
Abstract
Background:
This multicentre, open-label study evaluated the efficacy and safety of SPI-1620, an analogue of endothelin-1, administered in combination with docetaxel as second-line treatment for patients with advanced biliary tract cancer (ABTC).
Methods:
Eligible patients received continuous cycles of combination therapy with SPI-1620 (11 μg m−2) and docetaxel (75 mg m−2) intravenously every 3 weeks until disease progression (PD) or intolerable toxicity. Tumour response was evaluated using computed tomography or magnetic resonance imaging every 2 cycles (6 weeks). The primary efficacy end point was progression-free survival (PFS); secondary end points included overall response rate (ORR), duration of response, and overall survival (OS) that were estimated using the Kaplan–Meier method.
Results:
Of the 30 enrolled patients, 25 patients had qualifying events (PD or death), 1 patient was nonevaluable, and 4 patients were censored at the time of their last tumour assessment. Our primary end point of PFS ⩾5 months was not reached. Median PFS was 2.6 months (95% confidence interval (CI): 1.4–2.8), ranging from 0.7 to 8.4 months. The ORR was 10.3% (95% CI: 0.02–0.27). Eleven additional patients achieved stable disease. The OS was 4.87 months. The most common grade 3–4 toxicities were febrile neutropenia and neutropenia.
Conclusions:
The addition of docetaxel to SPI-1620 in second-line ABTC did not meet the pre-specified primary end point of PFS ⩾5 months in unselected patient population.
Similar content being viewed by others

Main
Biliary tract cancers are a heterogeneous group of invasive carcinomas that include intrahepatic and extrahepatic biliary tract cancers and carcinomas arising from the gallbladder and ampulla of Vater. In the United States, the incidence of extrahepatic biliary tract cancers is ∼11 420 per year, and two-thirds of these are gallbladder cancers. It is difficult to determine the incidence of intrahepatic cholangiocarcinoma, but an estimated 39 230 primary liver and intrahepatic cholangiocarcinomas are diagnosed annually (Siegel et al, 2016). At presentation, a majority of these cancers are at an advanced stage – either unresectable or metastatic – and carry a median survival time of ⩽1 year.
Chemotherapy remains the mainstay of treatment for a majority of patients with biliary cancers. For first-line treatment, a gemcitabine/cisplatin regimen is the preferred regimen based on data from the ABC-02 trial, the largest, randomised phase 3 trial for the treatment of advanced biliary cancer (Valle et al, 2010). However, no standard second-line treatment has been established. A number of studies have demonstrated modest single-agent activity of several chemotherapeutic agents, such as gemcitabine, fluorouracil, S-1, oxaliplatin, docetaxel and others, in the treatment of biliary cancer (Oh et al, 2011; Suzuki et al, 2013; He et al, 2014). Of these, single-agent docetaxel showed some activity as single agent in one trial with one complete response and three partial responses in 21 patients (Papakostas et al, 2001). However, another study reported no objective responses in 17 patients (Pazdur et al, 1999).
Additional supporting data are derived from retrospective analyses demonstrating disease control in 45% of patients (Walter et al, 2013), particularly in those patients with good performance status who responded to initial chemotherapy (Lee et al, 2012). There is a clear need for further study of more efficient and tolerable regimens in biliary cancers.
A potential mechanism for the modest efficacy of chemotherapy regimens in biliary tract cancers is the failure to achieve adequate cytotoxic concentrations at the tumour bed because of poor penetration and nonuniform drug distribution within the tumour (Jain, 1991). A new approach has been proposed that involves targeting receptors on the tumour vasculature. One such group of receptors is the endothelin (ET) receptor family that are widely distributed throughout the body and are involved in a variety of physiological functions (Brooks et al, 1995; Nelson et al, 2003). The ET receptors exert their effects by binding to two distinct types of cell surface receptors, ETA and ETB. It has been shown that both ETA and ETB receptors are overexpressed in breast cancer (Rajeshkumar et al, 2005) and prostate (Rajeshkumar et al, 2007) tumour models in rats and melanoma and ovarian tumour models in mice (data on file). Consequently, it may be possible to use selective ETB receptor agonists to transiently increase tumour blood perfusion and enhance delivery of antineoplastic agents to solid tumours.
