
Abstract
Trypanosoma brucei is the causative agent of African sleeping sickness. Available treatments are ineffective, toxic and susceptible to resistance by the parasite. Here we show that various endogenous neuropeptides act as potent antitrypanosome agents. Neuropeptides exerted their trypanolytic activity through an unusual mechanism that involves peptide uptake by the parasite, disruption of lysosome integrity and cytosolic accumulation of glycolytic enzymes. This promotes an energetic metabolism failure that initiates an autophagic-like cell death. Neuropeptide-based treatment improved clinical signs in a chronic model of trypanosomiasis by reducing the parasite burden in various target organs. Of physiological importance is the fact that hosts respond to trypanosome infection producing neuropeptides as part of their natural innate defense. From a therapeutic point of view, targeting of intracellular compartments by neuropeptides suppose a new promising strategy for the treatment of trypanosomiasis.
Similar content being viewed by others
Main
African trypanosomes cause sleeping sickness in humans and severe epidemics in livestock. The disease is a major health threat to rural Africans, with an annual incidence in humans of about 500 000 people infected per year and 60 million people at a risk of infection.1 Available drugs are antiquated, ineffective, toxic and susceptible to resistance by the parasite,2 and the lack of prospect for a vaccine against Trypanosoma brucei has generated the urgent need for the development of new therapeutic approaches that should include agents with non-traditional modes of action. In this sense, an almost unexplored field for the treatment of parasitic diseases is the use of antimicrobial peptides that are effective against other pathogens such as bacteria, fungi and viruses.3 Despite their broad structural diversity, these antimicrobial peptides share common properties and mechanisms of actions. They have high positive charge and amphipathic and α-helical structures that promote the initial binding of the peptides to the negatively charged outer leaflet of the plasma membranes of pathogens and the insertion into the cell membrane. This leads to a rapid loss of the cell homeostasis and eventually death of pathogen by membrane disruption.4 As other pathogens, trypanosomes have a highly negative charged coat.5 In fact, some cationic antimicrobial peptides, such as cecropin and mellitin (of insect origin) or defensins and cathelicidins (produced by leukocytes and epithelial cells), kill trypanosomatids by disrupting plasma membrane integrity and leading to osmotic instability and lysis.6, 7 Based on the assumption that our organism might respond to parasite infections by secreting antimicrobial-like agents able to act against trypanosomes in a similar way, we here investigate the trypanolytic capacity of a panel of neuropeptides produced by the neuroendocrine and immune systems that are highly abundant in our defense barriers. Despite their broad physiological and source diversity, vasoactive intestinal peptide (VIP), urocortin (UCN), adrenomedullin (AM), corticotropin-releasing hormone, ghrelin and melanocyte-stimulating hormone share some structural characteristics that are common to antimicrobial peptides, such as small size (<6 kDa) and the presence of clusters of hydrophobic and cationic residues arranged into an amphipathic and α-helical structure (Table 1). Indeed, the antibacterial capacities for some of these neuropeptides have been described recently.8, 9 In this report, we show that these endogenous peptides act as potent antitrypanosome agents, showing an unusual trypanolytic mechanism.
Results
Neuropeptides kill bloodstream form T. brucei
We first investigated the ability of these neuropeptides to kill infective bloodstream forms (BSFs) of T. brucei brucei, the parasite life-cycle stage present in mammalian hosts. All of them exhibited a potent capacity to kill BSFs (Figure 1a), with LD50 in the range of those antiparasitic drugs used in clinic (∼2–3 μM, Table 1). A second administration of the peptides (at the LD50) 24 h after initiation of the culture caused 100% mortality (not shown). The neuropeptides were able to kill the monomorphic and pleomorphic forms of T. brucei isolated from the blood of infected individuals in a similar manner as they did with cultured forms (Supplementary Figure 1). The decrease in the parasite numbers and viability was accompanied by morphological alterations. Untreated trypanosomes showed the typical elongated slender morphology with the flagellum attached to the undulating membrane (Figure 1b) that confers them a high mobility (Supplementary Video S1). However, neuropeptide-treated parasites showed a swollen and round morphology (Figure 1b, 6–12 h), increased cell size and complexity (Figure 1b, flow cytometry), presence of vacuolar-like structures (Figure 1b, 2–6 h), detached flagellum (Figure 1b, arrows at 12 h) and highly reduced mobility (Supplementary Videos S2 and S3). Moreover, neuropeptides induced alterations in the normal sequence of cell division of trypanosomes. These parasites have a single mitochondrion, and the mitochondrial DNA is arranged in the kinetoplast, a large disk-like structure. Under normal conditions, there is a well-coordinated kinetoplast cycle starting shortly before the nuclear S phase. VIP treatment increased the numbers of aberrant parasites containing two nuclei and one kinetoplast, or two nuclei and two kinetoplasts without cytokinesis (Figure 1c). This suggests that VIP induces cell cycle arrest and blocks cytokinesis in T. brucei, although not preventing nuclear re-entry into another cell cycle or the replication of the kinetoplast.

Neuropeptides kill BSF Trypanosoma brucei. (a) Time- and dose-dependent trypanocidal activity of neuropeptides. BSF T. brucei were cultured with medium (control) or with different concentrations of VIP, AM or UCN (25 μg/ml in upper panel). After different times (24 h in lower panel), parasite viability (% of untreated control samples) was determined. (b) Morphological alterations of VIP-treated parasites. DAPI (blue) stains nucleus (N) and kinetoplast (K). f: flagellum; v: vacuolar-like structures. Scale bars: 2.5 μm. Parasites were analyzed by flow cytometry for cell size (FSC) and complexity (SSC). (c) Quantification of cells with different numbers of nuclei and kinetoplasts after 12 h of VIP treatment. Data are mean±S.D. of 150–250 parasites in each group. (d) Differential trypanolytic activity of neuropeptide fragments (12.5 μg/ml, 48 h). Data are mean±S.D. of eight experiments. *P<0.001 versus controls
The rate and conformational orientation of positive charges and hydrophobic residues in the molecule seem to be structural requirements for their trypanolytic activity. Thus, although the VIP fragment VIP1–12 corresponding to the first 12 N-terminal amino acids was completely ineffective, the VIP10–28 and AM20–50 fragments, both containing clusters of residues that could potentially interact with the parasite membrane (Table 1), were even more efficient as trypanolytic factors than native VIP or AM (Figure 1d).
