
Abstract
Background:
Cabazitaxel is approved for treatment of castration-resistant metastatic prostate cancer. The current dosing strategy of cabazitaxel is based on body surface area (BSA). Body surface area is known as a poor predictor for total systemic exposure to drugs, since it does not take into account variability in activity of metabolising enzymes, necessary for clearance of drugs. As exposure to cabazitaxel is related to treatment response, it is essential to develop a better individualised dosing strategy.
Methods:
Ten patients with metastatic castration-resistant prostate cancer, who received cabazitaxel dosed on BSA as a part of routine palliative care, were enrolled in this study. Midazolam was administered as phenotyping probe for cytochrome P450 isoenzyme 3A (CYP3A). The relationship between midazolam and cabazitaxel clearance was investigated using non-linear mixed effects modelling.
Results:
The clearance of Midazolam highly correlated with cabazitaxel clearance (R=0.74). Midazolam clearance significantly (P<0.004) explained the majority (∼60%) of the inter-individual variability in cabazitaxel clearance in the studied population.
Conclusions:
Metabolic phenotyping of CYP3A using midazolam is a promising strategy to individualise cabazitaxel dosing. Before clinical application, a randomised study is warranted.
Similar content being viewed by others

Main
Cabazitaxel is a novel drug from the class of taxanes that is approved for treatment of men with castration-resistant metastatic prostate cancer who have previously been treated with docetaxel (de Bono et al, 2010). Like other taxanes, cabazitaxel acts by binding to tubulin, and thereby stabilising the microtubules which leads to inhibition of the mitotic cellular functions (NDA, 2013). Cabazitaxel is 80–90% metabolised by the cytochrome P450 isoenzyme 3A (CYP3A), and a part of the remaining drug is metabolised by CYP2C8. Seventy-six per cent of the inactive metabolites are excreted via the faecal route and 4% by the renal system. Only a small fraction of the active drug, 2.3%, is found unchanged in the urine (NDA, 2013).
Treatment with cabazitaxel is often associated with considerable and dose-limiting side-effects, like neutropenia and diarrhoea. In daily practice, diarrhoea and neutropenia seem to be less pronounced than mentioned in the registration trial (Heidenreich et al, 2013; Nieuweboer et al, 2016). A post hoc analysis of the registration trial showed that the occurrence of neutropenia during treatment with cabazitaxel is associated with survival benefit (Meisel et al, 2016). Although this finding has not been confirmed prospectively, it does indicate that there is a fine balance between toxic and subtherapeutic exposure. This is supported by the finding that cabazitaxel-induced grade ⩾3 neutropenia is positively correlated with systemic exposure (NDA, 2013). Similar to other chemotherapeutics, cabazitaxel is dosed based on body surface area (BSA), while BSA only partly explains the variability in cabazitaxel clearance (NDA, 2013; Ferron et al, 2013). In 1958, Pinkel et al, proposed to use BSA as the criterion of dosage in chemotherapy (Pinkel, 1958). Although widely used, the appropriateness of BSA-based dosing for anticancer agents is under heavy debate (Anderson and Holford, 2009; Chatelut et al, 2012; Bins et al, 2014). For example, BSA is a poor predictor of clearance in obese and underweight patients (Anderson and Holford, 2008). Furthermore, variability in activity and expression of metabolising enzymes and drug transporters are only partially explained by body size (Undevia et al, 2005; Mathijssen et al, 2014). Therefore, other dose individualisation strategies with cytotoxic drugs have been employed, such as ‘therapeutic drug monitoring’ (TDM) (de Jonge et al, 2005), where dose adaptations are performed based on measurement of drug concentrations after administration of the drug. As early toxicity or subtherapy is unwanted, the inherent downside of therapeutic drug monitoring is that dose individualisation cannot take place until a therapeutic dose has been administered, and steady state has been established. An attractive alternative to individualise dosing of cytostatic drugs is by means of metabolic phenotyping (Mathijssen and van Schaik, 2006; Opdam et al, 2012): drug disposition of an anticancer drug can be predicted by studying the disposition of a relatively harmless probe drug. The pharmacokinetics of this phenotypic probe drug can then be used to individualise dosing from the first dose onwards. Metabolic phenotyping to individualised dosing has proven to be successful for different cytotoxic agents, including the taxane docetaxel (Opdam et al, 2012). The activity of the isoenzyme CYP3A is notoriously variable between individuals (de Wildt et al, 1999), and can be metabolically phenotyped with the probe drug midazolam (Oneda et al, 2009). Up to 90% of the metabolism of cabazitaxel is accounted for by CYP3A, with CYP2C8 likely to be responsible for the majority of the remaining metabolism (Ridoux et al, 2015). Furthermore, although it is known that cabazitaxel may be a substrate for efflux transporters from the ATP-binding cassette family, it is in a lesser extent than its structural analogue docetaxel, which may also partly explain why cabazitaxel is still efficacious in patients who have progressive disease after docetaxel treatment (Oprea-Lager et al, 2013; Duran et al, 2015; Kathawala et al, 2015; Tang et al, 2015).
