
Abstract
Background:
Tumour-derived exosomes (TEXs) have a potential for application in cancer vaccines. Whether TEXs after induction by interferon regulatory factor 1 (IRF-1) are capable of enhancing the antitumour response remains to be determined.
Methods:
Exosomes released by tumour cells infected with IRF-1-expressing adenovirus (IRF-1-Exo) or treated with interferon-γ (IFN-Exo) were isolated via ultracentrifugation. The IRF-1 target proteins IL-15Rα and MHC class I (MHC-I) were analysed by western blot. Exosomes along with CpG adjuvant were injected into tumour models to assess the antitumour effects. Tumours were harvested for immunofluorescence staining. Splenocytes from tumour-bearing mice were co-cultured with tumour cells. The IFNγ-positive and granzyme B-positive CD8α+ splenocyte cells were quantified by flow cytometry.
Results:
The IRF-1-Exo or IFN-Exo displayed increased IL-15Rα and MHC-I expression. Injection of IRF-1-Exo or IFN-Exo combined with CpG had improved antitumour effects in mice. This effect may be a result of increased infiltration of tumours by CD4+ and CD8α+ T cells. Antibody-mediated depletion of CD4+ or CD8+ T cells abrogated the antitumour effects. Splenocytes isolated from CpG+IRF-1-Exo-injected Hepa 1–6 tumour mice had increased IFNγ-positive and granzyme B-positive CD8+ cells after co-culturing with Hepa 1–6 cells as compared with MC38 cells.
Conclusions:
The IRF-1 priming of TEXs enhances antitumour immune response.
Similar content being viewed by others

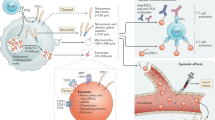

Main
Tumour vaccine therapy promotes a tumour-specific T-cell immune response as a result of tumour-associated antigens (TAAs) as well as immune-stimulatory signals. However, TAAs differ between tumour types, and more importantly between patients (Escors, 2014), and this is a primary hurdle for the use of tumour vaccine therapy. Therefore, collection of personalised TAAs from each tumour sample is required in order to develop effective individualised therapies. Tumour-released extracellular vesicles (EVs) carry not only TAAs, but also immunoregulatory factors that can be used as an additional strategy for the development of personalised tumour vaccines (Zhang et al, 2015).
Exosomes are 50–100 nm EVs that are released from several types of cells (Stoorvogel et al, 2002), and are generated by the fusion of multiple vesicular bodies (MVBs) with the cell membrane (Sahoo et al, 2011). Exosomes contain lipids, proteins, RNA, and DNA, and carry information that identifies their cellular origin (O'Brien et al, 2013). They can present antigens (Martin et al, 2014), and also regulate immune responses through their surface proteins, including major histocompatibility complex (MHC) class I (MHC-I), MHC class II (MHC-II), CD86, interleukin 15 receptor subunit-α (IL15Rα), and heat shock protein 70 (HSP70) (Viaud et al, 2009; Robbins and Morelli, 2014). Tumour- derived exosomes (TEXs) can resemble their host cells with expression of tumour-associated antigens such as CEA, HER-2/neu, gp100, EGFR2, methoselin, as well as HSP family members including HSP90 and HSP70 (Andre et al, 2002; Dai et al, 2005; Gastpar et al, 2005; Clayton et al, 2008).
The effects of TEXs in tumour immunity are controversial. Some studies have shown that TEXs can suppress T-cell activity (Andreola et al, 2002), inhibit IL-2 induced T-cell proliferation, promote regulatory T cell (Treg) cell function (Clayton et al, 2007), or induce production of myeloid-derived suppressor cells (Xiang et al, 2009). In contrast, other studies have shown that TEXs have antitumour effects (Dai et al, 2005; Cho et al, 2009; Xie et al, 2010). These data indicate that TEXs may differentially regulate tumour immunity and, furthermore, that TEXs have the potential to be engineered in order to enhance antitumour effects.
Interferon regulatory factor 1 (IRF-1) is a transcription factor that can regulate expression of target genes such as MHC-I (Taniguchi et al, 2001), IL-15Rα (Ogasawara et al, 1998), and interferon-α (IFNα) or IFNβ (Harada et al, 1990; Matsuyama et al, 1993). By regulating expression of these target genes, IRF-1 plays an important role in antitumour (Connett et al, 2003; Stang et al, 2007; Armstrong et al, 2015) and antiviral immunity, immune system development, and inflammation. The IRF-1 has been shown to be a tumour suppressor gene, and cancer patients with tumours expressing IRF-1 have improved immune responsiveness (Wang et al, 2002; Galon et al, 2006) and survival (Zeimet et al, 2009; Sakai et al, 2014). In a murine tumour model, irradiated mice exhibit compromised tumour growth inhibition in AdIRF-1-infected tumours. Therefore, IRF-1 has the potential to not only decrease the malignant phenotype of a tumour, but also increase tumour recognition (Yim et al, 1997).
As IRF-1 promotes antitumour immunity, and TEXs contain potential IRF-1 target gene products (IL-15Rα and MHC-I), we hypothesised that TEXs can be primed with IRF-1 to enhance the antitumour effects. Indeed, our results show that, following transduction with IRF-1, TEXs elicit tumour-specific antitumour immunity.