Interestingly, all peptides failed to kill the procyclic culture forms (PCFs) of T. brucei brucei, the parasite life-cycle stage present in the tsetse vector midguts (Figure 2a), suggesting that differences between the two life stages of the parasite are determinant of the trypanolytic activity of these peptides. Importantly, for a future application, none of the neuropeptides affected the viability of mammalian cells at the effective antiparasitic dose (Figure 2b).

Neuropeptides do not affect viability of T. brucei procyclic forms and mammal cells. T. brucei procyclic forms (PCFs), bloodstream forms (BSFs) and murine peritoneal macrophages were incubated with medium (control) or with 12.5 μg/ml of VIP, AM, UCN or ghrelin (GHR). After 48 h, cell viability was determined and expressed as percentage of survival of untreated controls. Cell number and morphological changes induced on PCFs and macrophages after neuropeptide treatment were visualized by microscopy (optical magnification: × 200). Data are mean±S.D. of four experiments performed in duplicate
Neuropeptides are endocytosed by parasites and disrupt intracellular compartments
According to the way of action of other antimicrobial peptides,4, 6 trypanosome lysis might be a consequence of binding of the neuropeptides to the parasite surface and subsequent formation of pores in the plasma membrane. To investigate this possibility, we used carboxifluorescein (FAM)-labeled VIP (FAM-VIP). Interestingly, we found that T. brucei progressively accumulated VIP in intracellular compartments (Figure 3a). VIP entry occurred very fast (<15 min) through the flagellar pocket, an invagination of the plasma membrane through which all intracellular trafficking of trypanosomes is directed. Later, VIP redistributed in multiple intracellular vesicles initially localized in endomembrane compartments between the nucleus and the kinetoplast (Figure 3a). Between its initial endosomal localization and its multivesicular distribution, VIP seems to be partially released to the cytosol (Figure 3a, 1 h). Identical results were obtained with FAM-UCN (Figure 3a). Interestingly, the PCFs did not uptake VIP or UCN (Figure 3b). To simplify the figures, further experiments will only show data corresponding to VIP, although similar results were obtained with the other neuropeptides.

Neuropeptides are endocytosed by parasites. BSF (a) and PCF (b) T. brucei cultured with medium (control), FAM-VIP or FAM-UCN for different times. DAPI (blue) stains nuclear (N) and kinetoplast (K). (c) BSF T. brucei cultured at 4 or 17°C with FAM-VIP. Bars: 2.5 μm. Images are representative of five independent experiments
To determine whether endocytosis of VIP is critical for its trypanolytic activity, we treated the parasites with VIP at various temperatures that are related with different cellular events. At 4°C, a temperature that inhibits membrane molecular movements and endocytosis, BSF trypanosomes showed a uniform cell surface distribution of FAM-VIP, although they did not endocytose it (Figure 3c). At 17°C, a temperature that inhibits the endosomal fusion with the lysosome, parasites accumulated FAM-VIP in endosomes close to the flagellar pocket, but the vesicular redistribution of VIP was impaired (Figure 3c). Consequently, VIP did not show any trypanolytic activity at 4 or 17°C (not shown). Therefore, neuropeptide uptake by the parasite is a necessary, but not sufficient, step for the trypanolysis, which also requires the intracellular accumulation and extensive distribution of the peptide. Indeed, VIP removal after an initial contact (for 1 h) with the parasites aborts the trypanolysis (not shown).
We observed that once endocytosed, VIP alters the normal intracellular trafficking of the parasite. Most of the untreated trypanosomes showed staining for rhodamine-labeled dextran (a bulk-phase endocytic marker) and p67 (a lysosomal membrane marker) in one to two vesicles located between the kinetoplast and the nucleus (Figure 4a and b), corresponding to the typical endosomal–lysosomal localization in trypanosomes.10 VIP treatment progressively increased the number of dextran-containing and p67+ vesicles, and ∼75% of trypanosomes had more than three lysosome-like vesicles after 4 h of treatment. This correlates with the increased cell complexity induced by VIP (Figure 1c). Importantly, in addition to this multivesicular scenario, a significant proportion of the VIP-treated parasites showed overall diffused dextran+ and p67+ staining in the cytosol (Figure 4a and b), suggesting that their endosomal–lysosomal vesicles are disrupted. Increasing the culture pH by the addition of NH4Cl blocked the VIP-induced multivesicle scenario (Figure 4c) and reversed the antiparasitic effect (not shown), but not endocytosis of VIP (Figure 4c). This suggests that neuropeptides need the formation of the lysosome and/or their localization in an acidic compartment to exert the trypanolytic activity. Interestingly, coinciding with the disruption of lysosomes, VIP was partially released to the cytoplasm (Figure 3a, 1 h) and then accumulated in different cytoplasmic non-lysosome vesicles (Figure 4d).