As cabazitaxel is mainly metabolised by the hepatic CYP3A isoenzyme, metabolic phenotyping with midazolam could be a good strategy to improve individualised dosing. Therefore, we performed a proof-of-concept study to investigate the correlation of midazolam clearance with cabazitaxel clearance and to compare the performance of CYP3A metabolic phenotype and BSA to predict pharmacokinetic variability in cabazitaxel clearance, and thus systemic exposure.
Materials and methods
This proof-of-concept pharmacokinetic study was performed at Meander Medical Center in Amersfoort, the Netherlands. The study was approved by the medical ethics committee (MEC-U, Nieuwegein, The Netherlands) and all patients provided their informed consent. Ten men with metastasised castration-resistant prostate cancer, who were already scheduled to receive cabazitaxel as a part of routine clinical care, were included in the study. Cabazitaxel was administered at the approved dose of 25 mg m−2. Body surface area for each patient was calculated according to the Dubois and Dubois formula (du Bois and du Bois, 1916). For purposes of metabolic phenotyping, the patients received a flat dose of 2.5 mg as an intravenous shot of midazolam, followed by a shot of 5 ml of NaCl 0.9%, at 1-7 days prior to chemotherapy treatment. The intravenous route was chosen deliberately, as it bypasses first-pass metabolism and better resembles the metabolic fate of cabazitaxel. Just prior to the infusion of midazolam (time point 0) and at 30, 60, 120, 240 and 360 min after, blood samples were collected from a venous cannula in the opposite arm. On the day of cabazitaxel treatment, several blood samples were collected before (time point 0) and at 30, 60, 120, 240, 360, 600 min after infusion of cabazitaxel. In addition, a blood sample was collected 24 hours after the cabazitaxel infusion. For the determination of plasma drug concentrations, blood samples of 5 ml were obtained to collect at least 2 ml of plasma in a vacuum collection tube with EDTA as an anticoagulant. Immediately after blood collection, the blood plasma was separated from the erythrocytes by means of centrifugation and plasma was stored at −30 °C until further analysis.
Bioanalytics and pharmacokinetic analysis
Frozen plasma concentrations of cabazitaxel and midazolam were shipped to the laboratory of Translational Pharmacology (Rotterdam, The Netherlands) and quantified using validated liquid chromatography tandem mass spectrometry assays (de Bruijn et al, 2012). The limits of quantification for midazolam and cabazitaxel were 0.4 ng ml−1 and 1 ng ml−1, respectively. The inter- and intra-run precisions were less than 12.5% for midazolam and less than 9% for cabazitaxel. The accuracies for both assays were within 12% of the nominal concentrations.
Modelling was performed by means of non-linear mixed effects modelling, using the software program NONMEM V7.3.0, interfaced with Perl Speaks Nonmem, R and Xpose (Keizer et al, 2013). The first order conditional estimation method with interaction was used and parameter precision was obtained using the covariance step. Throughout model building, standard goodness of fitness plots and prediction-corrected visual predictive checks (Bergstrand et al, 2011) were explored to assess the developed model. First, two separate pharmacokinetic models for midazolam and cabazitaxel were developed that captured the inter-individual variability in clearance. Thereafter, the individual empirical Bayes estimates for midazolam clearance were investigated as covariate for cabazitaxel clearance.
