O-octadecyl-sn-glycerocarbonate) when added along with S18986 led to increased cell death over S18986 alone; however, there was no increase in cell death when inhibitors of PI3K and Akt were added along with BDNF in comparison to BDNF alone (Figure 2A). In addition, western blot analysis of cortical cultures performed at 24 h after addition of either S18986 or BDNF showed an increase in the level of pERK and PI3K but not PKC (Figure 2B and C).
S18986 and BDNF reduce cell death in GAP-43(+/+) cultures but not in GAP-43(−/−) cultures
To determine whether GAP-43 was a novel component of BDNF-mediated neuroprotection, cortical cultures were prepared from GAP-43(−/−) and GAP-43(+/+) mice and S18986 or BDNF was added to the cultures. TUNEL combined with βIII tubulin immunocytochemistry was performed after 24 h. First, there was a 2.1-fold increase in level of cell death in control GAP-43(−/−) cultures compared to GAP-43(+/+) cultures (Figure 2D and E). Second, addition of S18986 or BDNF resulted in a significant decrease in neuronal cell death in GAP-43(+/+) cortical cultures, although it had no effect on the neuronal cell death seen in GAP-43(−/−) cultures (Figure 2D and E). The optimal BDNF concentration for reducing cell death in the wild–type (WT) cultures was determined and increasing the concentration of BDNF did not lead to a neuroprotective effect in the absence of GAP-43. To show that the inability of BDNF to rescue cell death in the absence of GAP-43 was indeed specific to BDNF several other growth factors were tried. IGF, GDNF and PDGF were found to confer a neuroprotective effect on cortical cells in the absence of GAP-43 (Figure 2D).
BDNF signaling components are overexpressed in GAP-43(−/−) neuronal cultures
To determine whether the increased basal cell death and the lack of neuroprotection observed in GAP-43(−/−) cultures in response to S18986 or BDNF was a primary effect of the absence of GAP-43 or whether it was a secondary effect because of the absence of other signaling components important for BDNF-mediated neuroprotection in GAP-43(−/−) cultures, several components of the BDNF signaling were analyzed for their presence in the GAP-43(−/−) cortices and neuronal cultures.
In basal conditions, western blot showed that BDNF and TrkB levels were significantly increased in the GAP-43(−/−) cortex as compared to GAP-43(+/+) cortex and that BDNF and PI3K levels were significantly increased in the GAP-43(−/−) cultures as compared to GAP-43(+/+) cultures whereas other signaling components such as GluR1, PKC and ERK were present at similar levels in both genotypes (Figure 3a and b). Therefore, the increased basal levels of cell death was not because of the absence of BDNF in the GAP-43(−/−) cells and furthermore the inability of either S18986 or BDNF to reduce cell death in GAP-43(−/−) cultures was not because of the absence of TrkB, the receptor for BDNF, AMPA-type glutamate receptor GluR1, the receptor for S18986 or of the downstream components of the BDNF signaling pathway, ERK, PI3K and PKC that were previously shown to be important for BDNF-mediated neuroprotection.

Expression of S18986 downstream signaling components. (a) Western blot of cortical extracts from P0 mice. Relative optical density (OD) values normalized to tubulin are shown as mean±S.E.M. from two independent experiments. (b) Western blot of cultured neurons. Relative OD values normalized to tubulin are shown as mean±S.E.M. from five independent experiments. Black bar=GAP-43(+/+) and white bar=GAP-43(−/−). (c) Western blot of cortical cultures induced with BDNF for 24 h(+). Relative OD values for pERK and pTrkB normalized to total ERK and total TrkB, respectively, are shown as mean±S.E.M. from five independent experiments (*P<0.05, **P<0.01, ***P<0.001 in one-way ANOVA compared to the unstimulated (−) condition with Bonferroni's test). Lanes 1 and 2=GAP-43(+/+) and lanes 3 and 4=GAP-43(−/−). Lanes 1 and 3=unstimulated control and lanes 2 and 4=BDNF stimulation for 24 h. (d) Western blot of cortical cultures probed after induction with BDNF and BDNF along with 500 nm of bisindolylmaleimide (Bis). Relative OD values for pERK normalized to total ERK are shown as mean±S.E.M. from four independent experiments. (***P<0.001 compared to control condition in one-way ANOVA with Bonferroni's test). Lane 1=unstimulated control, lane 2=BDNF and lane 3=BDNF+Bis
Following BDNF exposure, western blot showed that pTrkB and pERK levels were significantly increased in the GAP-43(+/+) cultures as compared to basal conditions (Figure 3c). However, in GAP-43(−/−) cultures, BDNF exposure increased levels of pTrkB but not of pERK as compared to basal conditions (Figure 3c). These data suggest that, in the absence of GAP-43, upon BDNF exposure, TrkB receptor gets fully activated but that there is a blockade in the signaling pathway between TrkB receptor activation and ERK activation, although basal levels of ERK and pERK are conserved as compared to WT neurons.
It has been shown previously that inhibition of PKC in cortical neurons leads to decreased phosphorylation of ERK,24 suggesting that PKC is required to get optimal ERK phosphorylation. To test whether PKC is also required for ERK phosphorylation in the current study, we added PKC inhibitor along with BDNF in GAP-43(+/+) cells. We found that when PKC was inhibited ERK phosphorylation was no longer induced (Figure 3d).
We know from previous studies that GAP-43 is localized in caveolin containing lipid rafts as is TrkB.25, 26 We confirmed these data in our GAP-43(+/+) cultures (Figure 4a). We therefore hypothesized that GAP-43 is required for ERK phosphorylation because GAP-43 in caveolin containing lipid rafts is required for activated PKC to be recruited to the same domain as ERK. To test this hypothesis, we added BDNF to GAP-43(+/+) and GAP-43(−/−) cultures to see if it induced the localization of both PKC and pERK to regions of the cell that contained caveolin. Addition of BDNF led to increased colocalization of pERK and PKC with caveolin (Figure 4b and c). However, this did not occur in GAP-43(−/−) cultures (Figure 4b and c). This data is consistent with the hypothesis that the presence of GAP-43 in lipid rafts leads to the localization in the same spatial domain of activated PKC and ERK and that this is required for the phosphorylation of ERK upon TrkB activation.