SPI-1620, previously known as IRL 620 or N-Suc-[Glu9, Ala11,15]ET-1(8–21), is an investigational, highly selective peptide agonist of ETB receptors expressed on endothelial cell lining the interior surface of blood vessels (Yanagisawa et al, 1988). Preclinical studies of SPI-1620 in several different tumour models suggest that it stimulates ETB receptors on tumour vascular, selectively and transiently increasing tumour blood flow and improving delivery of anticancer agents to the tumour (Rajeshkumar et al, 2005, 2007). The SPI-1620 has been shown to enhance the anticancer activity of paclitaxel and cisplatin in animal studies (Rai et al, 2005; Rajeshkumar et al, 2005).
A phase 1, open-label study of SPI-1620 in combination with docetaxel was conducted in 36 patients with recurrent or progressive carcinoma, including 2 patients with refractory cholangiocarcinoma (Tolcher et al, 2011). In cycle 1, patients received escalating doses of SPI-1620 ranging from 0.5 to 15.1 μg m−2 (no intrapatient escalation) on days 1 and 8 alone, and on day 15, patients were given SPI-1620 followed by docetaxel on day 1 every 21 days. Based on the results of this study, the SPI-1620 maximum tolerated dose of 11 μg m−2 was selected for further study. One of the two patients with biliary cancer who had lung and liver metastases and had previously been treated with gemcitabine, cisplatin, and 5-fluorouracil exhibited a complete disappearance of lung metastases and a partial response of the liver metastases. Based on these early results and the preclinical rationale, a phase 2 study of SPI-1620 and docetaxel in the second-line treatment of patients with refractory bile duct tumours was conducted.
PAtients and Methods
This phase 2, multicentre, nonrandomised, open-label clinical study was designed to evaluate the efficacy and safety of SPI-1620 administered in combination with docetaxel in patients with advanced biliary tract cancers who had failed one prior chemotherapy regimen. Before study initiation, the clinical study protocol and written informed consent form (ICF) were reviewed and approved by an independent ethics committee or internal review board, as required by law. The investigators agreed to comply with the World Medical Association Declaration of Helsinki concerning written informed consent and the protection and rights of human subjects.
Eligibility criteria
Eligible patients had histologically confirmed biliary tract or gallbladder cancer that had relapsed or was refractory after one prior gemcitabine-based chemotherapy regimen for advanced disease. Patients were allowed to have received adjuvant treatment if their relapse had occurred within 1 year post gemcitabine-based chemotherapy regimen. Measurable disease was required. Other eligibility criteria included a Zubrod performance status of 0–1; adequate bone marrow function indicated by a leukocyte count ⩾3000 per μl, absolute neutrophil count ⩾1000 per μl, and platelet count ⩾75,000 per μl; adequate hepatic function with a total bilirubin up to 2.5 × the institutional upper limit of normal; aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels ⩽2.5 the upper limit of normal. Patients with congestive heart failure or myocardial infarction and significant circulatory disorders in the past 6 months, stroke, significant ventricular arrhythmia, or inadequately controlled hypertension were excluded.
The evaluable population was defined as all patients who received at least one dose of both SPI-1620 and docetaxel and had at least one post-baseline tumour response assessment using Response Evaluation Criteria in Solid Tumours (RECIST) versions 1.1. The safety population was defined as all patients who received at least one dose of SPI-1620 or docetaxel.