Midazolam clearance was investigated as a continuous covariate for cabazitaxel clearance using linear, power and exponential functions, as described previously (Joerger, 2012).
Midazolam clearance was considered for inclusion in the final model if the model fit improved significantly, and if it reduced the unexplained inter-individual variability in cabazitaxel clearance. In our analysis, P<0.05 was considered significant, corresponding with a reduction in objective function of 3.84 points or more as a critical χ2-value.
Results
Patient characteristics
Ten patients with metastatic castration-resistant prostate carcinoma treated with cabazitaxel were included in the study. All patients received prior treatment with docetaxel (Table 1). The median age of the participants was 67 (range 65–77) years. Median BSA of the patients was 1.95 m2 (range 1.76–2.34 m2). Median dose of administered cabazitaxel was 46.25 mg (range 38–50 mg). Hepatic function as measured with bilirubin was not decreased in any of the patients. During treatment, in 50% of the patients a decrease in PSA with 50% was seen. Also, 50% of the patients reported diarrhoea during treatment. None of these patients had to be admitted to the hospital due to diarrhoea, nor needed the dose to be adjusted. One patient had to be admitted due to neutropenic fever. None of the patients had to stop treatment because of toxicity.
Pharmacokinetic modeling
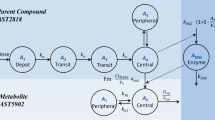
Midazolam pharmacokinetics were highly variable and best described with a first order one compartment linear pharmacokinetic model (Figure 1). The model parameter estimates are presented in Table 2A. Inter-individual variability in midazolam clearance and volume of distribution were high at 73.2% and 77.3%, respectively. Standard goodness of fit plots of the pharmacokinetic model for midazolam and the prediction-corrected visual predictive check are provided as Supplementary Files with this manuscript (Supplementary Figures 1–3). Cabazitaxel pharmacokinetics were best described with a linear two compartment pharmacokinetic model (see Figure 1 and Table 2B). Unexplained inter-individual variability in clearance of cabazitaxel was moderate at 24.8%. The limited eta-shrinkage (<10%) for midazolam and cabazitaxel clearance showed that data were informative to describe variability in these parameters (Savic and Karlsson, 2009).

Pharmacokinetic models for midazolam and cabazitaxel.Schematic representation of the pharmacokinetic models for midazolam and cabazitaxel. CLbase=clearance of cabazitaxel in the base model, CLCBZ=clearance of cabazitaxel, CLmdz=clearance of midazolam, CLmdz,i=clearance of midazolam individual, CLmdz,pop=Clearance of midazolam population, Q=intercompartmental clearance, V1=volume of distribution of the central compartment of cabazitaxel, V2=Volume of distribution of the peripheral compartment of cabazitaxel, V3=Volume of distribution of the central compartment of midazolam, θclearance=gradient of the correlation between the clearance of midazolam and cabazitaxel.
Upon visual inspection of the empirical Bayes estimates of cabazitaxel clearance vs midazolam clearance (Figure 2), a high correlation could be observed. When midazolam clearance was tested as a covariate for cabazitaxel clearance using linear, exponential and power functions, a linear function (Equation (1)) best described the relationship between midazolam clearance and cabazitaxel clearance: approximately 60% of the unexplained inter-individual variability of cabazitaxel clearance was explained, and it reduced significantly (P=0.004) from 24.8 to 10.2%. The parameter estimates for the cabazitaxel pharmacokinetic model, including midazolam clearance as a linear covariate, are presented in Table 2B. As observed, the shrinkage in cabazitaxel clearance increased, confirming that midazolam clearance accounted for the majority of the observed variability in cabazitaxel clearance.

Relationship between individual estimates for clearance of midazolam and cabazitaxel.Empirical Bayes estimates for midazolam and cabazitaxel clearance (black dots) and their model-predicted relationship (dotted line) from the final model.

In this Equation, CL is the cabazitaxel clearance (l h−1), CLbase is the typical value for cabazitaxel clearance (119 l h−1) in an individual with a midazolam clearance (CLmdz,i) that equals the population value for midazolam clearance (CLmdz,pop) of 26 l h−1 and θclearance is the gradient of 1.71 that describes the relationship between midazolam and cabazitaxel clearance.