Neuropeptides alter the intracellular trafficking. (a) BSF T. brucei cultured with medium (control) or with FAM-VIP in the presence of rhodamine-labeled dextran. (b) Lysosomal protein p67 was detected by immunofluorescence at indicated time points. Percentage of parasites with p67+ vesicles or diffuse fluorescence (disrupted vesicles) was determined at different time points. (c) NH4Cl was added to cultures before neuropeptide addition to block lysosomal acidification. Percentage of parasites with dextran-containing vesicles or diffuse cytosolic dextran was determined at 4 h. (d) Colocalization of VIP with in the endosomal–lysosomal compartment. Bars: 1.5 μm. Images are representative of five independent experiments
Neuropeptides induce irreversible metabolic changes in T. brucei and target glycosomes
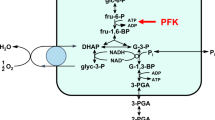
We next analyzed the cell death mechanisms involved in the antiparasitic effect of VIP and whether other cellular components were affected. We found that VIP failed to induce necrosis or apoptosis cell death (Figure 5a and b). However, the treatment with VIP resulted in a striking depletion in ATP levels in BSF trypanosomes (Figure 5c). This was followed by an important reduction in mitochondrial membrane potential (Figure 5d). This is interesting because, in contrast to PCFs, BSF trypanosomes rely entirely on glycolysis for their ATP supply. The glycolytic enzymes are localized in glycosomes, the specialized peroxisome-like organelles.11 As VIP induced a failure in the energetic metabolism of the parasite, we therefore asked whether the neuropeptide affected the glycosomes in some way. We used the glycosomal enzymes phosphoglycerate kinase (PGK) and aldolase as markers of the number and integrity of glycosomes. Although untreated trypanosomes showed the characteristic punctuate staining pattern of both glycosomal proteins, most of the VIP-treated parasites showed a faint and diffuse PGK and aldolase staining throughout the entire parasite body (Figure 5e). This indicates that a considerable fraction of the glycosomal enzymes relocated to the cytosol after VIP treatment. Western blot analysis of parasites treated with VIP showed a high content of both enzymes in the cytosolic fraction (Figure 5e), confirming the mislocalization of glycolytic enzymes out of the glycosome after treatment with neuropeptides. Interestingly, we observed that VIP mostly colocalized with the glycosomes that remained intact before the release of the glycolytic enzymes (Figure 5f), indicating that VIP seems to target glycosomes.

Cell death mechanisms involved in the trypanocidal effect of neuropeptides. (a–d) VIP induces energetic metabolism failure but not necrosis or apoptosis. BSF T. brucei were cultured with medium (control) or with VIP (12.5 μg/ml) for different times. Percentage of propidium iodide+ necrotic cells was analyzed by flow cytometry. Digitonin-treated parasites were used as positive controls (a). Apoptotic cells were determined by annexin V staining (b). Changes in intracellular ATP levels are expressed as relative luminescence to untreated controls (c). Percentage of cells with loss of mitochondrial membrane potential (ψm) was determined by tetramethylrhodamine uptake assay. Values for untreated control parasites (gray bars) did not change throughout the 24 h culture (d). (e) VIP treatment alters compartmentalization of glycosomal enzymes. BSF T. brucei cultured with medium (control) or with VIP for 12 h were immunostained for the glycosomal enzymes phosphoglycerate kinase (PGK) and aldolase (ALD), analyzed by confocal microscopy and quantified for intracellular localization of PGK as punctuated fluorescence (glycosomal PGK) or diffuse fluorescence (cytosolic PGK). Parasites were cell fractionated with digitonin and the cytosolic fraction analyzed by western blotting to show mislocalization of PGK and aldolase. (f) VIP mostly colocalized with PKG+ glycosomal vesicles at 8 h. (g) Electron micrographs showing morphological feature of autophagy-like cell death. Black arrows point sites of plasma membrane damage. N, nucleus; av, autophagic vacuoles; mls, myelin-like structures. Scale bars: 1.5 μm (e–f), 2 μm (g), 0.5 μm (g, 8 and 16 h′). Images are representative of three independent experiments. Data are mean±S.D. of six experiments. *P<0.001 versus control
Neuropeptides induce autophagic-like cell death in trypanosomes
We next investigated the nature of damage caused by the peptides within the parasite by electron microscopy (Figure 5g). We observed that VIP-treated trypanosomes underwent swelling with extensive intracellular degeneration and formation of multiple lysosome-like digestive vesicles containing an electrodense cytoplasmic material. These autophagic vesicles increased in number and size shortly after peptide contact, progressively surrounded parts of cytosol fusing with each other, resulting in a big vacuole containing abundant dispersed material enveloped by broken double membranes (Figure 5g). Only at the end, the cells showed some visible breaks in the plasma membrane, previous to cell disintegration (Figure 5g). Nuclear morphology was well preserved and no evidence of mitochondrial or flagellar pocket swelling accompanied the prominent cytoplasmic changes. Moreover, the subpellicular layer of microtubules appeared to be intact during peptide treatment. These results indicate that VIP-induced cell damage has features distinct from those of typical apoptosis or necrosis. The extensive vacuolization and the presence of cytoplasmic contents inside vacuoles clearly indicate that VIP-induced cell damage has features of autophagic-like death, similar to those observed after nutrient starvation of the cells.12, 13 Moreover, we investigated the presence of the microtubule-associated protein-1 light-chain-3 (LC3)-like proteins in cytoplasmic vacuoles of VIP-treated parasites. LC3 is an autophagosomal membrane marker in mammal cells that shows 60% homology with the recently identified proteins Tb07.10C21.40 and TcATG8.1 in T. brucei and T. cruzi, respectively.14, 15 TcATG8.1 has been functionally related with autophagy, although the role of Tb07.10C21.40 as an autophagic marker needs to be confirmed. Interestingly, VIP induced the appearance of multiple LC3-like+ spherical vesicles that progressively increased in size and fused each other; in some cases resulting in a unique big vesicle (Supplementary Figure 2). This correlates with the electron microscopy images showing the intermediate multivesicular scenario and a final huge vacuole.