Treatment
Eligible patients received 11 μg m−2 of SPI-1620 intravenously (IV) over 1 min on day 1 of each cycle, with dose reductions permitted to 9 and 7 μg m−2 for toxicity. At 10 min (±2) after SPI-1620 administration, patients received docetaxel 75 mg m−2 IV, with dose modification in accordance with the Taxotere (Sanofi-aventis Bridgewater, NJ, USA) prescribing information. This regimen was repeated in 3-week (21-day) cycles until progression or intolerable toxicity. Specific dose modification and treatment interruption criteria for docetaxel and SPI-1620 were applied. If docetaxel was held for specific toxicity, then SPI-1620 was held; however, if SPI-1620 was held because of toxicity, then docetaxel was given by itself. Docetaxel dose modification was permitted, as per Taxotere prescribing information. Two dose reductions were allowed for SPI-1620. The SPI-1620 dose reduction or discontinuations were necessary in the event of hypersensitivity reactions, hypotension, or bronchospasm. For mild-to-moderate infusion reactions, the infusion rate was decreased by 50% for the remainder of the dose. If severe infusion reactions occur, discontinuation of therapy was recommended.
Assessment of efficacy and toxicity and statistical considerations
The primary end point of the trial was progression-free survival (PFS) in patients with advanced gallbladder cancer or cholangiocarcinoma treated with docetaxel and SPI-1620. Progression-free survival was calculated as the time from the first dose of study treatment to the first observation of a qualifying PFS event (progression of disease or death). For patients without an event, PFS was censored at the time of the last evaluable disease assessment.
We hypothesised that combination therapy with SPI-1620 and docetaxel would have been of further interest if the median PFS had been ⩾5 months (PFS rate of ⩾44% at 6 months) and was deemed ineffective if the median PFS was ⩽3 months (PFS rate of ⩽25% at 6 months). With a significance level of 5% and power of 80%, 39 patients were planned to be treated. The study would have been considered positive if at least 15 patients had been progression free after 6 months.
Secondary efficacy end points were overall response rate (ORR) (proportion of patients achieving complete response (CR) or partial response (PR)); duration of response (time from first response to first documentation of progression of disease (PD) or death); and overall survival (OS) (time from the first dose of study treatment to death because of any cause). Distribution of PFS, duration of response, and OS were estimated using the Kaplan–Meier method. The safety end points were worst grade adverse events (AEs); deaths, other serious adverse events (SAEs), and AEs leading to discontinuation of study treatment; and worst grade laboratory abnormalities.
Patients were seen and evaluated on a weekly basis during cycle 1, and every 3 weeks in cycle 2 and beyond. Tumour response was evaluated using computed tomography (CT) or magnetic resonance imaging (MRI) in accordance with the RECIST (version 1.1). Disease assessments were performed at baseline and every two cycles (6 weeks) during the treatment period and every 6 to 8 weeks after discontinuation for reasons other than progressive disease (PD); assessments continued until documented PD or first subsequent therapy for biliary cancer, whichever occurred first. Survival follow-up was performed every 3 months following the last tumour assessment and continued until 2 years from the start of study treatment or until study closure. For patients without an event, PFS was censored at the time of the last evaluable disease assessment.
Measurable disease was defined as at least one lesion for which the longest diameter could be accurately measured as ⩾1 cm using spiral CT or MRI. Measurable lymph nodes had to have a short-axis measurement of ⩾1.5 cm. All other lesions, including ascites and pleural effusions, were considered non-measureable. Blood samples for tumour markers carcinoembryonic antigen (CEA) and cancer antigen (CA) 19-9 were collected at baseline and, if abnormal, every two cycles (6 to 7 weeks) thereafter.
Safety measurements included AEs, physical examinations, vital signs, clinical laboratory assessments, and electrocardiograms. Adverse events considered related to study procedures were collected from the time informed consent was obtained until the administration of the first dose of study treatment, after which all AEs were recorded through 30 days after the last dose of study treatment. From 31 days after the last dose of study treatment, only treatment-related AEs were recorded. Adverse events were graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), version 4.03, grouped by Medical Dictionary for Regulatory Activities (MedDRA) preferred term, and summarised by worst grade severity. Treatment-emergent AEs (TEAEs) were defined as AEs that occurred or worsened at the time of or after the first dose of study treatment, or any treatment-related AEs, regardless of the time of onset. Clinical laboratory results were classified according to the NCI CTCAE version 4.03.
