
Abstract
Familial hypocalciuric hypercalcemia (FHH) is caused by inactivating mutations in the calcium-sensing receptor (CaSR) gene. The loss of function of CaSR presents with rickets as the predominant skeletal abnormality in mice, but is rarely reported in humans. Here we report a case of a 16-year-old boy with FHH who presented with skeletal manifestations of rickets. To identify the possible pathogenic mutation, the patient was evaluated clinically, biochemically, and radiographically. The patient and his family members were screened for genetic mutations. Physical examination revealed a pigeon breast deformity and X-ray examinations showed epiphyseal broadening, both of which indicate rickets. Biochemical tests also showed increased parathyroid hormone (PTH), 1,25-dihydroxyvitamin D, and elevated ionized calcium. Based on these results, a diagnosis of FHH was suspected. Sequence analysis of the patient’s CaSR gene revealed a new missense mutation (c.2279T>A) in exon 7, leading to the damaging amino change (p.I760N) in the mature CaSR protein, confirming the diagnosis of FHH. Moreover, the skeletal abnormities may be related to but not limited to vitamin D abnormity. Elevated PTH levels and a rapid skeletal growth period in adolescence may have also contributed. Our study revealed that rickets-like features have a tendency to present atypically in FHH patients who have a mild vitamin D deficiency, and that CaSR mutations may have a partial role in the pathogenesis of skeletal deformities.
Similar content being viewed by others
Introduction
Familial hypocalciuric hypercalcemia (FHH) is an inherited disorder of mineral homeostasis that is transmitted as an autosomal dominant trait. FHH is characterized biochemically by lifelong modest elevation of the serum calcium concentration with a relatively mismatched hypocalciuria and normal or mildly elevated circulating parathyroid hormone (PTH) level that is not suppressed by the hypercalcemia.1,2 Individuals with FHH are usually asymptomatic, and the disorder is considered benign. FHH is associated with heterogeneous inactivating mutations in the calcium-sensing receptor (CaSR) gene.3 The CaSR protein is a G-protein-coupled receptor that belongs to family (or class) C, which includes seven exons and is mainly expressed in parathyroid and renal tubule cells, as well as in bones.4
CaSR helps maintain a nearly constant levels of extracellular calcium (Cao2+) in the blood and other extracellular fluids. By sensing even minute changes in the Cao2+ from its normal level, CaSR modulates the functions of key tissues, especially the parathyroid glands and the kidneys.5 In the parathyroid glands, CaSR senses perturbations in the Cao2+ level of only a few percent and then responds to the hypocalcemia by increasing PTH secretion, stabilizing mRNA and inducing the proliferation of parathyroid cells. In the kidneys, CaSR stimulates Ca2+ reabsorption in the cortical thick ascending limb in response to hypocalcemia, and 1-hydroxylation of 25-hydroxyvitamin D3 [25(OH)D3] in the proximal renal tubules.6,7
CaSR is also expressed in the skeleton, but the importance of CaSR in bone formation and resorption remains somewhat controversial. Studies in mice with a conditional knockout of the CaSR gene have indicated that CaSR has key roles in osteoblasts.1 The skeleton could mediate PTH independently with the homeostatical uptake or release of calcium during induced hypocalciuria, actions that may involve the CaSR gene. The calcium-sensing receptor is present in the skeleton, and the absence of exon 5 results in defective mineralization of cartilage and bone, which produces rickets as the predominant skeletal abnormality in mouse models.8 In case reports, human FHH caused by inactive mutations of CaSR rarely manifested as rickets. A recent report described a patient who developed severe adolescent rickets due to vitamin D deficiency.2 Another report considered that vitamin D deficiency modulates the severity of FHH.3
In this study, we report an FHH patient with typical skeletal manifestations. The patient demonstrated a fracture with transient hypophosphatemia, which confounded the plausible initial diagnosis of rickets based on the clinical findings. In addition to hypocalciuria, increased PTH and the an ultimately slightly elevated serum calcium eventually led to consideration of FHH. The identification of a CaSR mutation confirmed the FHH diagnosis. The patient also presented with a mild vitamin D deficiency, but that alone could not completely explain the manifestation of rickets. We proposed the possibility that a patient with CaSR mutation may tend to present with skeletal phenotype of rickets under conditions of vitamin D deficiency. Further, elevated PTH and pubescent skeletal growth and development modulate the manifestation of rickets in FHH patients.