Therapeutic effects of neuropeptides in T. b. brucei infection
We next studied the potential therapeutic effect of these neuropeptides in an experimental model of chronic trypanosomiasis. All the mice infected with the pleomorphic AnTat 1.1 strain of T. brucei brucei die before day 34 after infection (Figure 6a), as a consequence of two peaks of parasitemia (Figure 6b) and massive invasion of trypanosomes in several target organs after the second peak (Figure 6c). Intraperitoneal administration of VIP or VIP10–28 significantly protected infected animals. Neuropeptides decreased parasite burden in vivo and significantly delayed death of infected mice (Figure 6a and b, P<0.001). Thus, at the time when all untreated animals succumbed to parasitic infection, 83 and 91% of the mice treated with VIP and VIP10–28, respectively, were still alive. The absence of the second peak of parasitemia and the reduced trypanosome load in target organs were beneficial in alleviating local damage, mainly regarding the parasite entry to the brain, one of the key events in the pathogenesis of the disease (Figure 6b and c).

Therapeutic effects of neuropeptides in experimental chronic trypanosomiasis. C57BL/6 mice were infected with the pleomorphic AnTat 1.1 strain of T. brucei brucei. Saline (control), VIP (4 nM) or VIP10–28 (10 nM) was i.p. injected (three times/week) from day 1 after infection. (a) Survival was monitored daily. n=11–13 mice/group, four independent experiments. P<0.001, control versus VIP or VIP10–28. (b) Number of parasites in the blood of infected animals was counted daily. n=15–20 mice/group. (c) Number of parasites was determined in various organs after second peak of parasitemia (day 26 after infection). n=8 mice/group. *P<0.001, versus control. (d) VIP levels in the serum of non-infected and infected mice determined by ELISA. n=8 mice/group. *P<0.001, versus non-infected mice
Finally, we investigated whether animals produced antiparasitic neuropeptides in response to trypanosome infection. Interestingly, we observed a 20-fold increase in serum VIP levels in infected animals in comparison with uninfected mice (Figure 6d).
Discussion
Identification of new trypanolytic agents with non-traditional modes of action is critical for the design of more efficient therapeutic strategies for clinical intervention in African trypanosomiasis. In this study, we report that various endogenous neuropeptides, such as VIP, AM, UCN, CRH and ghrelin, show potent trypanolytic activity, with LD50 in the range of those antiparasitic drugs used in the clinic.16, 17 All these neuropeptides belong to different families of peptides, showing no homology between them, and their major cellular sources and physiological roles in the body are highly different.18 However, they share some structural characteristics, which include small sizes (8–50 amino acids) and amphipathic α-helical structures arranging clusters of hydrophobic and cationic residues. These characteristics are common for antimicrobial peptides. However, the present data show that the cell death mechanisms involved in the antiparasitic effect of neuropeptides substantially differ from those described for other antimicrobial peptides.19 For example, cationic peptides, such as cecropin, defensins or cathelicidins, lyse the parasite by pore formation and plasma membrane disruption.7, 20 Human histatin-5 translocates across the plasma membrane of the trypanosomatid Leishmania in a non-lethal manner and, once inside the cell, targets mitochondria and kills the parasite.21 Alternatively, trypanolysis mediated by TNF-α and human apolipoprotein-1 (apo-L1) requires uptake and processing of both factors. Although none of these possibilities can be completely excluded, our data suggest that neuropeptides exert their trypanolytic activity through an unusual mechanism that involves peptide uptake by the parasite, disruption of lysosome integrity and cytosolic accumulation of glycolytic enzymes. This promotes an energetic metabolism failure that initiates an autophagic-like cell death (Figure 7).

Mechanism underlying the trypanolytic activity of neuropeptides. (1) Cationic α-helical neuropeptides bind to highly negatively charged glycoproteins (VSG is shown as an example) that are widely coating BSFs. (2) Carried by surface glycoproteins, neuropeptides are quickly endocytosed through the flagellar pocket entering in the normal trafficking pathway of the parasite. (3) Neuropeptides pass to the lysosomal compartment where they, in an acidic environment, acquire the capacity to disrupt the membrane of these vesicles. (4) The release of digestive enzymes from the lysosomes could be enough to cause the death of the parasite. In addition, this could induce a general perturbation of the intracellular membrane compartment characterized by increased numbers of lysosome-like vesicles. At this point, neuropeptides are released into the cytosol. Once in the cytosol, neuropeptides could target glycosome, disrupt the glycosome membrane and release the glycolytic enzymes to cytosol. (5) Mislocalization of glycolytic enzymes to the cytosol disrupts energetic metabolism. (6) The fall in ATP levels in the parasite leads to alteration in the mitochondrial potential membrane (ψm) and to an autophagic-like process that kills the parasite
As cationic peptides, neuropeptides preferentially recognize and interact with the anionic residues exposed on the outer surface of the BSF trypanosome membrane, but not on the mammalian cell membrane, nor on the PCF trypanosome membrane. No targeting for mammalian cells could be partially explained because in higher eukaryotes, the anionic phospholipids are mostly confined to the inner cytoplasmic leaflet of the plasma membrane and they have higher membrane cholesterol amounts than trypanosomes. This could prevent interaction with the peptides and avoid host cell damage. Although minimal amounts of neuropeptides could be internalized by mammalian cells after their binding to specific receptors in a natural regulated process of desensitization, this process does not result in a toxic effect for the cells. This could be because the neuropeptides do not accumulate in the endosomal compartments and are cleared before their receptors are recycled again to the cell surface. The specific targeting for the BSFs, but not for the PCFs, could be due to various factors. First, the two parasite forms present different anchored membrane proteins in terms of their relative abundance, structure and glycosylation.22, 23 Second, PCFs show lower endocytic rates than the BSFs. Although endocytic recycling of the entire BSF coat is very rapid, and the entire flagellar pocket is internalized within ∼2 min, uptake in PCFs is unusually slow, with little turnover of surface proteins.24 These differences could be critically related to the differential trypanolytic activity of the neuropeptides in the two parasitic stages.