Figure 2 shows the model-derived relationship between the empirical Bayes estimates for midazolam and cabazitaxel clearance from the base models. As observed, midazolam clearance highly correlated with cabazitaxel clearance (R=0.74). Midazolam clearance was therefore retained in the final model as a covariate for cabazitaxel clearance. Figure 3 shows the prediction-corrected visual predictive check for the final model, including individual midazolam clearance as covariate for cabazitaxel clearance. As observed in this figure, the observed data correspond well with the simulated data (based on 1000 simulations), showing the internal validity of our model.

Prediction-corrected visual predictive check of cabazitaxel final model.The prediction-corrected simulated (shaded areas) and observed (circles and lines) cabazitaxel concentrations vs time (h), based on 1000 simulations are shown. The thick black line connects the observed median values per bin. The dotted grey lines connect the 5th and 95th percentiles of the observations. The light grey areas are the 95% confidence interval of the 5th and 95th percentiles. The dark grey area indicates the confidence interval of the median.
Discussion
The variability in pharmacokinetics of both midazolam and cabazitaxel are greatly explained by variability in the CYP3A enzymatic activity. Since both drugs act as substrates for CYP3A, we hypothesised that the clearance of cabazitaxel could be predicted by the clearance of midazolam. In cancer patients it is known that CYP3A4 activity varies about four-fold between patients (Lepper et al, 2005; Mathijssen et al, 2014), based on genetic and environmental factors. In our opinion individualised dosing strategies for drugs metabolised through this phase I enzyme should take this variation into account.
In our proof-of-concept study, we showed that midazolam clearance and cabazitaxel clearance were highly correlated. This indicates that midazolam can be a useful phenotypic probe to individualise cabazitaxel dosing. The unexplained variability of cabazitaxel clearance in our population was in the same order of magnitude as found in previous large population pharmacokinetic study (Ferron et al, 2013). In this study BSA and cancer type together accounted for only a small part (19%) of the observed inter-individual variability in cabazitaxel clearance. As metabolic phenotype appears to explain a much larger part of the inter-individual variability in cabazitaxel clearance, this suggests that midazolam has the potential to better individualise cabazitaxel clearance than BSA. As observed in Figure 2, the intercept with the Y-axis of our model-derived relationship suggests clearance of cabazitaxel when midazolam clearance is 0. Although the exact reason for this phenomenon remains uninvestigated, it may be partly accounted for by the fact that a small portion (<10%) of cabazitaxel is metabolised by the enzyme CYP2C8 (NDA, 2013; Ridoux et al, 2015), an enzyme that is not involved in midazolam metabolism.
Our findings are in line with previous studies that showed that midazolam clearance predicts the clearance of different anticancer drugs that share a metabolic pathway with midazolam (Goh et al, 2002; Mathijssen et al, 2004; de Wit et al, 2014), although the correlation between midazolam and these anticancer agents studied in these previous studies was weaker compared to the current study. Potentially, the role of CYP3A in cabazitaxel metabolism is of larger importance compared to the other compounds studied (i.e., irinotecan, docetaxel and sunitinib). Although the latter has been shown for several different chemotherapeutics (Mathijssen and van Schaik, 2006), dosing strategies based on BSA are still used daily in clinical practice. This has to do with a lack of confirming studies where the use of phenotype-based dosing strategy actually leads to better clinical results. However, although also a logistical challenge, therapeutic drug monitoring-based dosing strategy for cabazitaxel is currently tested in a prospective randomised phase II trial (CAINTA trial, EudraCT number: 2013-005504-34).
A limitation of our study was that the number of studied patients was small. For implementation of a metabolic phenotype-based dosing strategy, therefore, some hurdles have to be overcome. First, to obtain a more reliable estimate of the relationship between metabolic phenotype and cabazitaxel pharmacokinetics, the relationship should be studied in a larger population. Second, since sampling a full pharmacokinetic curve of midazolam to establish the metabolic phenotype is not very feasible in routine clinical practice, a limited sampling strategy should be developed, as previously proposed (Katzenmaier et al, 2010). Lastly, a prospective study comparing routine BSA-based dosing vs metabolic phenotype-based dosing should be performed on clinical and pharmacokinetic end points.