Materials and methods
Subject
A 16-year-old boy who was admitted to our hospital for knee pain that had lasted 2 years and who had suffered a femur fracture 4 months prior with subsequent nonunion presented with signs of rickets. The patient underwent clinically, biochemically, and radiologically evaluations. He was the child of a non-consanguineous couple, without any craniofacial or skeletal abnormality recorded in his family history.
Biochemical parameters
Fasting blood samples were collected, and serum was stored at −80 °C for measurement. The concentrations of serum calcium (Ca), serum phosphate (Pi), serum creatinine (Scr), serum alkaline phosphatase (ALP), 24-h urinary phosphate and urinary calcium were measured using the routine methods available at the central laboratory of Peking Union Medical College Hospital, an in-hospital laboratory accredited for quality supervision standards.
25-hydroxyvitamin D [25(OH)D], serum intact PTH, and β C-terminal telopeptide of bone type I collagen concentrations were measured with an automated electrochemiluminescence system (Roche Diagnostics, Basel, Switzerland). Serum 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] was measured with a 1,25-dihydroxyvitamin D [1,25(OH)2D] 125I RIA kit (DiaSorin, Stillwater, USA). Intact fibroblast growth factor 23 (FGF23; Kainos Laboratories Inc., Tokyo, Japan), sclerostin (Biomedica Medizinprodukte GmbH, Vienna, Austria), receptor activator for nuclear factor-κ B ligand (Biomedica Medizinprodukte GMBH), and osteoprotegerin (Biomedica Medizinprodukte GMBH) were measured using a two-site enzyme-linked immunosorbent assay according to the manufacturer’s instructions.
Molecular genetic analysis
Targeted next-generation sequencing
Genomic DNA of the patient and his parents was extracted from peripheral leukocytes using a QIAamp DNA Blood Mini kit (QIAamp DNA; Qiagen, Hilden, Germany). A customized targeted enrichment kit (Roche NimbleGen, Madison, USA) was used to capture the splice sites of all exons and the immediate intron-flanking sequences of 722 genes associated with rare genetic skeletal disorders or that were involved in the pathways related to skeletal development. A previously described pipeline was used for targeted sequence capture and for sequencing library preparation.9–11 Sequencing (PE101) was performed using an Illumina HiSeq2000 platform (Illumina, San Diego, USA) according to the standard manual.
Read mapping and variant detection, annotation, and interpretation
Illumina Pipeline (version 1.3.4) was used for image analysis, error estimation, base calling, and generation of the primary sequence data. The primary sequence data were analyzed using GAEA software, which was developed in-house at the Beijing Genomics Institute (BGI) and integrated the modules for quality control of raw data, reads alignment, SNV calling and INDEL calling. The annotation of SNVs and INDELs was accomplished using in-house scripts. The clinical significance of variants was interpreted according to the guidelines from American College of Medical Genetics and Genomics.12
Sequence analysis of the human CaSR gene
Sanger sequencing was used to confirm the potential mutations identified by targeted next-generation sequencing. PCR was performed to cover all exons and 50 bp on both sides of the exon–intron boundaries of CaSR. The samples were sequenced with an automated sequencer, and sequence alignment was performed using the basic local alignment search tool (BLAST) from the National Center for Biotechnology Information database. The bioinformatics tool PolyPhen was used to predict the protein function.
Radiography
Radiographic studies were performed in the Department of Radiology of Peking Union Medical College Hospital. Plain X-rays of both hands and lateral films of the thoracolumbar vertebra and femur were taken.