Results
Patient disposition
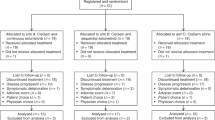
A total of 30 patients were enrolled in the study between August 2013 and July 2015. Reasons for study treatment discontinuation were: disease progression (n=13, 43.3%), withdrawal of consent (n=5, 16.7%), death (n=4, 13.3%), adverse events (n=3, 10.0%), other (n=3, 10.0%), physician decision (n=1, 3.3%), and sponsor decision (n=1, 3.3%).
Treatment exposure
All but 1 patient received at least one dose of SPI-1620 and docetaxel; therefore, the safety population consisted of 29 (96.7%) patients. The median total dose of SPI-1620 and docetaxel administered were 43 μg (range: 20–275 μg) and 292 mg (range: 111–1361 mg), respectively. The median number of cycles administered was two (range: 1–12).
Demographics and baseline characteristics
As shown in Table 1, slightly more male than female patients were enrolled (53.3% vs 46.7%). The majority of patients were not Hispanic or Latino (83.3%) and White (86.7%), and the median age was 64 years (range: 42–80 years). The majority of patients were ECOG Performance Status 1 (67%) or 2 (30%). Most patients (73%) had intrahepatic cholangiocarcinoma or extrahepatic cholangiocarcinoma (23%). The vast majority of patients (93%) had metastatic disease. Slightly more patients had received prior cancer surgery (53% vs 47%, no surgery). Finally, prior chemotherapy consisted of gemcitabine and cisplatin for 70%, gemcitabine plus oxaliplatin (20%), and gemcitabine monotherapy (10%).
Safety and tolerability
As shown in Table 2, all treated patients experienced at least one treatment-emergent AE, and most patients (n=28, 96.6%) experienced at least one treatment-related AE. Overall, 19 (65.5%) patients experienced at least one SAE, including 11 patients (37.9%) with SAEs assessed as related to study treatment (Table 4). Severe (grade 3) AEs were reported at least once in 24 patients (82.8%), and life-threatening (grade 4) AEs were reported at least once in 16 patients (55.2%). Eight patients (27.6%) experienced AEs leading to discontinuation of study treatment.
As shown in Table 3, the most commonly occurring treatment-related AEs were fatigue (n=18, 62.1%); anaemia, nausea, neutropenia, and leukopenia (n=8, 27.6% each); decreased appetite (n=7; 24.1%); diarrhoea (n=6; 20.7%), and febrile neutropenia, hot flush, and neutropenia (n=5, 17.2% each).
The most frequently reported grade 3 AEs (>2 patients) were anaemia and febrile neutropenia (each n=5, 17.2%); abdominal pain, ascites, fatigue, and neutropenia (n=4, 13.8%); and anorexia (n=3, 10.3%). Grade 4 AEs included neutropenia (n=8, 27.6%); white blood count (WBC) decreased (n=6, 20.7%); neutrophil count decreased (n=4, 13.8%); and anaemia, dyspnoea, lymphocyte count decreased, urinary tract infection, and vomiting (each n=1, 3.4%).
For 19 patients (65.5%), at least one event was an SAE. The most frequently reported SAEs (>1 patient) were febrile neutropenia (n=6, 20.7%), disease progression (n=4, 13.8%) and abdominal pain, ascites, dyspnoea, and neutropenia (each n=2, 6.9%). Treatment-related SAEs were largely haematologic, as presented in Table 4.
Sixteen patients (55.2%) died on study, all because of PD.
Efficacy
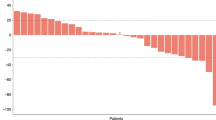
In the analysis of PFS, 25 patients had qualifying events (PD or death), and 4 patients were censored at the time of their last tumour assessment. The median PFS was 2.6 months (95% confidence interval (CI): 1.4–2.8), with individual patient PFS ranging from 0.7 to 8.4 months. The ORR (patients with either a CR or PR) was 10.3% (95% CI: 0.02–0.27), and 11 patients achieved stable disease. There were no complete radiographic responses. No disease-specific subset analyses were performed because of the small number of patients. Overall survival was 4.87 months (Figures 1 and 2).