Neutropenia⩾grade 3 was the most seen severe complication in the registration trial of cabazitaxel, with an incidence of 82% (de Bono et al, 2010). Although later performed safety studies showed drastically lower incidence numbers of neutropenia (7.2–33.9%), neutropenia was still the most frequent severe complication observed (Heidenreich et al, 2013; Bracarda et al, 2014). The occurrence of severe neutropenia is mentioned to correlate with the AUC of cabazitaxel (NDA, 2013). A post hoc analysis of the registration trial showed that the occurrence of grade⩾3 neutropenia was associated with an increased overall survival and progression free post hoc survival (Meisel et al, 2016). In this group of patients with grade⩾3 neutropenia, a greater decrease in PSA levels was also observed, compared with the patients with milder or no neutropenia (Meisel et al, 2016). This phenomenon, where the occurrence of neutropenia is a positive predictor for survival, has been described for various solid and haematological malignancies (Brosteanu et al, 2004; Di Maio et al, 2005), and is an indirect marker for the pharmacokinetic-pharmacodynamic relationship of cytotoxic agents. Because the results of those post hoc analyses have not been prospectively confirmed, and since neutropenia can also be harmful for the patient, these results should be interpreted with great caution. However, these studies indirectly show that relatively high exposure might lead to toxicity, such as neutropenia, but this high exposure is on the other hand also probably correlated with survival. Therefore, prevention of subtherapeutic exposure seems to be of utmost importance. Phenotypically guided dosing strategies may decrease the number of patients who are underdosed with the current BSA drug dosing paradigm.
In our proof-of-concept study, we have shown that midazolam-based metabolic phenotyping may be a valuable tool to individualise cabazitaxel dosing. Besides using metabolic phenotyping to predict the correct starting dose, other strategies like toxicity and drug level monitoring to guide following doses may be employed to further tailor cabazitaxel therapy. If future trials support the hypothesis that phenotype-based dosing increases outcome and decreases side-effects there will be a real alternative for the long used BSA dosing strategy.
Change history
09 May 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Anderson BJ, Holford NH (2008) Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol 48: 303–332.
Anderson BJ, Holford NH (2009) Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 24: 25–36.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO (2011) Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J 13: 143–151.
Bins S, Ratain MJ, Mathijssen RH (2014) Conventional dosing of anticancer agents: precisely wrong or just inaccurate? Clin Pharmacol Ther 95: 361–364.
Bracarda S, Gernone A, Gasparro D, Marchetti P, Ronzoni M, Bortolus R, Fratino L, Basso U, Mazzanti R, Messina C, Tucci M, Boccardo F, Carteni G, Pinto C, Fornarini G, Mattioli R, Procopio G, Chiuri V, Scotto T, Dondi D, Di Lorenzo G (2014) Real-world cabazitaxel safety: the Italian early-access program in metastatic castration-resistant prostate cancer. Future Oncol 10: 975–983.
Brosteanu O, Hasenclever D, Loeffler M, Diehl V Group German Hodgkin's Lymphoma Study (2004) Low acute hematological toxicity during chemotherapy predicts reduced disease control in advanced Hodgkin's disease. Ann Hematol 83: 176–182.
Chatelut E, White-Koning ML, Mathijssen RH, Puisset F, Baker SD, Sparreboom A (2012) Dose banding as an alternative to body surface area-based dosing of chemotherapeutic agents. Br J Cancer 107: 1100–1106.
de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L, Roessner M, Gupta S, Sartor AO Tropic Investigators (2010) Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 376: 1147–1154.
de Bruijn P, de Graan AJ, Nieuweboer A, Mathijssen RH, Lam MH, de Wit R, Wiemer EA, Loos WJ (2012) Quantification of cabazitaxel in human plasma by liquid chromatography/triple-quadrupole mass spectrometry: a practical solution for non-specific binding. J Pharm Biomed Anal 59: 117–122.