Ethics statement
Approval for this study was obtained from the local ethics committee in the Department of Scientific Research at Peking Union Medical College Hospital. The patient and his family provided informed consent to participate in this study.
Results
Clinical features
A 16-year-old Chinese boy was admitted to our hospital for a 2-year duration of knee pain. The patient also presented with a femur fracture that had occurred 4 months prior with subsequent nonunion, which rendered him wheelchair-bound.
The patient was born via natural labor, had been breast-fed, and had never developed rickets-like symptoms when he was a child. He started to sit and walk at the appropriate age and presented equal or higher height compared with his peers. He had no difficulty in feeding, walking, or running. His growth and pubertal development were normal. There was no history of delayed tooth eruption, bowed legs, waddling, tetany, or skeletal deformities either. The remaining developmental milestones were within the normal limits.
From the age of 10 years, he practiced the sport of fencing and gradually developed gonalgia at approximately 12–13 years old, which was considered a consequence of fencing. Then, he began to complain of more severe and localized pain in the knee related to exercise, which was relieved by rest. He also felt the occasional backache, which was accompanied by minor weakness.
The patient gradually developed the clinical signs of rickets (pectus carinatum) until 14 years of age. He presented with a slight, ameliorative pain after minor trauma to the right lower extremity when bicycling, without impaired physical mobility. However, two days later he felt severe localized pain around the right knee with aggravated symptoms and arthrocele. X-ray examination was performed at a local hospital and revealed fractures of the right distal femur and proximal fibula. An external fixator with traction did not resolve the problem, and the patient presented with delayed union and angulation deformities of the long bones. Laboratory evaluation showed hypophosphatemia and normal serum calcium, increased alkaline phosphatase, procollagen I N-terminal peptide, and C-terminal telopeptides of bone type I collagen.
He was treated for 2 months with oral 1,25(OH)2D 0.25 μg per day, and calcium carbonate 1 200 mg per day as the basic treatment for osteoporosis. Then, the patient underwent surgery for the placements of an internal fixator that was performed on the right femur. Interestingly, the patient’s biochemical indicators showed increased and nearly normalized serum phosphate one to two months later.
After being admitted to our hospital, a laboratory examination of the patient showed a normal serum phosphate concentration. The parents stated that the patient’s family history was negative for hypophosphatemia, rickets, osteomalacia, and other metabolic bone diseases. His height and weight were within the calculated mid-range (height of 168 cm, between the median and 1 s.d. of the same age and gender population; weight of 50 kg, between −1 s.d. and the median), and his body mass index was 17.7 kg·m−2.
Physical examination revealed a pigeon breast deformity, possible rachitic rosary, and bilateral widening of the wrists and ankles. Dental examination showed no dental caries or periodontal abscess, and the patient did not present with bowing of the lower extremities or other skeletal deformity.
X-ray examinations revealed signs of rickets, such as epiphyseal broadening in the distal femur, the proximal fibula, and the proximal phalanx, as well as a lower bone density.