Overall survival. NE=not estimated.
Discussion
In this multicentre, open-label phase 2 trial, we explored the efficacy and safety of SPI-1620, an analogue of endothelin-1, administered in combination with docetaxel in patients with ABTC. Ampullary carcinoma was excluded from this study, as it portends a more favourable prognosis of than other biliary tract cancers (Albores-Saavedra et al, 2009). The sponsor decided to close the study after 30 patients had been treated, owing to the failure to meet predetermined median PFS end point. Specifically, we had specified that SPI-1620 would be deemed to have good activity if 15 or more patients out of 39 were progression free after 6 months of treatment. However, after 30 patients, it was clear that our goal of 15 patients would not be reached, especially given that only 2 patients exhibited a PFS of >6 months.
This study was based on the hypothesis that the use of SPI-1620 in combination with docetaxel would transiently and selectively increase perfusion of the chemotherapeutic agent into tumour tissue without increasing chemotherapy exposure. Despite the robust preclinical data suggesting that using SPI-1620 can alter vascular permeability and enhance drug delivery in biliary tumours, this study failed to demonstrate significant activity in biliary cancer. Furthermore, toxicity was also a concern, with 38% of patients experiencing grade 3 toxicity. The median PFS and OS were 2.56 and 4.86 months, respectively. In retrospect, it is clear that our goal of achieving a primary end point of median PFS time of ⩾5 months was too ambitious. Current phase 3 data of second-line chemotherapy in advanced biliary cancer are lacking; therefore, the target median PFS or OS in the second-line setting is unclear. Available data are derived primarily from small prospective studies or retrospective data. The largest retrospective study conducted to date was from the Princess Margaret Hospital that included 378 patients with advanced biliary tumours. The authors evaluated the benefit of various second-line chemotherapy regimens and demonstrated a PFS of 2.8 months and an OS of 7.5 months (Walter et al, 2013). Another retrospective study involving 56 patients and evaluating various second-line chemotherapy regimens like gemcitabine and 5-fluorouracil reported a similar PFS of 2.8 months but a better median OS of 13.8 months (Rogers et al, 2014). Other small, prospective studies included small numbers of patients with PFS ranging from 1.6 to 5.4 months (Oh et al, 2011; Sasaki et al, 2012). However, no definitive conclusion can be drawn, as none of these studies were randomised. Currently, there is a prospective trial being conducted in the United Kingdom where patients are being randomised to receive either 5-fluorouracil/oxaliplatin or best supportive care alone in the second-line setting (Clinicaltrials.gov).
Bevacizumab, an antivascular endothelial growth factor monoclonal antibody, has been studied in phase 2 studies in advanced biliary cancer. Although generally the outcomes have been encouraging, these studies were primarily conducted in the first-line setting. For example, the combination of bevacizumab with gemcitabine and oxaliplatin achieved an OS of 10 months and a response rate (RR) of 44% (Zhu et al, 2010). Furthermore, one retrospective study evaluated the use of FOLFIRI plus bevacizumab in the refractory setting and demonstrated promising results, with a PFS of 8 months and an OS of 20 months (Guion-Dussere et al, 2015). However, once again, the results of these studies are limited because of their retrospective nature and small number of enrolled patients. Cabozantinib, a multikinase inhibitor with potent activity against vascular endothelial growth factor receptor 2 (VEGFR2) and MET, was performed for patients with advanced refractory cholangiocarcinoma in a small phase 2 study (Goyal et al, 2017). Similar to our study, the drug had limited activity in unselected patients with cholangiocarcinoma and significant toxicity.