Materials and methods
Cell lines and reagents
The Hepa 1–6 mouse hepatic cancer cell line (H-2b) was purchased from ATCC (Manassas, VA, USA). The MC38 mouse colorectal cancer cell line (H-2b) was a kind gift from Dr Michael Lotze (University of Pittsburgh, Pittsburgh, PA, USA). To derive exosomes from the two cell lines, exosome-free foetal bovine serum (exo-free FBS) was generated by centrifugation at 100 000 g for 16 h. Both cell lines were maintained in DMEM media with 10% FBS (growth media) or 10% exo-free FBS (exosome derivation media). Anti-MHC Class I (Abcam, Cambridge, MA, USA), IL15Rα (R&D, Minneapolis, MN, USA), CD63 (Santa Cruz, Dallas, TX, USA), β-actin (CST, Beverly, MA, USA), and Lamin A/C (CST) antibodies were used for western blot. Alexa Fluor 488-conjugated anti-mouse CD4 and Alexa Fluor 647-conjugated anti-mouse CD8α antibodies were purchased from Biolegend (San Diego, CA, USA). The PE-eFluor 610 anti-mouse granzyme B antibody was purchased from eBioscience (San Diego, CA, USA) and PE anti-mouse IFNγ antibody was purchased from BD Bioscience (San Jose, CA, USA). The adenoviral IRF-1 expression construct (AdIRF-1) was created by inserting a mouse IRF-1 cDNA into E1- and E3-deleted adenovirus, as previously described (Kim et al, 2009). The adenoviral β-galactosidase (AdLacZ) expression construct with the same vector backbone was used as a control. The AdIRF-1 and AdLacZ viruses were amplified at the University of Pittsburgh core laboratory. CpG oligonucleotides (5′-TCCATGACGTCCCTGACGTT-3′) were synthesised from Life Technologies (San Diego, CA, USA).
Exosomes isolated from tumour cell culture supernatant
The Hepa 1–6 and MC38 cells were cultured in growth medium for 24 h, and then infected with AdIRF-1, AdLacZ (50 MOI), or an equivalent volume of PBS as a control. Cells were cultured for another 24 h in growth medium, changed to exosome derivation media, and cultured for an additional 48 h. The cultured media were collected for exosome isolation. Cells were collected at time points of 24, 36, and 48 h post infection.
Exosomes were isolated from media as previously described (Valadi et al, 2007). Briefly, cell culture medium was sequentially centrifuged at 500 g for 10 min, 16 500 g for 20 min, and then filtered using a 0.22 μm filter (Millipore, Burlington, MA, USA). Exosomes were pelleted at 100 000 g for 70 min using a SWi48 rotor. The pellets were washed twice with 9 ml PBS and centrifuged at 100 000 g for 70 min. The exosome pellets were then suspended in a small volume of PBS for cell treatment or mouse injection. Alternatively, exosome pellets were lysed with buffer (0.5% Triton X-100, 0.25% CHAPS, 150 mM NaCl, 100 mM Tris-HCl (pH 8.0)) containing a complete protease inhibitor mixture (Roche, Basel, Switzerland), and detected using a BCA assay (Pierce, Thermo Scientific, San Diego, CA, USA) or western blot. Nuclear proteins were extracted from Hepa 1–6 cells, as previously described (Klune et al, 2012).
Exosome purification with gradient iodixanol ultracentrifugation
Exosomes were enriched in 8.4–12% iodixanol (Cantin et al, 2008), with adenovirus located between 25% and 40% of iodixanol under ultracentrifugation (Peng et al, 2006). Potential adenovirus contamination was eliminated with gradient iodixanol ultracentrifugation. The protocol is as follows: 2 ml of 40%, 25%, and 15% iodixanol in PBS was added in Ultra-Clear tubes (Beckman Coulter, Brea, CA, USA) sequentially, and then 2 ml of exosome pellets in PBS was gently added on top of the iodixanol. After centrifugation at 35 600 r.p.m. for 70 min using an SW41 Ti bucket, the top 4 ml of the solution was collected and washed three times with PBS under 100 000 g for 70 min to pellet exosomes.
Transmission electron microscopy (TEM)
Isolated exosomes were verified with TEM, as previously described (Kim et al, 2007). Briefly, 1–5 μl of exosome samples in PBS was spread evenly onto formvar-coated grids. The excess liquid was absorbed, a drop of 1% uranyl acetate was added for staining, excess liquid was absorbed again, and exosomes were viewed on a JEM-1011TEM (Center for Biologic Imaging, University of Pittsburgh).
Exosome nanosight analysis (NTA)
The exosome samples were analysed, as previously described (Ouyang et al, 2016). Briefly, exosome samples in PBS (protein concentration of ∼1 μg μl−1, as measured by BCA assay) were diluted 1 : 500 in particle-free water (W4502, Sigma, St Louis, MO, USA). After vortexing, the diluted samples were injected into the NTA LM-10 system continuously using a syringe pump. Particles were acquired by the machine and data were analysed with NTA particle analysis software. Exosome particles were normalised to protein concentration.
Mouse tumour model and experimental protocol
The University of Pittsburgh Institutional Animal Care and Use Committee (IACUC) approved all murine experiments. C57BL/6J (H-2b) female mice (4–6 weeks old) from Jackson Labs (Bar Harbor, ME, USA) were used, and 1 × 107 Hepa 1–6 cells or 1 × 106 cells MC38 cells suspended in 100 μl DMEM were injected into the right flank of each mouse. Tumour size was calculated every 3 days using calipers (Zeelenberg et al, 2008). Tumour size=L × W × ((L+W)/2). At 6 days after Hepa 1–6 cell and MC 38 cell injection, mice were injected with exosomes or other reagents directly into tumour (i.t.). Mice received a total of four doses in each group, every 3 days. Experimental groups were designed as follows:
-
1)
PBS group, 0.1 ml PBS was injected for each mouse.
-
2)
CpG group, 0.2 μg ml−1 CpG in 0.1 ml PBS was injected.
-
3)
CpG+blank (naive) Exo group (CpG+BLK-Exo)=CpG (0.2 μg ml−1) and exosomes isolated from tumour cells without adenovirus infection (10 μg ml−1) in 0.1 ml PBS were injected.
-
4)
CpG+LacZ-Exo group (CpG+LacZ-Exo)=CpG (0.2 μg ml−1) and exosomes isolated from tumour cells after AdLacZ infection (10 μg ml−1) in 0.1 ml PBS were injected,
-
5)
CpG+IRF-1-Exo group (CpG+IRF-1-Exo)=CpG (0.2 μg ml−1) and exosomes isolated from tumour cells after AdIRF-1 infection (10 μg ml−1) in 0.1 ml PBS were injected.