The experiments of propidium iodide exclusion and the electron microscopy images argue against plasma membrane disruption as a major mechanism involved in the trypanolytic activity of neuropeptides, as occurs with other cationic antimicrobial peptides.7, 20 Moreover, our data indicate that the endocytosis of the neuropeptides through the flagellar pocket is strictly required for their trypanocidal effect. This mechanism also differs from that described for human histatin, which translocates across the plasma membrane through the entire surface of the parasite in a non-lethal manner by inducing cell membrane depolarization.21
Once endocytosed, neuropeptides initially followed normal delivery pathways in T. brucei, being trafficked from endosome network to lysosome, where they seem to disrupt the endosomal–lysosomal vesicles. Other trypanolytic factors, such as TNF-α and apo-L1, bind to the flagellar pocket, internalize through endocytic vesicles and ultimately localize into large lysosome-like vesicles, where they form ion-permeable channels that induce osmotic swelling of the lysosome until the parasite is lysed.25, 26 Neuropeptides need the formation of the lysosomes and/or their localization in an acidic compartment to exert the trypanolytic activity. It remains unknown whether neuropeptides form ion-permeable pores in the lysosome, but their way of action could slightly differ from those described for TNF-α and apo-L1. Although TNF-α and apo-L1 lyse trypanosomes quickly (in ∼4 h), longer and continuous uptake and intracellular accumulation of the neuropeptides are required to maintain a sustained trypanocidal action that could depend on alteration of parasite intracellular trafficking. Why do neuropeptides lyse intracellular vesicles but not the plasma membrane of the trypanosome? Glycocalix that coats the parasite could make cell membrane inaccessible to neuropeptides. Alternatively, neuropeptides could be inactive outside the trypanosome and activate only when internalized, probably in an acidic environment.
Coinciding with the disruption of lysosome, neuropeptides are partially released to the cytoplasm and then accumulated in different cytoplasmic non-lysosome vesicles. A complete colocalization of VIP with glycosomes suggests that, once in the cytosol, neuropeptides target these peroxisome-like organelles. This event could have important consequences in the survival of the parasite, because BSF trypanosomes rely entirely on glycolysis for their ATP supply and maintain all glycolytic enzymes confined in the glycosome. Indeed, glycolysis has been perceived as an excellent target for the design of new antitrypanosome drugs, and the inhibition of glycolysis leads to rapid death of BSFs. The presence of intact glycosomes and the proper compartmentation of the enzymes within these organelles are essential for parasite survival, and BSFs do not tolerate even a slight mislocalization of glycosomal enzymes to the cytosol.27, 28 Our findings show that neuropeptides induce the accumulation of glycolytic enzymes in the cytosol of BSFs. We still do not know whether mislocalization of glycolytic enzymes is a consequence of the lysosomal lysis or whether, once released from the lysosomes to the cytosol, neuropeptides are able to disrupt the glycosome membrane, allowing the exit of their enzymes to the cytosol.
These findings could indicate that the loss of lysosome integrity and/or a rise in the level of the glycolytic enzymes above a critical threshold in the cytoplasm of the neuropeptide-treated trypanosomes initiate a bioenergetic catastrophe, characterized by a decline in ATP levels and loss of mitochondrial membrane potential, which seem to initiate an autophagic-like cell death program. The increase in intracellular lysosome-like vesicles observed after peptide treatment could be directly related to the formation of the autophagic-like vacuoles that lately rely on lysosome fusion. The concept of autophagy in trypanosomatids is new and has been mainly related to processes of differentiation and change of stage,29, 30 although recent reports have shown that other antiparasitic drugs could induce autophagy.13, 31 Various studies have recently described the functional involvement of the genes homologous to Atg8 and LC3 in the autophagy of Leishmania major and T. cruzi.14, 32 Although it needs confirmation, their phylogenetic proximity and homology presume a similar functional implication of the Atg8/LC3 ortholog gene Tb07.10C21.40 in T. brucei. In the absence of molecular functional markers, the best phenotypic characteristic that defines the autophagic process is the ultrastructural morphologic changes. Although our electron microscopy studies show that neuropeptides induce a process with phenotypic characteristics of autophagy, until autophagy is fully demonstrated and molecularly characterized in T. brucei, we only can say that neuropeptides induce an ‘autophagic-like’ process.
This study has different important implications. From a therapeutic point of view, these cationic neuropeptides emerge as new trypanolytic agents with non-traditional modes of action (Figure 7). This is critical for the design of more efficient therapeutic strategies for clinical intervention in African trypanosomiasis. Using agents that specifically target vital intracellular components, present only in trypanosomes, could overcome the emergence of resistance observed against conventional drugs, because redesign of these targets would be a costly solution for parasites. Importantly, contrary to current treatments, neuropeptides are not toxic for the host. However, treatment with neuropeptides was insufficient to completely cure a chronic infection and treated mice succumbed to late-stage parasitemia. Therefore, methodological strategies should be developed before clinical translation to increase the neuropeptide half-life or improve delivery in the target tissues, especially in long-term treatments.