Colocalization of signaling molecules with caveolin. (a) Immunocytochemistry of GAP-43(+/+) cultures. Cultures were stimulated for 24 h with BDNF. pTrkB or caveolin (red), GAP-43 (green), DAPI (blue). Scale bar=25 μm. (b) Immunocytochemistry of GAP-43(+/+) and GAP-43(−/−) cultures stimulated for 24 h with BDNF. pERK or PKC (red), caveolin (green), DAPI (blue). Scale bar=8 μm. (c) Quantitation of percentage colocalization of either pERK or PKC with caveolin. Bars represent mean±S.E.M. from 10 measurements for each genotype (***P<0.001 in two-way ANOVA genotype × treatment with Bonferroni's test)
Mitochondrial stress in GAP-43(+/+) but not GAP-43(−/−) cortical cultures can be reversed by either S18986 or BDNF
Cleaved caspase-3 is significantly upregulated in the mitochondrial pathway of cell death. As there was an increased level of cleaved caspase-3 in the absence of GAP-43, we next investigated the effect of GAP-43 as well as the effect of S18986 or BDNF-mediated neuroprotection on the mitochondria. JC-1 is a voltage sensitive fluorescence dye that fluoresces red when the mitochondrial membrane is healthy and polarized and fluoresces green when it is depolarized as is the case of apoptosis.
First, in contrast to GAP-43(+/+) cortical cells, in GAP-43(−/−) cortical cells JC-1 fluoresced green indicative of mitochondrial membrane depolarization (compare Figure 5a panel (i) to Figure 5b panel (i) suggesting that absence of GAP-43 increases mitochondrial stress. Next, to study whether S18986 or BDNF reduce cell death by reducing mitochondrial stress, we looked at the JC-1 fluorescence after adding S18986 or BDNF to GAP-43(+/+) cortical cells. Addition of BDNF or S18986 to the GAP-43(+/+) cortical cultures led to a decrease in mitochondrial stress as evident from the increase in red fluorescence (Figure 5a, panels i, v and ix). Importantly, there was no effect of S18986 or BDNF on mitochondrial membrane potential in the absence of GAP-43 (Figure 5b, panels i, ii and iii).

Effect of S18986 and BDNF on mitochondrial stress. Representative images of JC-1 staining from GAP-43(+/+) (a) and GAP-43(−/−) (b) cortical cultures under different experimental conditions. JC-1 is a voltage sensitive dye and fluoresces red when the mitochondrial membrane is polarized (a, panel i) and fluoresces green when the membrane is depolarized (b, panel i). (c) Representative fields of immunoreactivity with BAX, cytochrome c and DCF-DA live cell imaging in GAP-43(+/+) (panels i, iii and v, respectively) and GAP-43(−/−) (panels ii, iv and vi, respectively) cortical cultures. Scale bar=25 μm
Further, to see if PD98059, wortmannin or Akt inhibitor acts on the mitochondria to counter the effects of BDNF, these inhibitors were added along with S18986 or BDNF. JC-1 staining showed that indeed addition of PD98059 along with BDNF or S18986 led to an increase in green florescence (Figure 5a compare panels v and vi with ix and x). Interestingly, paralleling the observation that inhibition of PI3K and Akt affected only S18986 but not BDNF-mediated neuroprotection, it was observed that addition of PI3K and Akt inhibitor along with BDNF had a less severe effect on mitochondrial potential than when these inhibitors were given along with S18986 (Figure 5a compare panels vii and viii with xi and xii).
As it was seen that the absence of GAP-43 lead to increased mitochondrial stress, the downstream components of the mitochondrial apoptotic pathway were compared in GAP-43(+/+) and GAP-43(−/−) cells. Results showed that Bax, which is normally sequestered in the nucleus in GAP-43(+/+) cortical cells (Figure 5c, panel i), was translocated to mitochondria in GAP-43(−/−) cortical cells (Figure 5c, panel ii). In addition, cytochrome c, which is normally retained in the mitochondria showed diffused expression in the cytoplasm of GAP-43(−/−) cortical cultures whereas in GAP-43(+/+) cortical cultures the expression was seen mostly in the perinuclear mitochondrial region (Figure 5c, panels iii and iv). Live cell imaging using DCF-DA also showed increased fluorescence in the GAP-43(−/−) cortical cultures especially in cells that had smaller nuclei characteristic of neurons as compared to GAP-43(+/+) cultures, which indicated an increase in the reactive oxygen species (ROS) in the absence of GAP-43 (Figure 5c, panels v and vi).
Regulation of astrocyte morphology and glutamate transporter expression by S18986 and BDNF is important for neuroprotection
Astrocytes are important in maintaining glutamine-glutamate homeostasis and in clearing free radicals generated via glutamate toxicity. As there is both an increase in cell death and an increase in ROS in the absence of GAP-43, we first investigated the role of GAP-43 in the regulation of astrocytes. When compared to cortical cultures from GAP-43(+/+), cortical cultures from GAP-43(−/−) showed an exclusive presence of ‘reactive astrocytes’ morphology-43(−/−) (Figure 6a, white arrowheads). Quantitation of GFAP-positive cells in culture showed that there was also a significant increase in the number of astrocytes in the GAP-43(−/−) cultures (Figure 6b).