-
6)
CpG+Boiled-IRF-1-Exo group (CpG+Boiled-IRF-1-Exo)=CpG (0.2 μg ml−1) and exosomes isolated from tumour cells after AdIRF-1 infection (10 μg ml−1) and boiled for 10 min in 0.1 ml PBS were injected.
-
7)
CpG+IFN-Exo group (CpG+IFN-exo)=CpG (0.2 μg ml−1) and exosomes (10 μg ml−1), which were isolated from tumour cells after 48 h IFNγ (250 U ml−1, R&D) treatment, were injected in 0.1 ml PBS.
-
8)
CpG+cell lysis group (CpG+IFN-cell lysis): Hepa 1–6 or MC38 cells were treated with IFNγ (250 U ml−1) for 6 h, and then harvested in PBS, and repeatedly freeze/thawed five times. The CpG (0.2 μg ml−1) and cell lysate (10 μg ml−1) in 0.1 ml PBS were injected.
-
9)
CpG+iodixanol purified IRF-1-Exo group (CpG+pIRF-1-Exo)=CpG (0.2 μg ml−1) and iodixanol purified exosomes isolated from tumour cells after AdIRF-1 infection (10 μg ml−1) in 0.1 ml PBS were injected.
IFNγ detection in exosome pellets
The IFNγ was detected in the exosomes, as previously described, with slight modification (Konadu et al, 2016). Briefly, exosome pellets were resuspended in 1% Triton X-100 in PBS to cause exosome lysis. The IFNγ was detected in the lysates using a BD CBA soluble protein kit, according to the manufacturer’s protocol. The concentrations of standards varied from 500 to 2 pg ml−1. The concentration of IFNγ in exosomes was normalised to exosome proteins.
Depletion of CD4+ and CD8+ T cells in mice using antibodies
The CD4+ and CD8+ T cells were depleted in mice, as described previously (Xu et al, 1996). Briefly, 100 μg GK1.5 (anti-CD4 mAb) or YTS 169.4 (anti-CD8 mAb) was injected intraperitoneally beginning on day 3 of tumour inoculation and for every 3 days thereafter.
Tissue immunofluorescence
Tumour tissues were harvested 7 days after the last exosome injection and fixed with 4% paraformaldehyde. The CD8+ or CD4+ lymphocytes in tumours were stained, as previously described (Du et al, 2006). To evaluate the T-cell response against Hepa 1–6 tumour cells in vitro, spleens were harvested from Hepa 1–6 inoculated mice in each group 7 days after last exosome injection. Splenocytes were isolated and co-cultured with Hepa 1–6 cells (ratio of 10 : 1, respectively) and treated with mitomycin C (Fisher Scientific) or MC38.
Mitomycin C treatment was completed as follows: Hepa 1–6 and MC38 cells were treated with mitomycin C (50 μg ml−1) in media for 1 h, and then the culture medium was removed and cells were washed with PBS three times. During the co-culturing of splenocytes and tumour cells, 30 U ml−1 recombinant human IL-2 (R&D) was added to the media for 5 days. The cells were harvested and flow cytometry was used to analyse intracellular IFNγ and granzyme B in CD8+ cells. Cytofix/Cytoperm Fixation/Permeabilisation kit (BD Bioscience) was used to prepare cells for surface and intracellular flow cytometry staining, according to the protocol suggested by the manufacturer.
Statistics
All data were presented as mean±s.d. Flow cytometry data were analysed with Flowjo software (Ashland, OR, USA), and the cell percentages were analysed with one-way ANOVA and Tukey’s post hoc test. Tumour size across different groups was analysed with two-way ANOVA and Tukey’s post hoc test. Statistical analysis was performed with GraphPad Prism software (La Jolla, CA, USA).
Results
AdIRF-1 infection enhances exosome expression of IL-15Rα and MHC-I
Exosomes were isolated from tumour cell culture supernatant by ultracentrifugation. The TEM showed isolated exosomes ∼100 nm in diameter, with disc-like morphology (Figure 1A). The IRF-1 nuclear protein expression increased in a time-dependent manner in Hepa 1–6 murine liver tumour cells after AdIRF-1 infection, but did not increase with AdLacZ infection (Figure 1B, upper panel). As expected, IFNγ, an IRF-1 inducer, promoted IRF-1 expression (Figure 1B, lower panel). Interestingly, exosomes released from the AdIRF-1-infected cells or IFNγ-treated cells showed increased expression of the IRF-1 response genes IL-15Rα and MHC-I (Figure 1C and D). As expected, both pellet groups displayed high expression of exosome marker CD63 that is expressed independently of IRF-1 induction (Figure 1C and D).

Exosomes released by IRF-1-induced Hepa 1–6 cells have increased IL-15R α and MHC-I expression. (A) Under TEM, exosomes released by Hepa 1–6 cells have traditional disc-like shapes and are ∼100 nm in diameter. (B) Western blot of nuclear proteins from Hepa 1–6 cells at different time points following AdIRF-1 infection (50 MOI) or 250 U ml−1 IFNγ treatment. (C and D) Western blot of exosomes released by Hepa 1–6 cells. Blots shown are representative of three similar experiments.
Exosomes released by IRF-1-induced tumour cells exhibit enhanced antitumour effects in vivo
As CpG oligonucleotides have been shown to prime antitumour functions of exosomes (Chaput et al, 2004), we injected tumour-bearing B6 mice with CpG oligonucleotides along with exosomes released from cells that were infected by adenovirus. The Hepa 1–6 and MC38 tumour volumes were not influenced by injection of CpG alone or CpG+ naive exosomes (Figure 2A and C). However, injection of exosomes released by AdIRF-1-infected Hepa 1–6 or MC38 cells with CpG significantly decreased tumour volumes in vivo beginning on days 21–24 (Figure 2A and C). Exosomes released by IFNγ-treated cells also inhibited tumour growth (Figure 2A and C). The IFNγ-treated tumour cell lysates also inhibited tumour growth when compared with the PBS-injected group, but this effect was not as robust as that seen with IRF-1-Exos (Figure 2A and C). To confirm that the antitumour effect was due to an IRF-1-mediated response, the exosomes were boiled to deactivate proteins that abrogated the antitumour effect (Figure 2A and C). Furthermore, we excluded the possibility of direct AdIRF-1 contamination in TEXs. The CpG+ pIRF-1-Exo group, which should not have adenovirus in the exosomes following iodixanol purification, had similar antitumour effects as CpG+IRF-1-Exo group (Figure 2B and D).