Laboratory evaluation revealed a normal to lower serum ionized calcium of 1.03–1.23 mmol·L−1 (normal range, 1.13–1.23 mmol·L−1) with a serum level of total calcium of 2.36–2.46 mmol·L−1 (normal, 2.13–2.70 mmol·L−1), whereas the serum concentration of phosphorus was normal or mildly increased (1.48–1.77 mmol·L−1; normal range, 0.81–1.45 mmol·L−1). The patient showed a mildly high level of alkaline phosphatase of 287 –394 U·L−1 (normal range, 42–390 U·L−1), which led to the suspected diagnosis of rickets. The serum concentration of 25(OH)D3 was 17.6 ng·mL−1 (normal, 8–50 ng·mL−1), which seemed low but was considered normal in the Chinese population. The intact PTH concentrations (90.7–117 pg·mL−1; normal, 12–65 pg·mL−1) were increased, and the patient also showed an extraordinarily elevated serum concentrations of 1,25(OH)2D: 213.95 pg·mL−1 (normal range, 19.6–54.3 pg·mL−1). Therefore, the vitamin D deficiency and likely secondary hyperparathyroidism were never effectively treated during the patient’s course of treatment (to avoid vitamin D overdosage). Moreover, the 24-h urine calcium of the patient was significantly decreased, ranging from 0.31 to 0.82 mmol (normal, 2.5–7.5 mmol per 24 h), which was not consistent with rickets. Bone turnover markers were also assessed. The receptor activator for nuclear factor-κ B ligand concentration was significantly elevated, up to 0.688 pmol·L−1, and serum osteoprotegerin concentrations were decreased to 1.3 pmol·L−1. β C-terminal telopeptide of type I collagen was also increased, which indicated increased osteoclast activity. However, FGF23 and sclerostin concentrations were normal, reaching 4.096 1 pg·L−1 and 26.5 pmol·L−1, respectively.
One year later after hospitalization, the patient was pain-free with no aggravated skeletal deformity. Biochemical examination showed a mildly elevated serum ionized calcium of 1.32 mmol·L−1 (normal range, 1.13–1.23 mmol·L−1), with a total calcium concentration of 2.48 mmol·L−1. The 24-h urine calcium (0.4 mmol) was significantly lower compared with the normal range.
In all, during the patient’s course of the disease, he presented with laboratory values of an elevated serum ionized calcium, a continuously high PTH levels and a significantly decreased 24-h urine calcium, and the clinical diagnosis of FHH was highly suspected and later established. Negative radiological result of the parathyroid and significantly repetitive low 24-h urine calcium suggested that the diagnosis was unlikely to be primary hyperparathyroidism.
Imaging features
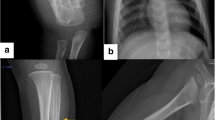
X-ray of the hands indicated epiphyseal broadening. X-ray films of the femur and the lumbar vertebra also demonstrated decreased bone density and sparse trabecula of the bones (Figure 1).

Radiographs and bone scans of a Chinese patient with familial hypocalciuric hypercalcemia. Radiographs of (a) orthophoria of the femur (postoperation), (b) lateral film of the femur (nonunion of fracture), and (c) lateral film of the lumbar vertebra showing osteoporosis. X-ray of the hands indicating epiphyseal broadening before (d) and after (e) epiphyseal closure. (f) Bone scan showing no specific foci of abnormal tracer deposition.
Medical imaging results showed that the bone mineral density (BMD) of the lumbar vertebra 1–4 was 0.820 g·m−2 (Z score, −1.3) and the BMD of the femoral neck was 0.460 g·m−2 (Z score, −2.7), whereas that of the total femur was 0.467 g·m−2 (Z score, −3.0). The anteroposterior delayed phase of the bone scan showed no specific foci of abnormal tracer deposition except on the right distal femur (Figure 1f).
One year later, the BMD of the spine at L1–4 had increased to 1.015 g·m−2 (Z score, 0.6), the BMD of the femoral neck was 0.599 g·m−2 (Z score, −2.2), and the BMD of the total femoral was 0.401 g·m−2 (Z score, 3.3). X-ray of the hands still indicated epiphyseal broadening even after the epiphyseal closure.
Mutation analysis
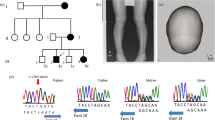
The targeted next-generation sequencing and direct sequencing of the coding regions of genetic hypophosphatemic rickets-related genes were normal. Both the next-generation sequencing and the Sanger sequencing data of amplified genomic DNA showed a heterozygous mutation on exon 7 of CaSR (c.2279T>A, p.I760N; NP_000379.2). The patient’s father was found to be a carrier but did not exhibit clinical symptoms of FHH. The sequences of the patient’s mother were normal (Figure 2a–c). We have excluded the single-nucleotide polymorphism reported in The National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/). We also performed direct sequencing of CaSR in 50 Chinese healthy controls, and 2279 T>A was not found in this group.