From a physiological point of view, neuropeptides could act as natural antiparasitic agents. We observed that the hosts respond to trypanosome infection by elevating the peripheral levels of VIP in an attempt to decrease parasite load. Although the concentrations required for an efficient in vitro trypanolytic activity are two or three orders of magnitude higher than the endogenous neuropeptide levels induced during trypanosomiasis, they correspond to the neuropeptide concentrations found in certain microenvironments, such as in the proximity of nerve endings.33 In addition to nerves, the neuropeptides described in this study are produced by leukocytes and epithelial cells and are elevated in skin and mucosal barriers of the respiratory, genital and gastrointestinal tract,18 which are in constant contact with the pathogens. Interestingly, human leukocytes were found to produce several VIP fragments, including VIP10–28, but their roles were completely unknown.34 Our data could give now a physiological significance to the production of these fragments. Most of these neuropeptides appear very early in the evolution and their sequences are well conserved, suggesting that they must play an ancient role, probably as important components of host innate defense.
Finally, from the point of view of the parasite, the induction of trypanolytic neuropeptides during infection could mean an evolutionary advantage for the trypanosome. Thus, parasites could promote the release of neuropeptides by cells of the host leading to elimination of most of the parasite population and selection of the fittest parasites, favoring parasite transmission for a longer time by keeping the host alive. Importantly, the induction of programmed cell death seems to play a crucial role in this strategy of the parasite.35, 36
Materials and Methods
Cell culture and in vitro trypanolysis assays
Parasite BSFs included the pleomorphic strain T. brucei brucei AnTat 1.1 (Institute of Tropical Medicine, Belgium) and the monomorphic strain MiTat 1.2 (variant surface glycoprotein 221). PCFs T. brucei (clone 221) were grown at 28°C in SDM-79 medium/10% fetal bovine serum. BSFs were cultured in axenic culture conditions at 37°C with 5% CO2 in HMI-9 medium. Exponentially growing parasites were incubated (4 × 105 cells/ml) with different concentrations of neuropeptides obtained from American Peptides (Sunnyvale, CA, USA) or Phoenix Pharmaceuticals (Burlingame, CA, USA) at 37°C (for BSFs) or 28°C (for PCFs) for various time periods. In some experiments, parasites were treated with a second administration of neuropeptide after 24 h culture. To analyze the importance of peptide uptake in the trypanolytic activity, parasites were cultured with peptides (25 μg/ml) for 1 h at 17 or 4°C, washed and then incubated for 4 h at 37°C. To increase pH and inhibit the acidification of endosomes or lysosomes, parasites were preincubated with 8 mM NH4Cl for 30 min at 4°C and then treated with neuropeptides for 4 h at 37°C.
Trypanolytic activity of neuropeptides was determined by measuring cell viability by using the chromogenic substrate 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT; Sigma, St Louis, MO, USA). In brief, parasites were incubated with MTT (5 mg/ml, 10 μl/well) for an additional 4 h period at either 28 or 37°C. The reduced substrate was then solubilized with 20% SDS (90 μl) overnight at 37°C and absorbance measured at 550 nm. Relative viability (%) was determined as (A550 neuropeptide-treated parasites/A550 control parasites) × 100.
Measurement of mitochondrial membrane potential
After treatment, the parasites were incubated with 25 nM tetramethylrhodamine (TMRE; Molecular Probes, Carlsbad, CA, USA) in HEPES buffer (10 mM, pH 7.4; containing 140 mM NaCl and 5 mM CaCl2) for 30 min at 37°C and analyzed by flow cytometry. Cells treated with the ionophore valinomycin (100 nM) were used as positive controls for a complete depolarization of the inner mitochondrial membrane.
Determination of ATP levels
ATP was extracted by incubating parasites (2 × 106) with 45 μl of ATP buffer assay (100 mM Tris-HCl, 2 mM EDTA, pH 7.5), 4 μl EGTA (50 mM) and 12 μl phenol:chloroform:isoamilic acid for 2 min. After centrifugation at 13 000 r.p.m., supernatant (5 μl) was added to ATP buffer (195 μl) and ATP assay mix (50 μl) containing luciferin/luciferase system (Sigma). After 3 min, luminescence was read in a FB12 luminometer.
Determination of necrosis
Plasma membrane disruption was assessed using propidium iodide (5 μg/ml) staining and flow cytometry. Parasites treated with digitonin (6 μM) were used as positive controls.
Determination of apoptosis
Apoptotic cells were assayed by measuring exposed phosphatidylserine on the outer membrane. After neuropeptide treatment, cells were washed in HEPES buffer (10 mM, pH 7.4; containing 140 mM NaCl and 5 mM CaCl2) and incubated for 15 min with annexin V at 4°C. Fluorescence was measured using FACS analysis.
Fluorescence microscopy
For FAM-VIP and FAM-UCN labeling, cells were cotreated with 12.5 μg/ml of peptides and 0.02 nM of fluorescent peptides (from Phoenix Pharmaceuticals). After washing with PBS, the cells were fixed in 2% formaldehyde and mounted in DAPI-containing Vectashield medium (Vector Laboratories, Burlingame, CA, USA). For indirect immunofluorescence assays, parasites were fixed in 2% formaldehyde, spread on poly-L-lysine-coated slides and permeabilized with 0.2% Triton X-100 in washing solution (PBS/1% BSA). Cells were then blocked with 2% goat serum and incubated with the corresponding primary antibodies against p67, rat LC3 (MBL laboratories), PGK-C and aldolase. Slides were then washed, incubated with Alexa-594-labeled goat anti-mouse or anti-rabbit antibodies (Molecular Probes), mounted as above and imaged by a microscope (AX10 Imager A1; Zeiss) or by a confocal microscope (LCS SP5; Leica).
To measure endocytosis, we used tetramethylrhodamine-labeled dextran (Molecular Probes; Mr=10 000; neutral charge) as a fluid-phase marker. Parasites were preincubated with 3 mg/ml dextran at 37°C for 30 min before the addition of neuropeptides (25 μg/ml), and then incubated at 37°C for 2–4 h. When used, NH4Cl was added 30 min at 4°C before dextran incubation. After incubation, the cells were fixed in 1% formaldehyde, mounted onto poly-L-lysine slides and processed for microscopy.