The Hepa 1–6 and MC38 tumour volumes after exosome injections. (A and B) 1 × 107 Hepa 1–6 cells were inoculated in female C57BL/6 mice. At 6 days after inoculation, CpG (0.20 μg ml−1) and exosomes released from Hepa 1–6 cells (10 μg ml−1) were dissolved in PBS, and injected into mice (0.1 ml) every 3 days for a total of four doses. (C and D) 1 × 106 MC38 cells were inoculated in female C57BL/6 mice. At 6 days after inoculation, the same concentration and volume of CpG and exosomes released from MC38 cells were injected every 3 days for a total of four doses. Tumours were measured every 3 days. *P<0.05. A full color version of this figure is available at the British Journal of Cancer journal online.
The IFNγ has antitumour effects itself, and cytokines can be encapsulated by exosomes. Accordingly, we detected IFNγ in the exosome pellets. The IFNγ was not detectable (<2 pg in 1 μg of exosome protein) in the LacZ-Exo or IRF-1-Exo groups. These data suggest that the observed antitumour effects were not a result of IFNγ action. We also detected particles in 1 μg of exosome protein from the LacZ-Exos and IRF-1-Exos with NTA. As shown in Figure 3, the particles did not result in a significant difference in the number of released exosomes (Figure 3B). Taken together, these data show that IRF-1-primed exosomes have an antitumour effect.

Tumour-derived exosomes (TEXs) analysed by NTA. (A) The TEX size distribution. (B) Particle analysis of exosomes released from AdLacZ- or AdIRF-1-infected cells (in 1 μg exosome protein). N=5. A full color version of this figure is available at the British Journal of Cancer journal online.
Exosomes released from AdIRF-1-infected cells promote infiltration of tumours by CD4+ and CD8α+ cells
To determine the mechanism of the antitumour effect elicited by IRF-1, we examined tumour-infiltrating T cells in the tumour-bearing mice. The Hepa 1–6 tumours were harvested 7 days after the last injection of exosomes and CpG. Immunofluorescence staining for CD4 and CD8α was completed in tumour tissues. The number of CD4+ and CD8α+ tumour-infiltrating cells was significantly increased in the CpG+IRF-1-Exo group compared with the PBS control group (Figures 4 and 5A and B). Heat inactivation of the proteins (by exosome boiling) abrogated the CD4+ and CD8α+ cell infiltration. These results suggest that the antitumour effects of IRF-1 are mediated, at least in part, by a CD4+ and CD8α+ T-cell immune response.

Haematoxylin and eosin (H&E) and immunofluorescence staining of Hepa 1–6 tumours. 1 × 107 Hepa 1–6 cells were inoculated in female C57BL/6 mice. At 6 days after inoculation, CpG (0.20 μg ml−1) and exosomes released from Hepa 1–6 cells (10 μg ml−1) were dissolved in PBS and injected into mice (0.1 ml) every 3 days for a total of four doses. At 7 days after the last injection, tumours were harvested, fixed, and stained with H&E and anti-CD4 and anti-CD8 antibodies. Results shown are representative of five similar experiments. A full color version of this figure is available at the British Journal of Cancer journal online.

Quantification of CD4+ and CD8+ T-cell tumour infiltration, and the effects of IRF-1-primed TEXs on tumour growth after depletion of CD4+ and CD8+ T cells. (A and B) Quantification of CD4+ and CD8+ T cells per high-power field (HPF, 20 ×). (C) Tumour growth after CD4+ T-cell or CD8+ T-cell depletion with antibodies in vivo. *P<0.05. A full color version of this figure is available at the British Journal of Cancer journal online.
Depletion of CD4+ or CD8+ T cells in vivo compromises the effects of exosomes released by AdIRF-1-infected cells
To confirm that the mechanistic effects of IRF-1-primed TEXs involve CD4+ and CD8+ T cells, we used antibodies to deplete CD4+ and CD8+ T cells in mice. Exosomes were injected 6 days after tumour inoculation. Depletion of CD8+ T cells in mice resulted in larger tumour volumes compared with the CpG+IRF-1-Exo-injected group and the PBS-injected control group (Figure 5C). Depletion of CD4+ T cells in mice only partially abrogated the effects of CpG+IRF-1-Exo injection (Figure 5C). These data further confirm that the effects of CpG+IRF-1-Exo involve, at least in part, the CD4+ and CD8+ T-cell pathways.
Mice injected with exosomes released from AdIRF-1-infected cells have a higher CD8α+ antitumour response in peripheral lymphocytes
To evaluate peripheral T-cell activation, we harvested spleens from Hepa 1–6 tumour-bearing mice 7 days after tumour injection. After isolation, splenocytes were co-cultured for 5 days with Hepa 1–6 or MC38 cells treated with mitomycin C. The mitomycin C was used to inhibit proliferation of Hepa 1–6 and MC38 tumour cells. We then examined granzyme B (Figure 6) and IFNγ (Figure 7) expression in CD8α+ splenocytes. The flow cytometry analysis shows that co-culture of Hepa 1–6 cells with splenocytes from CpG+IRF-1-Exo-injected mice results in a higher percentage of granzyme B (13.1%) and IFNγ (12.2%) expression within the CD8α+ cell population compared with the CpG+LacZ-Exo group (6.9% and 6.2%, respectively). Co-culture with MC38 cells also increased the percentage of CD8α+ cells that expressed granzyme B and IFNγ, but to a lesser extent than that co-culture with Hepa 1–6, suggesting that IRF-1-transduced exosomes trigger tumour-specific antitumour immunity. Quantification of the flow cytometry showed that both Hepa 1–6 and MC38 co-culture increased granzyme B- or IFNγ-positive T cells when compared with PBS (Figure 8).