Astrocyte phenotype in the absence of GAP-43. (a) GFAP immunoreactivity in 7-day-old cortical cultures. Scale bar=50 μm. (b) Percentage of GFAP-positive cells over total number of cells. Bars represent mean±S.E.M. of GFAP-positive cells as a percentage of the total number of cells from three independent experiments. Black bar=GAP-43(+/+) and white bar=GAP-43(−/−). (c) Representative western blot of GLT-1, GLAST, GFAP and tubulin. Lanes 1, 2 and 3 are GAP-43(+/+) and lanes 4, 5 and 6 are GAP-43(−/−). Lanes 1 and 4=control, lanes 2 and 5=S18986 stimulation for 24 h, lanes 3 and 6=BDNF stimulation for 24 h. (d) Relative optical density (OD) value of GLT-1 and GLAST normalized to tubulin under control conditions are shown as mean±S.E.M. from three independent experiments. (e) Relative OD value of GLT-1 and GLAST normalized to GFAP under control conditions and stimulation with S18986 and BDNF are shown as mean±S.E.M. from three independent experiments Asterisks indicate statistically significant difference from controls GAP-43(+/+) groups (*P<0.05, **P<0.01 in one-way ANOVA with Bonferroni's test comparing S18986 or BDNF with controls within each genotype)
Next, the effect of S18986 or BDNF on astrocytes was examined. In control conditions, there were more astrocytes in GAP-43(−/−) cultures than in GAP-43(+/+) cultures (Figure 6a and b). Addition of S18986 or BDNF to cortical cultures for 7 days had no effect on the astrocyte number in GAP-43(+/+) and (−/−) cultures (Figure 6a and b). However, there was a significant shift in astrocyte morphology from immature polygonal to the mature stellate phenotype in GAP-43(+/+) cells, although there was no effect on GAP-43(−/−) cells potentially because the mature stellate morphology was already present in the GAP-43(−/−) cultures (Figure 6a). To correlate the change in astrocyte morphology upon addition of S18986 or BDNF with reduction in mitochondrial stress and increased cell survival, the expression of glutamate transporters in the cortical cultures was examined. In control conditions, there was no difference in the expression of GLT-1 and GLAST in GAP-43(−/−) cultures when compared to GAP-43(+/+) cultures (Figure 6c and d). In GAP-43(+/+) cultures S18986 and BDNF led to a significant increase in GLT-1 and GLAST expression after 48 h (Figure 6c and e). In contrast, S18986 and BDNF had no detectable effects on GLT-1 and GLAST expression in GAP-43(−/−) cultures (Figure 6c and e).
Neuronal remodeling by S18986 or BDNF requires the presence of GAP-43
The above evidence shows that BDNF has no effect on neuronal survival in the absence of GAP-43. However, one aspect of neuronal cell survival is the restoration of connections and functionality. This involves the ability to extend a new process or support a sprouting response, events that enable neurons to form new synapses and regain their physiological role. These processes have previously been shown to involve both BDNF and GAP-43. Therefore, we looked at whether S18986 or BDNF resulted in cytoskeletal changes and neurite outgrowth and whether these responses such as neuronal survival also required the presence of GAP-43.
To study neurite outgrowth, cultures were processed for F-actin and tubulin IHC (Figure 7). Immunocytochemistry showed that complexity of neuritic morphology was higher in GAP-43(+/+) than that of GAP-43(−/−) neurites (Figure 7a). Further, the addition of either S18986 or BDNF resulted in a change in morphology of the neurons. However, this was true only for the GAP-43(+/+) cultures (Figure 7a). Analysis of the neurite cytoskeleton showed that GAP-43(−/−) cortical cultures had growth cones that were depleted of F-actin as compared to the GAP-43(+/+) cultures and in addition GAP-43(−/−) cortical cultures had growth cones that were enriched in tubulin in a region that normally was enriched in F-actin as is seen in GAP-43(+/+) cultures (Figure 7b, panels i and iii; high power in panels ii and iv). To quantitate the observed differences in morphology, neurite length, the number of neurites extending out of the cell body (primary neurites), and the number of branches (neurites extending out of a primary neurite) were quantitated. First, absence of GAP-43 had no effect on neurite length. Further, S18986 and BDNF did not affect neurite length in either GAP-43(+/+) or in GAP-43(−/−) animals (Figure 7c). When the number of primary neurites were quantitated, although there was no significant difference between GAP-43(−/−) and GAP-43(+/+) control cultures, addition of S18986 or BDNF resulted in a significant increase in the number of primary neurites only in the GAP-43(+/+) cultures but not in the GAP-43(−/−) cultures (Figure 7d). In addition, under control conditions the number of branches in GAP-43(+/+) was significantly higher compared to GAP-43(−/−). Addition of S18986 or BDNF to GAP-43(+/+) cultures led to an increase in neurite branching however there was no increase in branching in GAP-43(−/−) cultures (Figure 7e). The observed differences in growth cone morphology (Figure 7b, ii and iv) were quantitated. Results showed the growth cone area in the absence of GAP-43 was reduced by almost 50% (Figure 7f). In addition when the ratio of the intensity of F-actin was calculated with respect to α tubulin, it was observed that it was significantly reduced in the absence of GAP-43 (Figure 7g).

Effect of S18986 and BDNF on neuronal morphology and cytoskeleton. (a) GAP-43(+/+) (left) and GAP-43(−/−) (right) cortical cultures fixed and processed for immunoreactivity to tubulin (green) and F-actin (red) to reveal number of neurites, neurite branching and length of neurites. Scale bar=25 μm. (b) F-actin (red), tubulin (green) in GAP-43(+/+) (i and ii) and GAP-43(−/−) (iii and iv) cortical cells. Unlike in GAP-43(+/+) cells, in the absence of GAP-43, F-actin is reduced and tubulin is found to be transported to the tips of the process. Panels ii and iv show a magnified view of a neurite indicated by white arrows in panels i and iii. Scale bar=12.5 μm. (c) Quantitation of neurite length measurements. (d) Quantitation of number of neurites. (e) Quantitation of neurite branching. For graphs (c–e), bars represent mean±S.E.M. from three independent experiments. (***P<0.001 in two-way ANOVA – genotype × treatment – with Bonferroni's test). (f) Quantitation of the actin-rich growth cone area. Bars represent mean±S.E.M. for 15 growth cones in each genotype. (**P<0.01 in Student's t-test). (g) Quantitation of the intensity of F-actin normalized to the intensity of tubulin. Bars represent mean±S.E.M. for 45 F-actin intensity values normalized against their respective tubulin intensity values in each genotype. (***P<0.001 in Student's t-test) Black bar=GAP-43(+/+) and white bar=GAP-43(−/−)
PSA-NCAM is an important regulator of cell shape and thus plays a role in cell growth and migration.27 PSA-NCAM expression on cells allows for the dynamic changes in cell membrane contacts and thus is an important molecule in mediating brain plasticity and repair.28 In addition PSA-NCAM and GAP-43 are coexpressed in regions undergoing morphological plasticity.29 In addition, several proteins associated with the synaptic vesicles such as synaptophysin are upregulated during neuronal remodeling.30 Therefore, we looked at both synaptophysin and PSA-NCAM as markers of neuronal remodeling and plasticity in the current study. Western blot of cortical extracts showed that NCAM and synaptophysin were dramatically reduced in GAP-43(−/−) mice (Figure 8a and b). Immunocytochemistry corroborated the results of the western blot (Figure 8c).