Genetic analysis of CaSR in the proband and his parents. (a) Mutation analysis revealed a missense mutation of exon 7 (c.2279T>A, p.I760N) on the CaSR in the proband and his father (b, c). Sequences of his mother were normal. (d) Prediction of the impact of p.I760N mutation on the structure and function of the CaSR protein using PolyPhen2.
Pathogenicity prediction
The bioinformatics tool PolyPhen (http://genetics.bwh.harvard.edu/pph) was used to predict the effects of the missense mutation on protein function. The HumVar score was 0.913, indicating a possibly damaging mutation (Figure 2d).
Discussion
We have described a case of FHH with a new heterozygous CaSR mutation, I760N, in a patient who did not exhibit any obvious clinical symptom until the age of 16 years when he developed a spontaneous fracture and adolescent rickets. Laboratory evaluation revealed an elevated serum ionized calcium level, a continuously high PTH level, and a significantly decreased 24-h urine calcium, which are consistent with FHH.
We found a new mutation in exon 7 of CaSR in this patient, which is located in an important region of the transmembrane domain (TMD) of CaSR, which contains seven trans-intracellular helices, three relatively short extracellular loops and three intervening intracellular loops. The mutation in this patient was located in the second loop of the TMD. This domain might have a role in the activation of cellular signaling through multiple downstream signaling pathways: Gq/11, Gi/o, and G12/13. These signal pathways are closely related to the pathogenesis of FHH,13 because the mutation of GNA11 causes FHH type 2.14 In this patient, mutation I760N was located in the second extracellular loop of the TMD, which contains multiple reported mutations (Figure 3). In particular, the reported mutation of I761del is actually equivalent to the mutation of I760del, because the amino acids located in 760 and 761 were both isoleucine. In other words, although I760N was a new mutation, I760del was a known mutation. Moreover, isoleucine was located in the main evagination of the domain and possessed a high conservatism during species evolution (Figure 3b). In light of this information, the isoleucine located in 760 was crucially important for the normal function of CaSR, although function experiments in this location was not performed.

Sites of known mutations and protein sequence alignment in the second extracellular loop of the model of CaSR. (a) Extracellular domain (ECD) of CaSR was shown in color diagram according to protein structure homology-modeling server (http://swissmodel.expasy.org/). The seven membrane spanning helices of CaSR are shown, based on hydropathy plot analysis (www.casrdb.mcgill.ca/). (b) Protein sequence alignment of the I760N mutation among different species, as given in the NCBI database.
In addition, Chang and Shoback et al reported a conditional deletion of exon 7 in the CaSR in osteoblasts, leading to severely under-mineralized matrix and osteoid accumulation. A conditional deletion of exon 7 in chrondrocytes produced a rickets-like phenotype with expansion and reduced mineralization of the hypertrophic zone and decreased abundance of mature markers. This result indicated that the mutation in exon 7 of CaSR may be functionally significant in the skeleton.15 Moreover, the bioinformatics tool PolyPhen indicated a possibly damaging mutation. In addition, I760N was not found in the CaSR genes in 100 healthy chromosomal controls, and was not reported as a single-nucleotide polymorphism.
Gene analysis has shown that the mutation location was related to the clinical manifestation. It has been suggested that mutation receptors of this type could more effectively downstream effects in the kidney than in the parathyroid glands, which produces a greater “resistance” to Ca2+o in the latter than in the former.16 However, little data have focused on the difference between skeletal and other manifestations, and one study reported that there was no relationship between plasma alkaline phosphatase, PTH, and regional BMD in FHH patients, and U-NTx/creatinine ratio was inversely correlated to the BMD of the forearm.13 This finding might indicate that the severity in different organs varies.