Western blot analysis
BSF trypanosomes (2 × 107) were lysed in a buffer containing 25 mM Tris-HCl (pH 7.4), 0.3 M sucrose, digitonin (6 μg/ml, a concentration that lyses plasma membrane, but not glycosomal membrane) and proteinase inhibitor cocktail (Sigma) for 45 min on ice. After centrifugation (12 000 × g, 10 min), the proteins were separated on 10% SDS-PAGE gels, transferred onto PVDF membranes, blocked with 5% skimmed milk in PBS and incubated with primary antibodies against PGK-C or aldolase (dilution 1 : 8000), followed by HRP-conjugated anti-rabbit antibody (1 : 5000; Dako) and chemoluminescence was detected with the ECL Plus system (GE Healthcare).
Transmission electron microscopy
Trypanosomes were fixed with 1.5% glutaraldehyde/1% formaldehyde in 0.05 M sodium cacodylate buffer for 2 h at 4°C, washed and postfixed in 1% osmium tetroxide containing 1% potassium ferricyanide. Samples were stained in 2% uranyl acetate, dehydrated in ethanol and embedded in Epon according to standard procedures. Ultrathin sections were collected onto grids, stained with uranyl acetate, contrasted with citrate and observed in a Zeiss EM10C electron microscope at 80 kV.
Experimental model of chronic trypanosomiasis
Animal experimental protocols were approved by the Ethics Committee of the Spanish Council of Scientific Research (CSIC). Male C57BL/6 mice (7- to 9-week-old; Jackson Laboratories) were i.p. injected with 5000 parasites from the pleomorphic strain AnTat 1.1 of T. brucei brucei. VIP (13 μg/mouse) or VIP10–28 (23 μg/mouse) were i.p. administered on alternate days starting 1 day after infection. Control mice received PBS. Survival was monitored every day. Parasitemia was determined every other day by counting the number of trypanosomes in tail-vein blood with an optical microscope. Some animals were killed after the first (day 7) and second (day 26) wave of parasitemia, and serum was collected by cardiac puncture and the number of parasites determined in cell suspensions prepared from lymph nodes, spleen, liver, brain and lungs. VIP levels in the sera were determined by competitive ELISA (Phoenix Pharmaceuticals).
Statistical analysis
Significance for in vivo survival studies was evaluated by Fisher's exact test. Analysis of variance followed by Bonferroni post-test was used for in vivo parasitemia comparisons. Trypanosome numbers in tissues and in vitro viability studies were evaluated by using the Mann–Whitney U test. The differences were considered significant when the P-value was <0.05. All results are expressed as mean±S.D.
Abbreviations
- AM:
-
adrenomedullin
- apo-L1:
-
apolipoprotein-1
- BSF:
-
bloodstream forms
- FAM:
-
carboxifluorescein
- PCF:
-
procyclic forms
- PGK:
-
phosphoglycerate kinase
- UCN:
-
urocortin
- VIP:
-
vasoactive intestinal peptide
- VSG:
-
variant surface glycoprotein
References
Barrett MP, Burchmore RJ, Stich A, Lazzari JO, Frasch AC, Cazzulo JJ et al. The trypanosomiases. Lancet 2003; 362: 1469–1480.
Legros D, Ollivier G, Gastellu-Etchegorry M, Paquet C, Burri C, Jannin J et al. Treatment of human African trypanosomiasis: present situation and needs for research and development. Lancet Infect Dis 2002; 2: 437–440.
Zasloff M . Antimicrobial peptides of multicellular organisms. Nature 2002; 415: 389–395.
Huang HW . Molecular mechanism of antimicrobial peptides: the origin of cooperativity. Biochim Biophys Acta 2006; 1758: 1292–1302.
Souto-Padron T . The surface charge of trypanosomatids. An Acad Bras Cienc 2002; 74: 649–675.
Haines LR, Hancock RE, Pearson TW . Cationic antimicrobial peptide killing of African trypanosomes and Sodalis glossinidius, a bacterial symbiont of the insect vector of sleeping sickness. Vector Borne Zoonotic Dis 2003; 3: 175–186.
McGwire BS, Olson CL, Tack BF, Engman DM . Killing of African trypanosomes by antimicrobial peptides. J Infect Dis 2003; 188: 146–152.
Charnley M, Moir AJ, Douglas CW, Haycock JW . Anti-microbial action of melanocortin peptides and identification of a novel X-Pro-d/l-Val sequence in Gram-positive and Gram-negative bacteria. Peptides 2008; 29: 1004–1009.
Allaker RP, Grosvenor PW, McAnerney DC, Sheehan BE, Srikanta BH, Pell K et al. Mechanisms of adrenomedullin antimicrobial action. Peptides 2006; 27: 661–666.
Brickman MJ, Balber AE . Trypanosoma brucei rhodesiense: membrane glycoproteins localized primarily in endosomes and lysosomes of bloodstream forms. Exp Parasitol 1993; 76: 329–344.
Opperdoes FR . Compartmentation of carbohydrate metabolism in trypanosomes. Annu Rev Microbiol 1987; 41: 127–151.
Dunn Jr WA . Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol 1990; 110: 1935–1945.
Merkel P, Beck A, Muhammad K, Ali SA, Schonfeld C, Voelter W et al. Spermine isolated and identified as the major trypanocidal compound from the snake venom of Eristocophis macmahoni causes autophagy in Trypanosoma brucei. Toxicon 2007; 50: 457–469.
Alvarez VE, Kosec G, Sant’Anna C, Turk V, Cazzulo JJ, Turk B . Autophagy is involved in nutritional stress response and differentiation in Trypanosoma cruzi. J Biol Chem 2008; 283: 3454–3464.