Expression of NCAM and synaptophysin. (a) Representative western blot of the levels of NCAM and synaptophysin protein present in P0 cortical extracts from GAP-43(+/+) and GAP-43(−/−) mice. (b) Relative optical density value of NCAM and synaptophysin normalized to tubulin under control conditions are shown as mean±S.E.M. from two independent experiments. Black bar=GAP-43(+/+) and white bar=GAP-43(−/−). (*P<0.05 in Student's t-test shows significant difference compared to GAP-43(+/+) cultures). (c) Immunocytochemistry of GAP-43(+/+) and GAP-43(−/−) cultures. NCAM or synaptophysin (green), βIII tubulin (red), DAPI (blue). Scale bar=8 μm.
Discussion
We have shown that the positive modulator of AMPA-type glutamate receptor S18986 and BDNF have a neuroprotective effect in cortical cultures that is dependent on the presence of GAP-43. Further, our results point toward two important mechanisms by which the neuroprotective effect of positive AMPA receptor modulator BDNF is mediated: through preventing mitochondrial dysfunction and regulating glutamate transporters on astrocytes. In addition, we have shown that GAP-43 is essential for both neuroprotection and neuronal plasticity. These results held true for cerebellar granule cells (data not shown), suggesting that the basic mechanism of action of positive AMPA receptor modulator (S18986) and BDNF are the same in different neuronal populations.
As previously described,9, 10 our data support the hypothesis that the effect of positive AMPA receptor modulators are mediated via increased BDNF expression. In addition, several lines of evidence suggest that one of the downstream effectors of positive AMPA receptor modulator BDNF could be GAP-43. Among them is the fact that both BDNF and GAP-43 are upregulated in response to neuronal injury and that GAP-43 levels are upregulated in response to BDNF in culture.23 By using cortical cells from GAP-43(−/−) animals, we now directly show that when GAP-43 is not present, the effects of BDNF on neuroprotection are lost. This was not because of the absence of signaling components in the GAP-43(−/−) cells, in fact, all the signaling components of BDNF are present in the GAP-43(−/−) cells, at normal or increased levels. Interestingly, in the absence of GAP-43, BDNF was able to induce TrkB phosphorylation but not ERK phosphorylation. As both TrkB and GAP-43 are components of lipid rafts,25, 26 our data suggest the possibility that disruption of lipid rafts in the absence of GAP-43 leads to inefficient BDNF signaling through decreased recruitment of PKC that is necessary for BDNF-induced ERK activation.
Mitochondrial dysfunction and depolarization is a major common pathway leading to neuronal death31 and events leading to mitochondrial dysfunction include disruption of calcium homeostasis32 and cellular stress caused by destabilization or changed dynamics of cytoskeletal elements especially F-actin.33 The present study suggests that increased GAP-43 expression upon BDNF stimulation may lead to regulation of F-actin polymerization–depolymerization cycle that is an important factor in maintaining mitochondrial integrity and preventing cell death. However, increased GAP-43 expression may also be required to maintain calcium levels under conditions of cellular stress, a possibility that needs further investigation.
Our observation that BDNF leads to an induction of glutamate transporters points to the role of astrocytes in mediating the neuroprotective effects of BDNF. Although not in all cases,34 BDNF can induce the expression of GLT-1.35 In addition, the fact that stellate astrocytes specifically express GLT-136 supports our observation that there is an increase in both stellate morphology of astrocytes and GLT-1 expression upon BDNF stimulation. We also find an increase in the expression of GLAST that is in agreement with studies showing the increase of both GLT-1 and GLAST upon treatment with growth factors.37 Both these effects are dependent on the presence of GAP-43, suggesting that downregulated glutamate transporters and resulting glutamate excitotoxicity may be one of the cause of increased cell death in GAP-43(−/−) cells and one of the reasons that BDNF is unable to rescue the phenotype.
Previous studies have pointed to a role of BDNF and GAP-43 in axonogenesis and repair.38 In the present study, we directly show that the effect of positive AMPA receptor modulator-BDNF on neurite outgrowth and neurite branching requires the presence of GAP-43. Therefore, GAP-43 is a common mediator of the neuroprotective as well as plasticity effects of positive AMPA receptor modulator and BDNF, both of which are required for successful therapy that would involve functional recovery. The present study identifies GAP-43 as a novel target for neuroprotective strategies aiming at modulating neuronal cell death and plasticity in neurodegenerative diseases.
Materials and Methods
Chemicals
Brain-derived neurotrophic factor, IGF, GDNF and PDGF were purchased from R&D Systems (Minneapolis, MN, USA), wortmannin and Akt inhibitor from Sigma-Aldrich (St. Louis, MO, USA), JC-1 and DCF-DA from Molecular Probes (Carlsbad, CA, USA), PD98059 from New England Biolabs (Ipswich, MA, USA) and blocking anti-BDNF antibody from Chemicon (Temecula, CA, USA). Positive AMPA receptor modulator S18986 was synthesized as previously described39. The concentrations of factors and reagents used were as follows: BDNF (50 ng/ml), IGF (100 ng/ml), GDNF (100 ng/ml), PDGF (100 ng/ml), positive AMPA receptor modulator S18986 (50 μM), blocking anti-BDNF antibody (10 μg/ml), PD98059 (30 μM), wortmannin (200 nM) and Akt inhibitor (30 μM).
