
Abstract
Background:
Non-small-cell lung cancer (NSCLC) is the main type of lung cancer with high mortality rates in worldwide. There is a need to identify better biomarkers to detect NSCLC at an early stage as this will improve therapeutic effect and patient survival rates.
Methods:
Two lectins (AAL/AAGL and AAL2/AANL), which specifically bind to tumour-related glycan antigens, were first used to enrich serum glycoproteins from the serum of early NSCLC patients, benign lung diseases subjects and healthy individuals. The samples were investigated by using iTRAQ labelling and LC-MS/MS.
Results:
A total of 53 differentially expressed proteins were identified by quantitative proteomics and four glycoproteins (AACT, AGP1, CFB and HPX) were selected for further verification by western blotting. Receiver operating characteristic analysis showed AACT was the best candidate for early NSCLC diagnosis of the four proteins, with 94.1% sensitivity in distinguishing early tumour Stage (IA+IB) from tumour-free samples (healthy and benign samples, HB). The GlcNAcylated AACT was further detected by lectin-based ELISA and has better advantage in clinical application than total AACT. The GlcNAcylated AACT can effectively differentiate Stage I from HB samples with an AUC of 0.908 and 90.9% sensitivity at a specificity of 86.2%. A combination of GlcNAcylated AACT and carcinoembryonic antigen (CEA) was able to effectively differing Stage I from HB samples (AUC=0.914), which significantly improve the specificity of CEA. The combination application also has the better clinical diagnostic efficacy in distinguishing cancer (NSCLC) from HB samples than CEA or GlcNAcylated AACT used alone, and yielded an AUC of 0.817 with 93.1% specificity.
Conclusions:
Our findings suggest that the GlcNAcylated AACT will be a promising clinical biomarker in diagnosis of early NSCLC.
Similar content being viewed by others


Main
Lung cancer is the most common primary malignant tumour in the world and is the leading cause of mortality in cancer-related deaths due to metastases to other organs (Parkin et al, 2005), especially in China with increasing incidence (Yang et al, 2004). About 80% of lung cancer cases in clinicopathology are non-small-cell lung cancer (NSCLC), which includes the three major histological subtypes, adenocarcinomas (AD), squamous cell carcinomas (SCC) and large cell carcinomas (Wallace, 2009). As the aetiology of lung cancer is not well understood, there remains at present a lack of effective early diagnostic markers. The 5-year survival rate for late-phase patients in the clinic is very low and is at less than 10% (Hoffman et al, 2000). However, the 5-year survival rate in early detection of NSCLC at the IA stage can be achieved at 80% (Mulshine and Sullivan, 2005). Therefore, there is an urgent need to pursue efforts for screening useful biomarkers to improve the survival of patients with early lung cancer.
In a clinical setting, computed tomography (CT) scans are commonly used in the diagnosis of lung cancer patients, although this method has been improved in sensitivity, imaging diagnosis remains problematic due to its poor specificity, high cost and radiation effects (Hassanein et al, 2012). Importantly, CT scanning identifies suspicious lesions that may be benign lesions by tissue diagnosis undergoing surgery and unambiguous tumours may be at late phase resulting in missed optimum times for therapy. Several clinical serum biomarkers currently used in lung cancer diagnosis include carcinoembryonic antigen (CEA), cytokeratin 19-fragments (CYFRA 21-1), neuron-specific enolase and squamous cell carcinoma antigen. Current biomarkers lack sufficient sensitivity and specificity. The CEA is the most commonly biomarker used in lung cancer clinic diagnosis and has been in use since 1980 (Ferrigno et al, 1994). Carcinoembryonic antigen has no specificity in the detection of lung cancer and has only 40–70% sensitivity in NCSLC, resulting in poor clinic diagnosis efficiency at early stage cancer development (Molina et al, 2003). Any combination between CEA with other proteins used in diagnosis of early lung cancer will not improve the diagnostic efficiencies significantly (Plebani et al, 1995; Tarro et al, 2005). Inaccurate or missed diagnosis of lung cancer detection may lead to inappropriate therapy and even death. There remains a need for the identification of novel biomarkers for early diagnosis of lung cancer.