The role of CaSR in bone development in vivo has been controversial, although several studies have concluded that it mediates Ca2+o sensing in osteoblasts. In 2001, an unexpected skeletal phenotype of rickets was presented in CaSR-absent mice, indicating that calcium-sensing receptor was present in the skeleton and its absence resulted in the defective mineralization of cartilage and bone.1 However, the lack of apparent skeletal defects in global CaSR−/− and PTH−/− mice indicated a bone disorder resulted from the direct effects of PTH.17–19 Recent studies have focused on the function of CaSR in both the chondrocytes in growth plates and osteoblast-lineage cells. For growth plates, CaSR has been detected in maturing chondrocytes,19 and knockdown of CaSR impaired cell differentiation and matrix mineralization.2,20 For the osteoblast lineage, transgenic mice confirmed the biological significance of CaSR in bone development. After deleting the CaSR in early OBs, mice showed multiple skeletal fractures, soft bones, and large amounts of immature osteoid.2,19 In adulthood, their bones were severely osteoporotic,2,21,22 together with the increased expression of receptor activator of nuclear factor kappa-B ligand and decreased expression of osteoprotegerin. The symptom and bone turnover manifestation were consistent with our patient.23,24
However, few reports have discussed the skeletal performance of patients with a CaSR mutation, especially in patients with FHH. One case analyzed an older asymptomatic FHH patient who suffered from osteoporosis and fracture with subsequent nonunion,21 and whose presentation was consistent with the patient in this study. Other reports have described adolescent rickets of FHH patients, which is mainly attributed to elevated PTH or vitamin D deficiency.2,3 However, in this study the vitamin D concentration (13.7–18.6 ng·mL−1) was approximated to the mean 25(OH)D level in the Chinese population (19.4±6.4 ng·mL−1 and 13.2±5.4 ng·mL−1).25,26 The elevated PTH, attributed to both FHH and secondary hyperparathyroidism induced by low concentration of 25(OH)D, may contribute to the clinical manifestation of rickets.
In addition, other factors affect the development of rickets. Rickets is not limited to infancy or early childhood; but is also reported in adolescents. However, rickets occur during the period of rapid bone growth, with increased metabolic demands, it may develop long before any physical findings or radiological evidence are obtained.20,27,28 Hypocalcemic tetany and limb pain were the most common presenting symptoms in adolescents, whereas radiological evidence was not present in every case.29,30 The reported cases of FHH with clinical manifestations of rickets also occurred in adolescence, a period of rapid skeletal growth and mineralization.2,3 For this patient, the skeletal pain gradually appeared during his puberty, and his symptoms alleviated after the epiphyseal closure (Figure 1e). Furthermore, PTH directly affects bone mineralization, decreases mineralization in mature cells, and promotes bone turnover, which may lead to osteomalacia.13,31,32 There have been cases reported that primary hyperparathyroidism could masquerade as rickets, especially in childhood or in adolescence.33–35 The possible explanation is that atypical clinical manifestations of FHH may coexist with vitamin D deficiency, and might be further aggravated by elevated PTH levels. Moreover, there may be deleterious effect of continuously high level of PTH that acts directly on chondrocytes.
Conclusion
We have reported the case of a 16-year-old male with a heterozygous CaSR mutation, who developed skeletal abnormalities of spontaneous fracture with subsequent nonunion, and adolescent rickets. However, the skeletal abnormalities could not be simply explained by the known causes of a vitamin D deficiency or an increased PTH concentration. It has been suggested that rickets-like features may tend to present as uncommon characteristics in FHH patients with mild vitamin D deficiency, pubescent skeletal growth and epiphyseal closure, which may work to modulate the manifestation of rickets. There are some limitations in our study. We did not conduct a functional analysis of the mutant CaSR protein of the patient and could not provide a direct mechanism-based explanation of the relationship between CaSR and rickets. However, rickets and fractures have been rarely described in humans, although they have always been present in CaSR-absent mice. This case and other previous reports confirmed the clinical performance of rickets and fracture in FHH patients, and we hope to arouse concern about the symptoms of rickets or fractures in adolescent FHH patients with vitamin D deficiency, which might be ignored in clinical practice. Moreover, further studies are needed to determine the accurate function of CaSR in maintaining phosphate homeostasis and bone mineralization.