For immunocytochemistry, anti-ERK and anti-cleaved caspase-3 antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA), anti-p-ERK, anti-PSD-95, anti-PI3K, anti-PKC, anti-p-AKT, anti-Bax, anti-cytochrome c and anti-caveaolin-1 from Santa Cruz (Santa Cruz, CA), anti-BDNF and anti-p-TrkB from Calbiochem (San Diego, CA, USA), anti-mGLUR1 and anti-synaptophysin from Chemicon, anti-GFAP from Dako (Glostrup, Denmark), anti-GLT-1 (Alpha Diagnostics, TX, USA), anti-GLAST from Abcam (Cambridge, UK), anti-pan-tubulin and anti-NF-160 from Sigma-Aldrich, anti-βIII tubulin from Promega (Madison, WI, USA) and anti-pan-GAP-43 was generously provided by Dr. Karina Meiri, Tufts university. All secondary antibodies were purchased from Vector Laboratories (Burlingame, CA, USA).
Mice
Growth-associated protein-43 (GAP-43)-deficient mice were generated from targeted CJ7 ES cells in isogenic 129S3/imJ mice (genetic designation +Mgf-SIJ, JAX stock number 002448) and backcrossed for 8–12 generations with C57 BL/6N. Homozygotes (−/−) mice were identified by PCR genotyping as described previously. The morning when the vaginal plug was detected was defined as embryonic day (E) 0.5 and the day of birth as postnatal day (P) 0. All animals were maintained in specific pathogen-free conditions according to the regulation of the Institutional Animal Ethics Committee, India.
Western blot from cortex
Brain from P0 WT and knock out (KO) mice were taken out. The meninges were removed and the olfactory bulb and subcortical tissues were separated. The tissue was cut into small pieces, washed with PBS and then sonicated in lysis buffer (50 mM Tris, pH7.4, 150 mM NaCl, 50 mM NaF, 1 mM NaV, 1 mM EDTA and 2% SDS). The quantification of protein was carried out by standard BCA (Bicinchoninic acid-Sigma) method. The protein was separated using SDS-PAGE and transferred to methanol treated PVDF membrane. The membrane was blocked in 5% lactogen (milk protein) in Tris-buffered saline with Tween-20 (TBST). Following this, the membrane was incubated with the primary antibody (BDNF, 1 : 200, Santa Cruz; TrkB, 1 : 1000, Cell Signaling Technology; GluR1, 1 : 2000, Upstate; β-tubulin 1 : 1000, Santa Cruz) in TBST overnight at 4°C. The membrane was then incubated with the appropriate alkaline phosphatase-conjugated secondary antibody for 1 h and developed with NBT/BCIP (Roche, Basel, Switzerland). The reaction was terminated upon visualization the control band. The bands were quantified by obtaining integrated density values using Chemi Genius2 from SynGene software.
Cortical culture
Cortices were cultured similar to the published protocol for the postnatal cerebellum,40 with slight modifications. Briefly, P0 or P5 pups were killed by hypothermia; their cortex dissected aseptically in CMF-Tyrode solution. Meninges were removed, tissue were chopped into smaller pieces and collected in CMF-Tyrode. These were treated with trypsin DNAse and then dissociated in the same solution by triturating to make a single cell suspension, pelleted and resuspended in Dulbecco's modified Eagle's medium-F-12 (DMEM-F-12) containing 15 mM HEPES, L-glutamine (2 mM), pyroxidine hydrochloride (Invitrogen, Carlsbad, CA, USA), N2 supplement (Invitrogen), 10% fetal calf serum, 25 mM KCl and penicillin–streptomycin. Cells were plated at a density of 2 × 104 cells per cm2 onto poly-D-lysine-coated Labtek chamber (Nunc, Roskilde, Denmark). After 24 h of incubation at 37°C in 0.5% CO2, the serum-containing medium was removed. To allow for downregulation of serum-stimulated intracellular signaling pathways, cells were rested for 1 h incubation in DMEM-F-12 with antibiotics alone. For experimental treatments, the resting medium was exchanged for DMEM-F-12 with N2 supplement, 25 mM KCl and antibiotics. Cells were fixed in 4% PFA for 20 min for immunocytochemistry.
Immunostaining of cultures
Cultures were permeabilized with 0.05% Triton-X100 for 5 min. Primary antibodies were applied for 14–16 h at 4°C. Signals were developed by using fluorescent secondary for 1 h at room temperature. Stained slides were mounted in mounting medium-containing DAP1 to reveal nuclei and enable counting. Rhodamine phalloidin (Sigma-Aldrich) staining was performed strictly according to manufacturer's protocol to detect F-actin.
TUNEL assay
TUNEL kit was purchased from Roche and assay was performed according to manufacturer's suggestions. Briefly, sections and cultured cells were incubated for 5 min in 0.1% Triton-X consisting 0.1% sodium citrate, washed in PBS and incubated in labeling mixture consisting of terminal nucleotidyl transferase and labeled nucleotide mixture for 1 h at 37°C in a humidified chamber. The cultures were double labeled with TUJ1 antibody to identify neuronal cell death. Quantification of TUNEL-positive cells in cultures were performed by counting TUNEL+Tuj1+DAPI+ cells from a total of 10 random fields from two wells each.
Measurement of mitochondrial membrane potential
Primary cortical cultures were given different treatments and then incubated with 5 μM JC-1 fluorescence dye for 30 min in the CO2 incubator and washed several times with PBS prewarmed at 37°C. Mitochondrial membrane potential was evaluated qualitatively under a fluorescence microscope using a 468 nm filter.