Herman M, Gillies S, Michels PA, Rigden DJ . Autophagy and related processes in trypanosomatids: insights from genomic and bioinformatic analyses. Autophagy 2006; 2: 107–118.
Docampo R, Moreno SN . Current chemotherapy of human African trypanosomiasis. Parasitol Res 2003; 90: S10–S13.
Burri C, Brun R . Eflornithine for the treatment of human African trypanosomiasis. Parasitol Res 2003; 90: S49–S52.
Gonzalez-Rey E, Delgado M . Anti-inflammatory neuropeptide receptors: new therapeutic targets for immune disorders? Trends Pharmacol Sci 2007; 28: 482–491.
McGwire BS, Olson CL, Tack BF, Engman DM . Killing of African trypanosomes by antimicrobial peptides. J Infect Dis 2003; 188: 146–152.
Boulanger N, Brun R, Ehret-Sabatier L, Kunz C, Bulet P . Immunopeptides in the defense reactions of Glossina morsitans to bacterial and Trypanosoma brucei brucei infections. Insect Biochem Mol Biol 2002; 32: 369–375.
Luque-Ortega JR, Van’t Hof W, Veerman EC, Saugar JM, Rivas L . Human antimicrobial peptide histatin 5 is a cell-penetrating peptide targeting mitochondrial ATP synthesis in Leishmania. FASEB J 2008; 22: 1817–1828.
Mehlert A, Zitzmann N, Richardson JM, Treumann A, Ferguson MA . The glycosylation of the variant surface glycoproteins and procyclic acidic repetitive proteins of Trypanosoma brucei. Mol Biochem Parasitol 1998; 91: 145–152.
Acosta-Serrano A, Vasella E, Liniger M, Kunz Renggli C, Brun R, Roditi I et al. The surface coat of procyclic Trypanosoma brucei: programmed expression and proteolytic cleavage of procyclin in the tsetse fly. Proc Natl Acad Sci USA 2001; 98: 1513–1518.
Field MC, Carrington M . Intracellular membrane transport systems in Trypanosoma brucei. Traffic 2004; 5: 905–913.
Magez S, Geuskens M, Beschin A, del Favero H, Verschueren H, Lucas R et al. Specific uptake of tumor necrosis factor-alpha is involved in growth control of Trypanosoma brucei. J Cell Biol 1997; 137: 715–727.
Perez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, Homble F et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 2005; 309: 469–472.
Blattner J, Helfert S, Michels P, Clayton C . Compartmentation of phosphoglycerate kinase in Trypanosoma brucei plays a critical role in parasite energy metabolism. Proc Natl Acad Sci USA 1998; 95: 11596–11600.
Kessler PS, Parsons M . Probing the role of compartmentation of glycolysis in procyclic form Trypanosoma brucei: RNA interference studies of PEX14, hexokinase, and phosphofructokinase. J Biol Chem 2005; 280: 9030–9036.
Besteiro S, Williams RA, Coombs GH, Mottram JC . Protein turnover and differentiation in Leishmania. Int J Parasitol 2007; 37: 1063–1075.
Herman M, Pérez-Morga D, Schtickzelle N, Michels PA . Turnover of glycosomes during life-cycle differentiation of Trypanosoma brucei. Autophagy 2008; 4: 294–308.
Bera A, Singh S, Nagaraj R, Vaidya T . Induction of autophagic cell death in Leishmania donovani by antimicrobial peptides. Mol Biochem Parasitol 2003; 127: 23–35.
Besteiro S, Williams RA, Morrison LS, Coombs GH, Mottram JC . Endosome sorting and autophagy are essential for differentiation and virulence of Leishmania major. J Biol Chem 2006; 281: 11384–11396.
Brogden KA, Guthmiller JM, Salzet M, Zasloff M . The nervous system and innate immunity: the neuropeptide connection. Nat Immunol 2005; 6: 558–564.
Summers MA, O’Dorisio MS, Cox MO, Lara-Marquez M, Goetzl EJ . A lymphocyte-generated fragment of vasoactive intestinal peptide with VPAC1 agonist activity and VPAC2 antagonist effects. J Pharmacol Exp Ther 2003; 306: 638–645.
Debrabant A, Lee N, Bertholet S, Duncan R, Nakhasi HL . Programmed cell death in trypanosomatids and other unicellular organisms. Int J Parasitol 2003; 33: 257–267.
Debrabant A, Nakhasi H . Programmed cell death in trypanosomatids: is it an altruistic mechanism for survival of the fittest? Kinetoplastid Biol Dis 2003; 2: 7.
Acknowledgements
We thank PA Michels and V Hannaert (Université Catholique de Louvain) and JD Bangs (University of Wisconsin Medical School) for providing invaluable antibodies for glycosome and lysosomal proteins. This study was supported by Spanish Ministry of Education and Science, FIS and RETICS.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Piacentini
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Rights and permissions
About this article
Cite this article
Delgado, M., Anderson, P., Garcia-Salcedo, J. et al. Neuropeptides kill African trypanosomes by targeting intracellular compartments and inducing autophagic-like cell death. Cell Death Differ 16, 406–416 (2009). https://doi.org/10.1038/cdd.2008.161
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.161
Keywords
This article is cited by
-
Cell death pathways in pathogenic trypanosomatids: lessons of (over)kill
Cell Death & Disease (2019)
-
Microbiota and gut neuropeptides: a dual action of antimicrobial activity and neuroimmune response
Psychopharmacology (2019)
-
Enteroendocrine cells-sensory sentinels of the intestinal environment and orchestrators of mucosal immunity
Mucosal Immunology (2018)
-
Cell death in parasitic protozoa: regulated or incidental?
Nature Reviews Microbiology (2013)
-
Vasoactive intestinal peptide: a neuropeptide with pleiotropic immune functions
Amino Acids (2013)