References
Garner SC, Pi M, Tu Q et al. Rickets in cation-sensing receptor-deficient mice: an unexpected skeletal phenotype. Endocrinology 2001; 142: 3996–4005.
Szczawinska D, Schnabel D, Letz S et al. A homozygous casr mutation causing a FHH phenotype completely masked by vitamin d deficiency presenting as rickets. J Clin Endocrinol Metab 2014; 99: E1146–E1153.
Zajickova K, Vrbikova J, Canaff L et al. Identification and functional characterization of a novel mutation in the calcium-sensing receptor gene in familial hypocalciuric hypercalcemia: modulation of clinical severity by vitamin d status. J Clin Endocrinol Metab 2007; 92: 2616–2623.
Hendy GN, Canaff L, Cole DE . The casr gene: alternative splicing and transcriptional control, and calcium-sensing receptor (CASR) protein: structure and ligand binding sites. Best Pract Res Clin Endocrinol Metab 2013; 27: 285–301.
Brennan SC, Mun HC, Leach K et al. Receptor expression modulates calcium-sensing receptor mediated intracellular ca2+ mobilization. Endocrinology 2015; 156: 1330–1342.
Conigrave AD, Ward DT . Calcium-sensing receptor (CASR): pharmacological properties and signaling pathways. Best Pract Res Clin Endocrinol Metab 2013; 27: 315–331.
Dong B, Endo I, Ohnishi Y et al. Calcilytic ameliorates abnormalities of mutant calcium-sensing receptor (CASR) knock-in mice mimicking autosomal dominant hypocalcemia (ADH). J Bone Miner Res 2015; 30: 1980–1993.
Finney B, Wilkinson WJ, Searchfield et al. An exon 5-less splice variant of the extracellular calcium-sensing receptor rescues absence of the full-length receptor in the developing mouse lung. Exp Lung Res 2011; 37: 269–278.
Asan, Xu Y, Jiang H et al. Comprehensive comparison of three commercial human whole-exome capture platforms. Genome Biol 2011; 12: R95.
Wei X, Ju X, Yi X et al. Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PLoS ONE 2011; 6: e29500.
Liu G, Wei X, Chen R et al. A novel mutation of the SLC25A13 gene in a Chinese patient with citrin deficiency detected by target next-generation sequencing. Gene 2014; 533: 547–533.
Richards CS, Bale S, Bellissimo DB et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions. Genet Med 2008; 10: 294–300.
Hendy GND, Souza-Li L, Yang B et al. Mutations of the calcium-sensing receptor (CASR) in familial hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mutat 2000; 16: 281–296.
Li D, Opas EE, Tuluc F et al. Autosomal dominant hypoparathyroidism caused by germline mutation in gna11: phenotypic and molecular characterization. J Clin Endocrinol Metab 2014; 99: E1774–E1783.
Chang W, Tu C, Chen TH et al. The extracellular calcium-sensing receptor (casr) is a critical modulator of skeletal development. Sci Signal 2008; 1: ra1.
Christensen SE, Nissen PH, Vestergaard P et al. Familial hypocalciuric hypercalcaemia: a review. Curr Opin Endocrinol Diabetes Obes 2011; 18: 359–370.
Christensen SE, Nissen PH, Vestergaard P et al. Skeletal consequences of familial hypocalciuric hypercalcaemia vs. Primary hyperparathyroidism. Clin Endocrinol 2009; 71: 798–807.
Liu J, Lv F, Sun W et al. The abnormal phenotypes of cartilage and bone in calcium-sensing receptor deficient mice are dependent on the actions of calcium, phosphorus, and pth. PLoS Genet 2011; 7: e1002294.