Granzyme B-positive CD8 α + splenocytes from Hepa 1–6 tumour-bearing mice after co-culturing with mitomycin C-treated tumour cells in vitro. 1 × 107 Hepa 1–6 cells were inoculated in female C57BL/6 mice. At 6 days after inoculation, CpG (0.20 μg ml−1) and exosomes released from Hepa 1–6 cells (10 μg ml−1) were dissolved in PBS and injected into mice (0.1 ml) every 3 days for a total of four doses. At 7 days after the last injection, mouse spleens were harvested. Splenocytes were co-cultured with mitomycin C-treated (50 μg ml−1) Hepa 1–6 cells or MC38 cells for 5 days. Cells were analysed for expression of CD8α+ cell surface marker and intracellular granzyme B by flow cytometry. Results shown are representative of five similar experiments. A full color version of this figure is available at the British Journal of Cancer journal online.

The IFN γ -positive CD8 α + splenocytes from Hepa 1–6 tumour-bearing mice after co-culturing with mitomycin C-treated tumour cells in vitro. 1 × 107 Hepa 1–6 cells were inoculated in female C57BL/6 mice. At 6 days after inoculation, CpG (0.20 μg ml−1) and exosomes released from Hepa 1–6 cells (10 μg ml−1) were dissolved in PBS and injected into mice (0.1 ml) every 3 days for a total of four doses. At 7 days after the last injection, mouse spleens were harvested. Splenocytes were co-cultured with mitomycin C-treated (50 μg ml−1) Hepa 1–6 cells or MC38 cells for 5 days. Cells were analysed for expression of CD8α+ cell surface marker and intracellular IFNγ by flow cytometry. Results shown are representative of five similar experiments. A full color version of this figure is available at the British Journal of Cancer journal online.

Quantification of IFN γ and granzyme B-positive CD8 α + splenocytes after co-culture with mitomycin C-treated Hepa 1–6 or MC38 cells. (A) Quantification of flow cytometry results for granzyme B and CD8α double-positive splenocytes from Figure 6. (B) Quantification of flow cytometry results for IFNγ and CD8α double-positive splenocytes from Figure 7. N=5 in each group. **P<0.01; ****P<0.0001.
Discussion
Many studies have examined the use of exosomes as tumour vaccines (Zitvogel et al, 1998; Wolfers et al, 2001). Whereas dendritic cell released exosomes (DEXs) have predominantly antitumour effects, the effects of TEXs remain controversial. Some studies show that TEXs have antitumour effects (Chen et al, 2011; Lee et al, 2011), whereas others show TEXs can promote tumour progression and metastasis (Andreola et al, 2002; Wieckowski et al, 2009). Specifically, TEXs with increased immune-activating molecules like chemokines (Chen et al, 2011) or MHC class II proteins (Lee et al, 2011) enhanced antitumour effects, whereas TEXs with Fas ligand (Andreola et al, 2002) or NKG2D ligands (Clayton et al, 2008) suppressed antitumour immunity. Hence, the antitumour effect of TEXs depends on TEX content. As an exosome is a miniature version of its host cell, several studies have shown that the contents of TEXs can be modified by manipulation of their mother tumour cells (Lee et al, 2011).
As IRF-1 can promote antitumour immunity, and TEXs have IRF-1 target genes (IL-15Rα and MHC-I), we hypothesised that TEXs can be engineered to activate IRF-1 targeted genes in order to further enhance their antitumour effects. The major and novel findings of this study are: (1) exosomes released by IRF-1-primed cells have increased expression of IRF-1 target genes IL-15Rα and MHC-I; (2) these exosomes promote antitumour immunity in part by increasing tumour infiltration by CD4+ and CD8+ T cells; (3) and the antitumour effects were tumour specific.
The IFNγ and its target gene, IRF-1, play important antitumour roles (Lorenzi et al, 2012; El Jamal et al, 2016) by promoting tumour cell apoptosis (Schwartz-Roberts et al, 2015) and enhancing immune responses (Murtas et al, 2013). Further studies are required to determine whether or not IRF-1-primed TEXs can induce Hepa 1–6 or MC38 apoptosis.
The IRF-1 is an early-immediate transcription factor that can regulate many immune activation genes, including MHC-I (Taniguchi et al, 2001), MHC-II, IL-15Rα (Ogasawara et al, 1998), and IFNα or IFNβ (Harada et al, 1990; Matsuyama et al, 1993). The IRF-1 can inhibit cancer growth by regulating microRNAs (Mao et al, 2015) that can be shuttled to immune cells by exosomes (van der Vos et al, 2016). Although we did not specifically detect RNA or DNA in IRF-1-induced TEXs, the antitumour effects observed with IRF-1 Exo injection were abrogated following heat inactivation, indicating that the antitumour effects were likely mediated by proteins within the TEXs. The loss of the antitumour effects with heat inactivation also excludes the possibility that the antitumour effects were a result of AdIRF-1 DNA shuttled by IRF-1-Exo. Although virus can be transmitted via exosomes (Longatti, 2015), the AdIRF-1 construct used in these studies is replication deficient, and therefore excludes any direct effects from adenovirus in the exosomes. Furthermore, similar antitumour effects were observed in TEXs purified using iodixanol, a method that excludes adenovirus from exosomes, and therefore direct contamination by AdIRF-1 is unlikely.
In our studies, Hepa 1–6 tumour size decreased even in the PBS control group. Hepa 1–6 cells are derived from a BW7756 hepatoma in C57/L mice, and are often used to inoculate C57L/J mice (Huang and Ohno, 2003). Therefore, Hepa 1–6 cells are not syngeneic with C57BL/6J, and C57BL/6J mice can reject Hepa 1–6 cell tumours as a result of minor mismatch. One study found that Hepa 1–6 tumour formation depends on the site of inoculation, and reported that subcutaneous Hepa 1–6 tumours did not grow as well as intrahepatic tumours (Hiotis et al, 2003). Therefore, to confirm that IRF-1-primed TEXs have antitumour effects, we employed a second tumour model utilising MC38 cells, which are syngeneic with C57BL/6J, and found that these tumours also grew slower when treated with IRF-1-primed TEXs.