Our study has several limitations, including the small patient population and its nonrandomised study design. Furthermore, the incidence of grade 3/4 toxicities was high and they were primarily haematologic in nature. This may have been attributable to the selection of docetaxel as the backbone chemotherapy to use in combination with SPI-1620. In the previous study of docetaxel in cholangiocarcinoma, 76% of the patients experienced grade 4 neutropenia and 65% experienced febrile neutropenia. Once again, in retrospect, docetaxel may have not been the best choice; however, as no standard second-line chemotherapy has been identified, it is unclear which chemotherapy may have yielded better results. Evidence does suggest that perhaps the selection of capecitabine, with its more favourable haematologic safety profile, may have improved efficacy and safety.
Conclusion
In conclusion, the combination of SPI-1620 and docetaxel did not meet its primary end point in an unselected population of patients with advanced biliary cancer. Future studies in the second-line setting should select patients based on tumour location, tumour biology, and biomarkers. Furthermore, randomised studies are desperately needed in the second-line setting to establish standards of care.

Progression-free survival.
Change history
11 July 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Albores-Saavedra J, Schwartz AM, Batich K, Henson DE (2009) Cancers of the ampulla of vater: demographics, morphology, and survival based on 5,625 cases from the SEER program. J Surg Oncol 100: 598–605.
Brooks DP, DePalma PD, Pullen M, Gellai M, Nambi P (1995) Identification and function of putative ETB receptor subtypes in the dog kidney. J Cardiovasc Pharmacol 26: S322–S325.
The Christie NHS Foundation Trust. Active symptom control alone or with mFOLFOX chemotherapy for locally advanced/metastatic biliary tract cancers (ABC06). NCT01926236. Available at https://clinicaltrials.gov/ct2/show/NCT01926236 (accessed on 10 April 2017).
Goyal L, Zheng H, Yurgelun MB, Abrams TA, Allen JN, Cleary JM, Knowles M, Regan E, Reardon A, Khachatryan A, Jain RK, Nardi V, Borger DR, Duda DG, Zhu AX (2017) A phase 2 and biomarker study of cabozantinib in patients with advanced cholangiocarcinoma. Cancer 123 (11): 1979–1988.
Guion-Dussere JF, Lorgis V, Vincent J, Bengrine L, Ghiringhelli F (2015) FOLFIRI plus bevacizumab as a second-line therapy for metastatic intrahepatic cholangiocarcinoma. World J Gastroenterol 21: 2096–2101.
He S, Shen J, Sun X, Liu L, Dong J (2014) A phase II FOLFOX-4 regimen as second-line treatment advanced biliary tract cancer refractory to gemcitabine/cisplatin. J Chemother 26: 243–247.
Jain RK (1991) Haemodynamic and transport barriers to the treatment of solid tumours. Int J Radiat Biol 60: 85–100.
Lee SC, Kim K, Kim H, Kim HJ, Kim SH, Bae S-B, Kim CK, Lee N, Lee KT, Park SK, Won J-H, Yun J, Hong DS, Park HS (2012) Prognostic factor analysis of second-line chemotherapy in advanced biliary tract cancer. J Clin Oncol 30, Abstract e14688.
Nelson J, Bagnato A, Battistini B, Nisen P (2003) The endothelin axis: emerging role in cancer. Nat Rev Cancer 3: 110–116.
Oh SY, Jeong CY, Hong SC, Kim TH, Ha CY, Kim HJ, Lee GW, Hwang IG, Jang JS, Kwon HC, Kang JH (2011) Phase II study of second line gemcitabine single chemotherapy for biliary tract cancer patients with 5-fluorouracil refractoriness. Invest New Drugs 29: 1066–1072.
Papakostas P, Kouroussis C, Androulakis N, Samelis G, Aravantinos G, Kalbakis K, Sarra E, Souglakos J, Kakolyris S, Georgoulias V (2001) First-line chemotherapy with docetaxel for unresectable or metastatic carcinoma of the biliary tract. A multicenter phase II study. Eur J Cancer 37: 1833–1838.
Pazdur R, Royce ME, Rodriguez GI, Rinaldi DA, Patt YZ, Hoff PM, Burris HA (1999) Phase II trial of docetaxel for cholangiocarcinoma. Am J Clin Oncol 22: 78–81.