Tu Q, Pi M, Karsenty G et al. Rescue of the skeletal phenotype in casr-deficient mice by transfer onto the gcm2 null background. J Clin Invest 2003; 111: 1029–1037.
Sun W, Liu J, Zhou X et al. Alterations in phosphorus, calcium and pthrp contribute to defects in dental and dental alveolar bone formation in calcium-sensing receptor-deficient mice. Development 2010; 137: 985–992.
Unuvar T, Buyukgebiz A . Nutritional rickets and vitamin d deficiency in infants, children and adolescents. Pediatr Endocrinol Rev 2010; 7: 283–291.
Mohammed R, Bray MB, Koch CA et al. Spontaneous rib fractures in a black woman with familial hypocalciuric hypercalcemia. Med Sci Monit 2008; 14: CS102–CS106.
Takacs I, Speer G, Bajnok E et al. Lack of association between calcium-sensing receptor gene "a986s" polymorphism and bone mineral density in hungarian postmenopausal women. Bone 2002; 30: 849–852.
Hendy GN, Guarnieri V, Canaff L . Calcium-sensing receptor and associated diseases. Prog Mol Biol Transl Sci 2009; 89: 31–95.
Hannan FM, Thakker RV . Calcium-sensing receptor (CASR) mutations and disorders of calcium, electrolyte and water metabolism. Best Pract Res Clin Endocrinol Metab 2013; 27: 359–371.
Yu S, Fang H, Han J et al. The high prevalence of hypovitaminosis d in china: a multicenter vitamin d status survey. Medicine 2015; 94: e585.
Zhao J, Xia W, Nie M et al. The levels of bone turnover markers in chinese postmenopausal women: Peking vertebral fracture study. Menopause 2011; 18: 1237–1243.24.
Wagner CL, Greer FR . Prevention of rickets and vitamin d deficiency in infants, children, and adolescents. Pediatrics 2008; 122: 1142–1152.
Balasubramanian K, Rajeswari J, Gulab et al. Varying role of vitamin D deficiency in the etiology of rickets in young children vs. Adolescents in northern India. J Trop Pediatr 2003; 49: 201–206.
Narchi H, El Jamil M, Kulaylat N . Symptomatic rickets in adolescence. Arch Dis Child 2001; 84: 501–503.
Mahalingam CD, Sampathi BR, Sharma S et al. MKP1-dependent PTH modulation of bone matrix mineralization in female mice is osteoblast maturation stage specific and involves P-ERK and P-p38 MAPKs. J Endocrinol 2013; 216: 315–329.
Ueyama T, Yamamoto Y, Ueda K et al. Is gastrectomy-induced high turnover of bone with hyperosteoidosis and increase of mineralization a typical osteomalacia? PLoS ONE 2013; 8: e65685.
Dutta D, Kumar M, Das RN et al. Primary hyperparathyroidism masquerading as rickets: diagnostic challenge and treatment outcomes. J Clin Res Pediatr Endocrinol 2013; 5: 266–269.
Shah VN, Bhadada SK, Bhansali A et al. Influence of age and gender on presentation of symptomatic primary hyperparathyroidism. J Postgrad Med 2012; 58: 107–111.
Bhadada SK, Bhansali A, Dutta P et al. Characteristics of primary hyperparathyroidism in adolescents. J Pediatr Endocrinol Metab 2008; 21: 1147–1153.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (nos. 81070687 and 81170805), Beijing Natural Science Foundation (no. 7121012), Ministry of Science and Technology of the People’s Republic of China (National Science and Technology Major Projects for ‘Major New Drugs Innovation and Development 2008ZX09312-016), Scientific Research Foundation of Beijing Medical Development (no. 2007-3029), and National Key Program of Clinical Science (WBYZ2011-873).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wu, B., Wang, O., Jiang, Y. et al. Atypical skeletal manifestations of rickets in a familial hypocalciuric hypercalcemia patient. Bone Res 5, 17001 (2017). https://doi.org/10.1038/boneres.2017.1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/boneres.2017.1