Tumour immunofluorescence staining demonstrated that the antitumour effects of IRF-1-Exo were associated with CD4+ and CD8α+ T-cell recruitment. Consistent with our results of increased IL-15Rα expression in TEXs, others have shown that IL-15 has higher antitumour activity because of an increase in tumour-reactive CD8α+ T cells (Klebanoff et al, 2004; Morris et al, 2014). A previous report also shows that increasing MHC-I expression promotes antitumour immunity (Wan et al, 2012). Induction of IRF-1 not only increased MHC-I expression in tumour cells (Lorenzi et al, 2012), but also increased MHC-I expression in exosomes, thereby promoting antitumour immunity. Our data may further explain why tumour cell lysate vaccines, which are generated with the IRF-1 inducer IFNα, can inhibit tumour growth (Omori et al, 2012).
Antitumour effects depend in part on cytotoxic T lymphocytes (CTLs), and previous research has shown that TEXs can prime CTLs (Wolfers et al, 2001). After co-culturing splenocytes from tumour-bearing mice with mitomycin C-treated tumour cells, we observed that splenocytes from CpG+IRF-1-Exo-injected mice displayed a higher percentage of IFNγ and granzyme B-positive CTLs (CD8+ T cells). Interestingly, these splenocytes had a higher response rate to Hepa 1–6 cells as compared with MC38 cells, indicating that IRF-1-primed TEXs can trigger tumour-specific immunity. In our experiments, we used the CpG as an exosome adjuvant that has been demonstrated previously (Chaput et al, 2004). The CpG can activate Toll-like receptors to promote an immune response. Our data verify that CpG can be used as an adjuvant for a TEX vaccine.
In order to generate patient-specific IRF-1-primed TEXs, cancer cells must first be derived from patient samples. After treatment with IFNγ or transduction with AdIRF-1, TEXs can be harvested from cell culture supernatant. Cancer cells can then be excluded from TEXs in via filtration, as TEXs can pass through a 0.22 μm filter. The use of TEXs as a patient-specific vaccine is more advantageous than the use of tumour lysate, because of the many kinds of damage-associated molecule patterns (DAMPs) that result from the use of tumour lysates.
In conclusion, we have demonstrated that exosomes released by IRF-1-induced tumour cells increase expression of IRF-1 target proteins and have specific antitumour effects. As each patient has unique tumour antigens, our results provide a potential strategy for a patient-specific tumour vaccine utilising IRF-1-primed TEXs.
Change history
09 January 2018
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P, Pomel C, Lhomme C, Escudier B, Le Chevalier T, Tursz T, Amigorena S, Raposo G, Angevin E, Zitvogel (2002) Malignant effusions and immunogenic tumour-derived exosomes. Lancet 360 (9329): 295–305.
Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P, Squarcina P, Accornero P, Lozupone F, Lugini L, Stringaro A, Molinari A, Arancia G, Gentile M, Parmiani G, Fais S (2002) Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med 195 (10): 1303–1316.
Armstrong MJ, Stang MT, Liu Y, Yan J, Pizzoferrato E, Yim JH (2015) IRF-1 inhibits NF-kappaB activity, suppresses TRAF2 and cIAP1 and induces breast cancer cell specific growth inhibition. Cancer Biol Ther 16 (7): 1029–1041.
Cantin R, Diou J, Belanger D, Tremblay AM, Gilbert C (2008) Discrimination between exosomes and HIV-1: purification of both vesicles from cell-free supernatants. J Immunol Methods 338 (1-2): 21–30.
Chaput N, Schartz NE, Andre F, Taieb J, Novault S, Bonnaventure P, Aubert N, Bernard J, Lemonnier F, Merad M, Adema G, Adams M, Ferratini M, Carpentier AF, Escudier B, Tursz T, Angevin E, Zitvogel L (2004) Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol 172 (4): 2137–2146.
Chen T, Guo J, Yang M, Zhu X, Cao X (2011) Chemokine-containing exosomes are released from heat-stressed tumour cells via lipid raft-dependent pathway and act as efficient tumor vaccine. J Immunol 186 (4): 2219–2228.
Cho JA, Lee YS, Kim SH, Ko JK, Kim CW (2009) MHC independent anti-tumor immune responses induced by Hsp70-enriched exosomes generate tumor regression in murine models. Cancer Lett 275 (2): 256–265.
Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z (2008) Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol 180 (11): 7249–7258.
Clayton A, Mitchell JP, Court J, Mason MD, Tabi Z (2007) Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res 67 (15): 7458–7466.
Connett JM, Hunt SR, Hickerson SM, Wu SJ, Doherty GM (2003) Localization of IFN-gamma-activated Stat1 and IFN regulatory factors 1 and 2 in breast cancer cells. J Interferon Cytokine Res 23 (11): 621–630.
Dai S, Wan T, Wang B, Zhou X, Xiu F, Chen T, Wu Y, Cao X (2005) More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-specific CTL response by immunization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin Cancer Res 11 (20): 7554–7563.
Du Q, Park KS, Guo Z, He P, Nagashima M, Shao L, Sahai R, Geller DA, Hussain SP (2006) Regulation of human nitric oxide synthase 2 expression by Wnt beta-catenin signaling. Cancer Res 66 (14): 7024–7031.
El Jamal SM, Taylor EB, Abd Elmageed ZY, Alamodi AA, Selimovic D, Alkhateeb A, Hannig M, Hassan SY, Santourlidis S, Friedlander PL, Haikel Y, Vijaykumar S, Kandil E, Hassan M (2016) Interferon gamma-induced apoptosis of head and neck squamous cell carcinoma is connected to indoleamine-2,3-dioxygenase via mitochondrial and ER stress-associated pathways. Cell Div 11: 11.
Escors D (2014) Tumour immunogenicity, antigen presentation and immunological barriers in cancer immunotherapy. New J Sci 2014: pii: 734515.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313 (5795): 1960–1964.
Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, Multhoff G (2005) Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res 65 (12): 5238–5247.
Harada H, Willison K, Sakakibara J, Miyamoto M, Fujita T, Taniguchi T (1990) Absence of the type I IFN system in EC cells: transcriptional activator (IRF-1) and repressor (IRF-2) genes are developmentally regulated. Cell 63 (2): 303–312.
Hiotis SP, Yee HT, Luan W, Burakoff SJ (2003) Site does matter: tumorigenesis of the murine hepatocellular carcinoma cell line Hepa 1–6 is dependent upon intrahepatic rather than subdermal implantation. J Surg Res 114(2): 274.
Huang L, Ohno T (2003) Protective antitumor immunity induced by fixed tumor cells in combination with adjuvant in a murine hepatoma model. Cancer Lett 202 (2): 153–159.
Kim KH, Dhupar R, Ueki S, Cardinal J, Pan P, Cao Z, Cho SW, Murase N, Tsung A, Geller DA (2009) Donor graft interferon regulatory factor-1 gene transfer worsens liver transplant ischemia/reperfusion injury. Surgery 146 (2): 181–189.
Kim SH, Bianco NR, Shufesky WJ, Morelli AE, Robbins PD (2007) MHC class II+ exosomes in plasma suppress inflammation in an antigen-specific and Fas ligand/Fas-dependent manner. J Immunol 179 (4): 2235–2241.
Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC, Tagaya Y, Rosenberg SA, Waldmann TA, Restifo NP (2004) IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci USA 101 (7): 1969–1974.
Klune JR, Dhupar R, Kimura S, Ueki S, Cardinal J, Nakao A, Nace G, Evankovich J, Murase N, Tsung A, Geller DA (2012) Interferon regulatory factor-2 is protective against hepatic ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol 303 (5): G666–G673.
Konadu KA, Huang MB, Roth W, Armstrong W, Powell M, Villinger F, Bond V (2016) Isolation of exosomes from the plasma of HIV-1 positive individuals. J Vis Exp 107.
Lee YS, Kim SH, Cho JA, Kim CW (2011) Introduction of the CIITA gene into tumor cells produces exosomes with enhanced anti-tumor effects. Exp Mol Med 43 (5): 281–290.
Longatti A (2015) The dual role of exosomes in hepatitis A and C virus transmission and viral immune activation. Viruses 7 (12): 6707–6715.
Lorenzi S, Forloni M, Cifaldi L, Antonucci C, Citti A, Boldrini R, Pezzullo M, Castellano, Russo, van der Bruggen P, Giacomini P, Locatelli F, Fruci D (2012) IRF1 and NF-kB restore MHC class I-restricted tumor antigen processing and presentation to cytotoxic T cells in aggressive neuroblastoma. PLoS One 7 (10): e46928.
Mao L, Zhang Y, Mo W, Yu Y, Lu H (2015) BANF1 is downregulated by IRF1-regulated microRNA-203 in cervical cancer. PLoS One 10 (2): e0117035.
Martin RK, Brooks KB, Henningsson F, Heyman B, Conrad DH (2014) Antigen transfer from exosomes to dendritic cells as an explanation for the immune enhancement seen by IgE immune complexes. PLoS One 9 (10): e110609.
Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kundig TM, Amakawa R, Kishihara K, Wakeham A (1993) Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell 75 (1): 83–97.
Morris JC, Ramlogan-Steel CA, Yu P, Black BA, Mannan P, Allison JP, Waldmann TA, Steel JC (2014) Vaccination with tumor cells expressing IL-15 and IL-15Ralpha inhibits murine breast and prostate cancer. Gene Therapy 21 (4): 393–401.
Murtas D, Maric D, De Giorgi V, Reinboth J, Worschech A, Fetsch P, Filie A, Ascierto ML, Bedognetti D, Liu Q, Uccellini L, Chouchane L, Wang E, Marincola FM, Tomei S (2013) IRF-1 responsiveness to IFN-gamma predicts different cancer immune phenotypes. Br J Cancer 109 (1): 76–82.
O'Brien K, Rani S, Corcoran C, Wallace R, Hughes L, Friel AM, McDonnell S, Crown J, Radomski MW, O’Driscoll L (2013) Exosomes from triple-negative breast cancer cells can transfer phenotypic traits representing their cells of origin to secondary cells. Eur J Cancer 49 (8): 1845–1859.
Ogasawara K, Hida S, Azimi N, Tagaya Y, Sato T, Yokochi-Fukuda T, Waldmann TA, Taniguchi T, Taki S (1998) Requirement for IRF-1 in the microenvironment supporting development of natural killer cells. Nature 391 (6668): 700–703.
Omori R, Eguchi J, Hiroishi K, Ishii S, Hiraide A, Sakaki M, Doi H, Kajiwara A, Ito T, Kogo M, Imawari M (2012) Effects of interferon-alpha-transduced tumor cell vaccines and blockade of programmed cell death-1 on the growth of established tumors. Cancer Gene Ther 19 (9): 637–643.
Ouyang Y, Bayer A, Chu T, Tyurin VA, Kagan VE, Morelli AE, Coyne CB, Sadovsky Y (2016) Isolation of human trophoblastic extracellular vesicles and characterization of their cargo and antiviral activity. Placenta 47: 86–95.
Peng HH, Wu S, Davis JJ, Wang L, Roth JA, Marini FC 3rd, Fang B (2006) A rapid and efficient method for purification of recombinant adenovirus with arginine-glycine-aspartic acid-modified fibers. Anal Biochem 354 (1): 140–147.
Robbins PD, Morelli AE (2014) Regulation of immune responses by extracellular vesicles. Nat Rev Immunol 14 (3): 195–208.
Sahoo S, Klychko E, Thorne T, Misener S, Schultz KM, Millay M, Ito A, Liu T, Kamide C, Agrawal H, Periman H, Qin G, Kishore R, Losordo DW (2011) Exosomes from human CD34(+) stem cells mediate their proangiogenic paracrine activity. Circ Res 109 (7): 724–728.