Measurement of reactive oxygen species
Non-fluorescent 5-(and-6)-carboxy-2′, 7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) permeates live cells and is deacetylated by nonspecific intracellular esterases. In the presence of ROS, which are produced throughout the cell, particularly during oxidative stress, the reduced fluorescein compound is oxidized and emits bright green fluorescence. Live cell imaging was used to identify production of ROS in cell culture. Neurons were discriminated from glia based on diameter of the nucleus. Previous staining with either Tuj1 or GFAP had shown that the nuclei of GFAP-positive cells were on the average three times larger than the nuclei of Tuj1-positive cells. Dissociated primary cortical cultures from GAP-43(+/+) and GAP-43(−/−) were incubated with 10 μM of DCFDA in serum-free media (SFM) for 1 h in a CO2 incubator and washed several times with PBS prewarmed to 37°C. Quantification of the number of DCFDA fluorescence-positive cells was assessed by excitation at 468 nm. Setting the exposure at the lowest point at which signal was detected on Zeiss Axioplan microscope captured images in the linear range of the signal. Parallel images were captured for Hoechst staining to reveal nuclei in live cells. Signal-to-noise ratio was normalized by subtracting the background fluorescence.
Western blotting
For protein studies, cells were plated in 60 mm dishes at a density of 2 × 106 cells per cm2 and collected at 24-h time point. The cells were scraped in the lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 2% SDS and a protease inhibitor mixture (one tablet of Roche Diagnostics (Mannheim, Germany) protease inhibitor mixture/25 ml)), collected and centrifuged at 14 000 r.p.m. for 5 min at 4°C. These were further boiled for 5 min, centrifuged at top speed in a microfuge for 5 min to remove any insoluble material, and stored at 80°C until use. Protein concentration was estimated by BCA method and 15 μg of total protein was loaded in each lane. Proteins were resolved on 11% SDS-polyacrylamide gels and electrophoretically transferred to Hybond C membranes (Amersham Biosciences, Piscataway, NJ, USA). The blots were washed (three times for 5 min each) with 0.1% TBST (25 mM Tris-Cl, 137 mM NaCl, 2.7 mM KCl, pH 7.5, and 0.1% Tween 20). A 5% solution of BSA (Fraction V; Sigma-Aldrich) in TBST was used as a blocking agent. Primary antibodies, and secondary HRP-conjugated anti-mouse (from Cell Signaling Technology, Beverly, MA) were diluted in TBST containing 5% BSA. Sodium fluoride (20 mM) and sodium orthovanadate (2 mM) were included in BSA-containing solutions to inhibit the possible contaminant phosphatases.41 Incubation with the blocking buffer was performed for 1 h room temperature, after which blots were incubated with the primary antibody overnight at 4°C. After washing with TBST (three times for 5 min each), blots were incubated with the secondary antibody for 1 h room temperature. Blots were again washed with TBST (three times for 5 min each), and signal was detected using the ECL reagent (Amersham Biosciences). Exposure of the film (Hyperfilm, Amersham Biosciences) was kept in the linear range, and band intensity was quantified using NIH Image software (National Institutes of Health, Bethesda, MD, USA).
Analysis of colocalization of immunocytochemical markers
Cortical cells from P0 GAP-43 WT(+/+) and KO(−/−) mice were cultured for 24 h in serum-containing media (SCM). After 24 h the media was changed to SFM with 50 ng/ml BDNF keeping the control along with it. After 24 h of BDNF treatment, the culture was fixed for 20 min in 4% paraformaldehyde (Merck). These neurons were double stained for caveolin-1 (Santa Cruz; 1 : 200) with pERK (CST; 1 : 200) and with PKC (Santa Cruz; 1 : 200). After staining the images were taken using Zeiss Apotome microscope and then colocalization analysis was carried out using AxioVision Release 4.6.3 software. A minimum of 10 cells were analyzed from WT and KO culture separately both in control and BDNF-treated culture. To measure the colocalization 24-bit image of fluorescently labeled cells were processed in the Axio Vision Release 4.6.3 software. The amount of colocalization between caveolin and pERK or caveolin and PKC was determined with the colocalization function of the software. The threshold value of the chanel 125 and chanel 225 was kept constant for all the cells analyzed. For colocalization analysis whole cell was taken into consideration and the pixel value of the whole image as well as the pixel value of the colocalized portion of the image were calculated. On the basis of total image, pixel value and the colocalized pixel value, the percentage of the area of colocalization was calculated using formula: Area (μm2)=number of pixels × X scale × Y scale.
Neurite length quantitation
Total neurite length, number of neurites as well as neurite branching was quantified on Zeiss Axioplan2 Software. A freehand line was traced over each neurite, extending from the neurite hillock to the tip of the neurite. All neurites from each cell were treated independently for final calculation of mean neurite length. A total of 200 cells were counted from at least three GAP-43(+/+) and three GAP-43(−/−) animals.
F-actin quantitation
Cortical cells were cultured for 48 h from P0 GAP-43 WT(+/+) and KO(−/−) mice. The culture was fixed for 20 min in 4% paraformaldehyde (Merck) in Kreb's buffer (50 mM KCl, 100 mM NaCl, 1.3 mM MgCl2, 20 mM HEPES and 1.2 mM CaCl2; pH 7.4) with 10 mM glucose to preserve the F-actin. These neurons were stained for F-actin using rhodamine-phalloidin (Invitrogen; 1 : 1000) and α-tubulin (Abcam; 1 : 500). After staining, the images were taken using Zeiss Apotome microscope keeping the image capture conditions constant. Analysis was carried out using AxioVision (Release 4.6.3) software. >25 individual cells were analyzed for each genotype. For each cell, multiple regions of interest were selected in the growth cones and the intensity of F-actin and α-tubulin fluorescence was measured in the same pixel.20 To measure the area of the F-actin-rich periphery, the freehand tool in the software was used to outline the region that was positive for F-actin immunostaining but negative for α-tubulin at the tip of the growth cone.