de Jonge ME, Huitema AD, Schellens JH, Rodenhuis S, Beijnen JH (2005) Individualised cancer chemotherapy: strategies and performance of prospective studies on therapeutic drug monitoring with dose adaptation: a review. Clin Pharmacokinet 44: 147–173.
de Wildt SN, Kearns GL, Leeder JS, van den Anker JN (1999) Cytochrome P450 3A: ontogeny and drug disposition. Clin Pharmacokinet 37: 485–505.
de Wit D, Gelderblom H, Sparreboom A, den Hartigh J, den Hollander M, Konig-Quartel JM, Hessing T, Guchelaar HJ, van Erp NP (2014) Midazolam as a phenotyping probe to predict sunitinib exposure in patients with cancer. Cancer Chemother Pharmacol 73: 87–96.
Di Maio M, Gridelli C, Gallo C, Shepherd F, Piantedosi FV, Cigolari S, Manzione L, Illiano A, Barbera S, Robbiati SF, Frontini L, Piazza E, Ianniello GP, Veltri E, Castiglione F, Rosetti F, Gebbia V, Seymour L, Chiodini P, Perrone F (2005) Chemotherapy-induced neutropenia and treatment efficacy in advanced non-small-cell lung cancer: a pooled analysis of three randomised trials. Lancet Oncol 6: 669–677.
du Bois D, du Bois EF (1916) Clinical calorimetry: tenth paper a formula to estimate the approximate surface area if height and weight be known. Arch Intern Med XVII: 863–871.
Duran GE, Wang YC, Francisco EB, Rose JC, Martinez FJ, Coller J, Brassard D, Vrignaud P, Sikic BI (2015) Mechanisms of resistance to cabazitaxel. Mol Cancer Ther 14: 193–201.
Ferron GM, Dai Y, Semiond D (2013) Population pharmacokinetics of cabazitaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol 71: 681–692.
Goh BC, Lee SC, Wang LZ, Fan L, Guo JY, Lamba J, Schuetz E, Lim R, Lim HL, Ong AB, Lee HS (2002) Explaining interindividual variability of docetaxel pharmacokinetics and pharmacodynamics in Asians through phenotyping and genotyping strategies. J Clin Oncol 20: 3683–3690.
Heidenreich A, Scholz HJ, Rogenhofer S, Arsov C, Retz M, Muller SC, Albers P, Gschwend J, Wirth M, Steiner U, Miller K, Heinrich E, Trojan L, Volkmer B, Honecker F, Bokemeyer C, Keck B, Otremba B, Ecstein-Fraisse E, Pfister D (2013) Cabazitaxel plus prednisone for metastatic castration-resistant prostate cancer progressing after docetaxel: results from the German compassionate-use programme. Eur Urol 63: 977–982.
Joerger M (2012) Covariate pharmacokinetic model building in oncology and its potential clinical relevance. AAPS J 14: 119–132.
Kathawala RJ, Wang YJ, Shukla S, Zhang YK, Alqahtani S, Kaddoumi A, Ambudkar SV, Ashby CR Jr, Chen ZS (2015) ATP-binding cassette subfamily B member 1 (ABCB1) and subfamily C member 10 (ABCC10) are not primary resistance factors for cabazitaxel. Chin J Cancer 34: 115–120.
Katzenmaier S, Markert C, Mikus G (2010) Proposal of a new limited sampling strategy to predict CYP3A activity using a partial AUC of midazolam. Eur J Clin Pharmacol 66: 1137–1141.
Keizer RJ, Karlsson MO, Hooker A (2013) Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol 2: e50.
Lepper ER, Baker SD, Permenter M, Ries N, van Schaik RH, Schenk PW, Price DK, Ahn D, Smith NF, Cusatis G, Ingersoll RG, Bates SE, Mathijssen RH, Verweij J, Figg WD, Sparreboom A (2005) Effect of common CYP3A4 and CYP3A5 variants on the pharmacokinetics of the cytochrome P450 3A phenotyping probe midazolam in cancer patients. Clin Cancer Res 11: 7398–7404.
Mathijssen RH, de Jong FA, van Schaik RH, Lepper ER, Friberg LE, Rietveld T, de Bruijn P, Graveland WJ, Figg WD, Verweij J, Sparreboom A (2004) Prediction of irinotecan pharmacokinetics by use of cytochrome P450 3A4 phenotyping probes. J Natl Cancer Inst 96: 1585–1592.