Sakai T, Mashima H, Yamada Y, Goto T, Sato W, Dohmen T, Kamada K, Yoshioka M, Uchinami H, Yamamoto Y, Ohnishi H (2014) The roles of interferon regulatory factors 1 and 2 in the progression of human pancreatic cancer. Pancreas 43 (6): 909–916.
Schwartz-Roberts JL, Cook KL, Chen C, Shajahan-Haq AN, Axelrod M, Warri A, Riggins RB, Jin L, Haddad BR, Kallakury BV, Baumann WT, Clarke R (2015) Interferon regulatory factor-1 signaling regulates the switch between autophagy and apoptosis to determine breast cancer cell fate. Cancer Res 75 (6): 1046–1055.
Stang MT, Armstrong MJ, Watson GA, Sung KY, Liu Y, Ren B, Yim JH (2007) Interferon regulatory factor-1-induced apoptosis mediated by a ligand-independent fas-associated death domain pathway in breast cancer cells. Oncogene 26 (44): 6420–6430.
Stoorvogel W, Kleijmeer MJ, Geuze HJ, Raposo G (2002) The biogenesis and functions of exosomes. Traffic 3 (5): 321–330.
Taniguchi T, Ogasawara K, Takaoka A, Tanaka N (2001) IRF family of transcription factors as regulators of host defense. Annu Rev Immunol 19: 623–655.
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9 (6): 654–659.
van der Vos KE, Abels ER, Zhang X, Lai C, Carrizosa E, Oakley D, Prabhakar S, Mardini O, Crommentujin MH, Skog J, Krichevsky AM, Stemmer-Rachamimov A, Mempel TR, El Khoury J, Hickman SE, Breakefield XO (2016) Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neurooncology 18 (1): 58–69.
Viaud S, Terme M, Flament C, Taieb J, Andre F, Novault S, Escudier B, Bobert C, Caillat-Zucman S, Tursz T, Zitvogel L, Chaput N (2009) Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Ralpha. PLoS One 4 (3): e4942.
Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF (2012) Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS One 7 (3): e32542.
Wang E, Miller LD, Ohnmacht GA, Mocellin S, Perez-Diez A, Petersen D, Zhao Y, Simon R, Powell JI, Asaki E, Alexander HR, Duray PH, Herlyn M, Restifo NP, Liu ET, Rosenberg SA, Marincola FM (2002) Prospective molecular profiling of melanoma metastases suggests classifiers of immune responsiveness. Cancer Res 62 (13): 3581–3586.
Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL (2009) Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J Immunol 183 (6): 3720–3730.
Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, Flament C, Pouzieux S, Faure F, Tursz T, Angevin E, Amigorena S, Zitvogel L (2001) Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med 7 (3): 297–303.
Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, Cheng Z, Shah SV, Wang GJ, Zhang L, Grizzle WE, Mobley J, Zhang HG (2009) Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer 124 (11): 2621–2633.
Xie Y, Bai O, Zhang H, Li W, Xiang J (2010) Tumor necrosis factor gene-engineered J558 tumor cell-released exosomes stimulate tumor antigen P1A-specific CD8+ CTL responses and antitumor immunity. Cancer Biother Radiopharm 25 (1): 21–28.
Xu H, DiIulio NA, Fairchild RL (1996) T cell populations primed by hapten sensitization in contact sensitivity are distinguished by polarized patterns of cytokine production: interferon gamma-producing (Tc1) effector CD8+ T cells and interleukin (Il) 4/Il-10-producing (Th2) negative regulatory CD4+ T cells. J Exp Med 183 (3): 1001–1012.
Yim JH, Wu SJ, Casey MJ, Norton JA, Doherty GM (1997) IFN regulatory factor-1 gene transfer into an aggressive, nonimmunogenic sarcoma suppresses the malignant phenotype and enhances immunogenicity in syngeneic mice. J Immunol 158 (3): 1284–1292.
Zeelenberg IS, Ostrowski M, Krumeich S, Bobrie A, Jancic C, Boissonnas A, Delcayre A, Le Pecq JB, Combadiere B, Amigorena S, Thery C (2008) Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res 68 (4): 1228–1235.
Zeimet AG, Reimer D, Wolf D, Fiegl H, Concin N, Wiedemair A, Wolf AM, Rumpold H, Muller-Holzner E, Marth C (2009) Intratumoral interferon regulatory factor (IRF)-1 but not IRF-2 is of relevance in predicting patient outcome in ovarian cancer. Int J Cancer 124 (10): 2353–2360.
Zhang H, Tang K, Zhang Y, Ma R, Ma J, Li Y, Luo S, Liang X, Ji T, Gu Z, Lu J, He W, Cao W, Wan Y, Huang B (2015) Cell-free tumor microparticle vaccines stimulate dendritic cells via cGAS/STING signaling. Cancer Immunol Res 3 (2): 196–205.
Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, Ricciardi-Castagnoli P, Raposo G, Amigorena S (1998) Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med 4 (5): 594–600.
Acknowledgements
This work was supported by the NIH Grant (HHSN276201200017C, to DAG). We thank Ming Sun from Center of Biologic Imaging for TEM examination.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yang, Mq., Du, Q., Varley, P. et al. Interferon regulatory factor 1 priming of tumour-derived exosomes enhances the antitumour immune response. Br J Cancer 118, 62–71 (2018). https://doi.org/10.1038/bjc.2017.389
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.389
Keywords
This article is cited by
-
Overexpression of OASL upregulates TET1 to induce aberrant activation of CD4+ T cells in systemic sclerosis via IRF1 signaling
Arthritis Research & Therapy (2022)
-
Extracellular vesicles deliver sodium iodide symporter protein and promote cancer cell radioiodine therapy
Scientific Reports (2022)
-
Roles of IFN-γ in tumor progression and regression: a review
Biomarker Research (2020)
-
Interferon regulatory factor 1 (IRF-1) and IRF-2 regulate PD-L1 expression in hepatocellular carcinoma (HCC) cells
Cancer Immunology, Immunotherapy (2020)