Real-time PCR
Wild-type and GAP-43(−/−) cells (n=5 in each group, experiments run in triplicate) were collected after 3 h of S18986 treatment and total RNA was isolated using TRI reagent (Sigma) according to the manufacturer's instructions. The concentrations of total RNA were determined by spectrophotometric measurement at 260 nm. Reverse transcription was performed using 1 μg of total RNA in final volume of 20 μl using first-strand cDNA synthesis kit (Roche). Real-time PCR was performed using SYBR green (Sigma or Bio-Rad). Briefly, 1 μl of cDNA, 0.2 μm of forward and reverse primer and 12.5 μl of 2 × SYBR green mix was taken in a total reaction volume of 25 μl. Each cDNA sample was carried out in triplicate. Negative controls (no cDNA) were also cycled in parallel with each run. The data were analyzed by the Bio-Rad ICycler IQ optical software version 3.0a. The PCR efficiencies and correlation coefficients were calculated from the standard curve generated from serial dilutions performed for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as internal control. Quantitation of gene expression was based on the starting quantity (SQ) value for each sample generated from the standard curve graph. SQ value of each gene was normalized against GAPDH. The primers were designed by using M-fold software developed by Prof. M. Zuker (Rensselaer Polytechnic Institute, USA) and primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) software. Primer sequences were as follows: 5′-AACAATGTGACTCCACTGCC-3′ and 5′-ACCGAAGTAAATAACCAT-3′ for BDNF; 5′-GGCCTTCCGTGTTCCTAC-3′ and 5′-TGTCATCATACTTGGCAGGTT-3′ for GAPDH. Data were analyzed with a Student's t-test.
Statistical analysis
The data were expressed as mean±S.E.M. Data were analyzed with a Student's t-test, a one-way or two-way ANOVA (GraphPad Prism version 4.01 for Windows, GraphPad Software, San Diego, CA). When ANOVA showed significant differences among multiple experimental groups, multiple comparisons between experimental groups were performed using Dunnett's or Bonferroni's post hoc test.
Abbreviations
- AMPA:
-
α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
- BDNF:
-
brain-derived neurotrophic factor
- ERK:
-
extracellular-regulated kinase
- GAP-43:
-
growth-associated protein-43
References
Arai A, Kessler M, Xiao P, Ambros-Ingerson J, Rogers G, Lynch G . A centrally active drug that modulates AMPA receptor gated currents. Brain Res 1994; 638: 343–346.
Staubli U, Perez Y, Xu FB, Rogers G, Ingvar M, Stone-Elander S et al. Centrally active modulators of glutamate receptors facilitate the induction of long-term potentiation in vivo. Proc Natl Acad Sci USA 1994; 91: 11158–11162.
Suppiramaniam V, Bahr BA, Sinnarajah S, Owens K, Rogers G, Yilma S et al. Member of the Ampakine class of memory enhancers prolongs the single channel open time of reconstituted AMPA receptors. Synapse 2001; 40: 154–158.
Ingvar M, Ambros-Ingerson J, Davis M, Granger R, Kessler M, Rogers GA et al. Enhancement by an ampakine of memory encoding in humans. Exp Neurol 1997; 146: 553–559.
Hampson RE, Rogers G, Lynch G, Deadwyler SA . Facilitative effects of the ampakine CX516 on short-term memory in rats: enhancement of delayed-nonmatch-to-sample performance. J Neurosci 1998; 18: 2740–2747.
Dicou E, Rangon CM, Guimiot F, Spedding M, Gressens P . Positive allosteric modulators of AMPA receptors are neuroprotective against lesions induced by an NMDA agonist in neonatal mouse brain. Brain Res 2003; 970: 221–225.
Danysz W, Parsons CG . Neuroprotective potential of ionotropic glutamate receptor antagonists. Neurotox Res 2002; 4: 119–126.
Danysz W . Positive modulators of AMPA receptors as a potential treatment for schizophrenia. Curr Opin Investig Drugs 2002; 3: 1062–1066.
Mackowiak M, O'Neill MJ, Hicks CA, Bleakman D, Skolnick P . An AMPA receptor potentiator modulates hippocampal expression of BDNF: an in vivo study. Neuropharmacology 2002; 43: 1–10.
Rex CS, Lauterborn JC, Lin CY, Kramar EA, Rogers GA, Gall CM et al. Restoration of long-term potentiation in middle-aged hippocampus after induction of brain-derived neurotrophic factor. J Neurophysiol 2006; 96: 677–685.
Staubli U, Rogers G, Lynch G . Facilitation of glutamate receptors enhances memory. Proc Natl Acad Sci USA 1994; 91: 777–781.
Marini AM, Rabin SJ, Lipsky RH, Mocchetti I . Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-D-aspartate. J Biol Chem 1998; 273: 29394–29399.
Courtney MJ, Akerman KE, Coffey ET . Neurotrophins protect cultured cerebellar granule neurons against the early phase of cell death by a two-component mechanism. J Neurosci 1997; 17: 4201–4211.
Thomas K, Davies A . Neurotrophins: a ticket to ride for BDNF. Curr Biol 2005; 15: R262–R264.
Rose CR, Blum R, Kafitz KW, Kovalchuk Y, Konnerth A . From modulator to mediator: rapid effects of BDNF on ion channels. Bioessays 2004; 26: 1185–1194.
Han BH, Holtzman DM . BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci 2000; 20: 5775–5781.
Hetman M, Kanning K, Cavanaugh JE, Xia Z . Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem 1999; 274: 22569–22580.
Nagappan G, Lu B . Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci 2005; 28: 464–471.
Bramham CR, Messaoudi E . BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol 2005; 76: 99–125.
Shen Y, Mani S, Donovan SL, Schwob JE, Meiri KF . Growth-associated protein-43 is required for commissural axon guidance in the developing vertebrate nervous system. J Neurosci 2002; 22: 239–247.
Frey D, Laux T, Xu L, Schneider C, Caroni P . Shared and unique roles of CAP23 and GAP43 in actin regulation, neurite outgrowth, and anatomical plasticity. J Cell Biol 2000; 149: 1443–1454.