Mathijssen RH, Sparreboom A, Verweij J (2014) Determining the optimal dose in the development of anticancer agents. Nat Rev Clin Oncol 11: 272–281.
Mathijssen RH, van Schaik RH (2006) Genotyping and phenotyping cytochrome P450: perspectives for cancer treatment. Eur J Cancer 42: 141–148.
Meisel A, von Felten S, Vogt DR, Liewen H, de Wit R, de Bono J, Sartor O, Stenner-Liewen F (2016) Severe neutropenia during cabazitaxel treatment is associated with survival benefit in men with metastatic castration-resistant prostate cancer (mCRPC): A post-hoc analysis of the TROPIC phase III trial. Eur J Cancer 56: 93–100.
NDA (2013) 201023 Review http://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/201023s000ClinPharmR.pdf.
Nieuweboer AJ, de Graan AM, Hamberg P, Bins S, van Soest RJ, van Alphen RJ, Bergman AM, Beeker A, van Halteren H, Ten Tije A, Zuetenhorst H, van der Meer N, Chitu D, de Wit R, Mathijssen RH (2016) Effects of budesonide on cabazitaxel pharmacokinetics and cabazitaxel-induced diarrhea: a randomized, open-label multicenter phase II study. Clin Cancer Res e-pub ahead of print 4 October 2016; doi:10.1158/1078-0432.CCR-16-2110.
Oneda B, Crettol S, Jaquenoud Sirot E, Bochud M, Ansermot N, Eap CB (2009) The P450 oxidoreductase genotype is associated with CYP3A activity in vivo as measured by the midazolam phenotyping test. Pharmacogenet Genomics 19: 877–883.
Opdam FL, Gelderblom H, Guchelaar HJ (2012) Phenotyping drug disposition in oncology. Cancer Treat Rev 38: 715–725.
Oprea-Lager DE, Bijnsdorp IV, Moorselaar VAN, Van Den Eertwegh RJ, AJ, Hoekstra OS, Geldof AA (2013) ABCC4 decreases docetaxel and not cabazitaxel efficacy in prostate cancer cells in vitro. Anticancer Res 33: 387–391.
Pinkel D (1958) The use of body surface area as a criterion of drug dosage in cancer chemotherapy. Cancer Res 18: 853–856.
Ridoux L, Semiond DR, Vincent C, Fontaine H, Mauriac C, Sanderink GJ, Oprea C, Kelly L, Clive S (2015) A phase I open-label study investigating the disposition of [14C]-cabazitaxel in patients with advanced solid tumors. Anticancer Drugs 26: 350–358.
Savic RM, Karlsson MO (2009) Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J 11: 558–569.
Tang SC, Kort A, Cheung KL, Rosing H, Fukami T, Durmus S, Wagenaar E, Hendrikx JJ, Nakajima M, van Vlijmen BJ, Beijnen JH, Schinkel AH (2015) P-glycoprotein, CYP3A, and plasma carboxylesterase determine brain disposition and oral availability of the novel taxane cabazitaxel (Jevtana) in mice. Mol Pharm 12: 3714–3723.
Undevia SD, Gomez-Abuin G, Ratain MJ (2005) Pharmacokinetic variability of anticancer agents. Nat Rev Cancer 5: 447–458.
Acknowledgements
This project was supported by a donation of Sanofi and a donation of Tele2 (a telecom company in The Netherlands).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
RHJ Mathijssen receives funding by Sanofi for research projects. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Janssen, A., Verkleij, C., van der Vlist, A. et al. Towards better dose individualisation: metabolic phenotyping to predict cabazitaxel pharmacokinetics in men with prostate cancer. Br J Cancer 116, 1312–1317 (2017). https://doi.org/10.1038/bjc.2017.91
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.91
Keywords
This article is cited by
-
The influence of single-nucleotide polymorphisms on overall survival and toxicity in cabazitaxel-treated patients with metastatic castration-resistant prostate cancer
Cancer Chemotherapy and Pharmacology (2020)