Rai A, Rajeshkumar NV, Shord S, Gulati A (2005) ET-B receptor agonist, IRL 1620, does not affect paclitaxel plasma pharmacokinetics in breast tumour bearing rats. J Pharm Pharmacol 57: 869–876.
Rajeshkumar NV, Matwyshyn G, Gulati A (2007) IRL-1620, a tumor selective vasodilator, augments the uptake and efficacy of chemotherapeutic agents in prostate tumor rats. Prostate 67: 701–713.
Rajeshkumar NV, Rai A, Gulati A (2005) Endothelin B receptor agonist, IRL 1620, enhances the anti-tumor efficacy of paclitaxel in breast tumor rats. Breast Cancer Res Treat 94: 237–247.
Rogers JE, Law L, Nguyen VD, Qiao W, Javle MM, Kaseb A, Shroff RT (2014) Second-line systemic treatment for advanced cholangiocarcinoma. J Gastrointest Oncol 5: 408–413.
Sasaki T, Isayama H, Nakai Y, Mizuno S, Yamamoto K, Yagioka H, Yashima Y, Kawakubo K, Kogure H, Togawa O, Matsubara S, Ito Y, Sasahira N, Hirano K, Tsujino T, Toda N, Tada M, Omata M, Koike K (2012) Multicenter phase Ii study of S-1 monotherapy as second-line chemotherapy for advanced biliary tract cancer refractory gemcitabine. Invest New Drugs 30: 708–713.
Siegel RL, Miller KD, Jemal A (2016) Cancer statistics, 2016. CA Cancer J Clin 66: 7–30.
Suzuki E, Ikeda M, Okusaka T, Nakamori S, Ohkawa S, Nagakawa T, Boku N, Yanagimoto H, Sato T, Furuse J (2013) A multicenter phase II study of S-1 for gemcitabine-refractory biliary tract cancer. Cancer Chemother Pharmacol 71: 1141–1146.
Tolcher AG, Gari V, Reddy G, Lenaz L, Tidmarsh G, Gulati A (2011) A phase I, open label, ascending dose study of the safety, tolerability, pharmacokinetics and pharmacodynamics of the endothelin B agonist, SPI-1620, in patients with recurrent or progressive carcinoma. The Twelfth International Conference on Endothelin Vol. 9. The British Pharmacological Society: Cambridge, UK.
Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira SP, Roughton M, Bridgewater J ABC-02 Trial Investigators (2010) Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 362: 1273–1281.
Walter T, Horgan AM, McNamara M, McKeever L, Min T, Hedley D, Serra S, Krzyzanowska MK, Chen E, Mackay H, Feld R, Moore M, Knox JJ (2013) Feasibility and benefits of second-line chemotherapy in advanced biliary tract cancer: a larger retrospective study. Eur J Cancer 49: 329–335.
Yanagisawa M, Kurihawa H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T (1988) A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332: 411–415.
Zhu AX, Meyerhardt JA, Blaszkowsky LS, Kambadakone AR, Muzikansky A, Zheng H, Clark JW, Abrams TA, Chan JA, Enzinger PC, Bhargava P, Kwak EL, Allen JN, Jain SR, Stuart K, Horgan K, Sheehan S, Fuchs CS, Ryan DP, Sahani DV (2010) Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: a phase 2 study. Lancet Oncol 11: 48–54.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr Song is an employee of Spectrum. The other authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Kim, R., Chiorean, E., Amin, M. et al. Phase 2 study of combination SPI-1620 with docetaxel as second-line advanced biliary tract cancer treatment. Br J Cancer 117, 189–194 (2017). https://doi.org/10.1038/bjc.2017.160
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.160
Keywords
This article is cited by
-
Cholangiocarcinoma: State of the Art
Journal of Gastrointestinal Cancer (2020)
-
Crystal structures of human ETB receptor provide mechanistic insight into receptor activation and partial activation
Nature Communications (2018)
-
A randomised phase II study of second-line XELIRI regimen versus irinotecan monotherapy in advanced biliary tract cancer patients progressed on gemcitabine and cisplatin
British Journal of Cancer (2018)