Husson I, Rangon CM, Lelievre V, Bemelmans AP, Sachs P, Mallet J et al. BDNF-induced white matter neuroprotection and stage-dependent neuronal survival following a neonatal excitotoxic challenge. Cereb Cortex 2005; 15: 250–261.
Segal RA, Pomeroy SL, Stiles CD . Axonal growth and fasciculation linked to differential expression of BDNF and NT3 receptors in developing cerebellar granule cells. J Neurosci 1995; 15 (7 Part 1): 4970–4981.
Hasegawa J, Takekoshi S, Nagata H, Osamura RY, Suzuki T . Sevoflurane stimulates MAP kinase signal transduction through the activation of PKC alpha and betaII in fetal rat cerebral cortex cultured neuron. Acta Histochemica et Cytochemica 2006; 39: 163–172.
He Q, Meiri KF . Isolation and characterization of detergent-resistant microdomains responsive to NCAM-mediated signaling from growth cones. Mol Cell Neurosci 2002; 19: 18–31.
Pereira DB, Chao MV . The tyrosine kinase Fyn determines the localization of TrkB receptors in lipid rafts. J Neurosci 2007; 27: 4859–4869.
Bonfanti L . PSA-NCAM in mammalian structural plasticity and neurogenesis. Prog Neurobiol 2006; 80: 129–164.
Gascon E, Vutskits L, Kiss JZ . The Role of PSA-NCAM in Adult Neurogenesis. Neurochem Res 31 May 2008 [e-Pub ahead of print] PMID 18516676.
Alonso G, Prieto M, Legrand A, Chauvet N . PSA-NCAM and B-50/GAP-43 are coexpressed by specific neuronal systems of the adult rat mediobasal hypothalamus that exhibit remarkable capacities for morphological plasticity. J Comp Neurol 1997; 384: 181–199.
Hou XE, Dahlstrom A . Synaptic vesicle proteins and neuronal plasticity in adrenergic neurons. Neurochem Res 2000; 25: 1275–1300.
Nicholls DG, Budd SL . Mitochondria and neuronal survival. Physiol Rev 2000; 80: 315–360.
Budd SL, Nicholls DG . Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem 1996; 67: 2282–2291.
Gourlay CW, Ayscough KR . A role for actin in aging and apoptosis. Biochem Soc Trans 2005; 33 (Part 6): 1260–1264.
Figiel M, Maucher T, Rozyczka J, Bayatti N, Engele J . Regulation of glial glutamate transporter expression by growth factors. Exp Neurol 2003; 183: 124–135.
Rodriguez-Kern A, Gegelashvili M, Schousboe A, Zhang J, Sung L, Gegelashvili G . Beta-amyloid and brain-derived neurotrophic factor, BDNF, up-regulate the expression of glutamate transporter GLT-1/EAAT2 via different signaling pathways utilizing transcription factor NF-kappaB. Neurochem Int 2003; 43: 363–370.
Perego C, Vanoni C, Bossi M, Massari S, Basudev H, Longhi R et al. The GLT-1 and GLAST glutamate transporters are expressed on morphologically distinct astrocytes and regulated by neuronal activity in primary hippocampal cocultures. J Neurochem 2000; 75: 1076–1084.
Zelenaia O, Schlag BD, Gochenauer GE, Ganel R, Song W, Beesley JS et al. Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-kappaB. Mol Pharmacol 2000; 57: 667–678.
Kobayashi NR, Fan DP, Giehl KM, Bedard AM, Wiegand SJ, Tetzlaff W . BDNF and NT-4/5 prevent atrophy of rat rubrospinal neurons after cervical axotomy, stimulate GAP-43 and Talpha1-tubulin mRNA expression, and promote axonal regeneration. J Neurosci 1997; 17: 9583–9595.
Lockhart B, Iop F, Closier M, Lestage P . (S)-2,3-dihydro-[3,4]cyclopentano-1,2,4-benzothiadiazine-1,1-dioxide: (S18986-1) a positive modulator of AMPA receptors enhances (S)-AMPA-mediated [3II]noradrenaline release from rat hippocampal and frontal cortex slices. Eur J Pharmacol 2000; 401: 145–153.
Gao WO, Heintz N, Hatten ME . Cerebellar granule cell neurogenesis is regulated by cell–cell interactions in vitro. Neuron 1991; 6: 705–715.
Sharma SK, Carew TJ . Inclusion of phosphatase inhibitors during western blotting enhances signal detection with phospho-specific antibodies. Anal Biochem 2002; 307: 187–189.
Acknowledgements
This study was supported by the Inserm, the Université Paris 7, PremUP, the National Brain Research Centre, an INSERM-ICMR Grant, and the International Associated Laboratory PROTECT.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by D Kaplan
Rights and permissions
About this article
Cite this article
Gupta, S., Mishra, R., Kusum, S. et al. GAP-43 is essential for the neurotrophic effects of BDNF and positive AMPA receptor modulator S18986. Cell Death Differ 16, 624–637 (2009). https://doi.org/10.1038/cdd.2008.188
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.188
Keywords
This article is cited by
-
Transcriptomes of Clusterin- and S100B-transfected neuronal cells elucidate protective mechanisms against hypoxia and oxidative stress in the hooded seal (Cystophora cristata) brain
BMC Neuroscience (2022)
-
Learning and memory impairment induced by 1,4-butanediol is regulated by ERK1/2-CREB-BDNF signaling pathways in PC12 cells
Metabolic Brain Disease (2022)
-
Telomerase increasing compound protects hippocampal neurons from amyloid beta toxicity by enhancing the expression of neurotrophins and plasticity related genes
Scientific Reports (2019)
-
Immunization with neural derived peptides plus scar removal induces a permissive microenvironment, and improves locomotor recovery after chronic spinal cord injury
BMC Neuroscience (2017)
-
Lentiviral-Mediated Netrin-1 Overexpression Improves Motor and Sensory Functions in SCT Rats Associated with SYP and GAP-43 Expressions
Molecular Neurobiology (2017)