Proteomic analysis is a powerful tool for the global evaluation of protein expression, and has been widely applied in disease biomarker discovery (Srivastava and Srivastava, 2005), especially due to the rapid technological developments of mass spectrometry (Indovina et al, 2013). Quantitative protein expression profiling allows the efficient identification of accurate and reproducible differential expressed proteins in multiple biological samples. Serum samples are readily accessible for analysis and diagnosis clinically. Through the comparison of protein expression profiles between patient’s sera and healthy controls, it enables the potential discovery of clinically useful serum biomarkers for detection and elucidation of mechanisms in cancer progression. Previous serum markers have been investigated as lung cancer biomarker candidates, for instance serum amyloid A in the metastatic pathogenesis of lung cancer (Sung et al, 2011), complement C9 diagnosed for SCC with 53% sensitivity (Narayanasamy et al, 2011). The authors however provide no data on the use of these markers in early diagnosis. There remains an urgent need for the identification of reliable biomarkers in early diagnosis and disease progression monitoring.
Glycosylation is one of the most common post-translational modifications found in secreted proteins and has been shown to play important roles in cancer progression (Dennis et al, 1999). The glycosylation of serum glycoproteins is altered in the development of tumours, with the sensitive detection of disease-associated glycosylation abnormalities, this has the potential to become a useful milestone in the development and identification of diagnostic glycoprotein biomarkers (Ahn et al, 2014). A typical case has been exemplified by α-fetoprotein (AFP) with core fucosylation in the diagnosis of HCC (Aoyagi et al, 1988), the fucosylated form of AFP (AFP-L3) has been demonstrated to have better diagnosis than unmodified AFP alone (Aoyagi et al, 1993) and AFP-L3 as the only diagnostic assay available for HCC has been approved by FDA in 2005.
Lectin is a protein/glycoprotein that can bind to glycans. At present, lectins is an effective tool for enrichment of serum glycoproteins to discover novel biomarkers for cancer diagnosis. Fucosylation and sialylation are the most common glycosylation modifications and have been shown to occur on terminal structures of carbohydrate chains and are involved in the progression of tumours. Lectins as Aleuria aurantia lectin (AAL), Sambucus nigra agglutinin (SNA) and Concanavalin A (Con A) were commonly used to extract fucosylation or sialylation of serum glycoproteins to detect breast cancer (Drake et al, 2012), hepatocellular carcinoma (Ahn et al, 2012) and lung cancer (Wen et al, 2012). In 2009, a novel group of tumour-associated terminal GlcNAc (β-N-acetyl-D-glucosamine) was found to accumulate in lung cancer tissues (Satomaa et al, 2009), and these identified glycans have the potential to be used as a novel class of tumour biomarkers. To date, no lectins have been effectively used to capture terminal GlcNAc glycans in lung cancer.
In this study, the two lectins AAL/AAGL and AAL2/AANL from edible mushroom Agrocybe aegerita have been purified and published in other papers by our group (Zhao et al, 2003; Jiang et al, 2012), and they were first used to extract Gal- and GlcNAc- terminated glycoproteins from the serum of NSCLC patients in order to identify new sensitive early tumour biomarkers. The enriched proteins by lectin affinity chromatography were then labelled with iTRAQ reagents and analysed by LC-MS/MS. Among the identified differentially expressed proteins, four proteins (AACT, AGP1, CFB and HPX) were selected for further validation as serological markers for NSCLC and our results show that the expression level of AACT decreased significantly in early-stage NSCLC and was the best candidate for early diagnosis of NSCLC in the four tested proteins. Finally, we show that GlcNAcylated AACT has the effectively diagnostic performance in early NSCLC and can improve the specificity of CEA when combined with CEA. The GlcNAcylated AACT will be used as useful biomarkers to diagnose early NSCLC in a medical examination setting.
Materials and methods
Clinical serum sample collection
Serum samples were collected from patients with NSCLC and healthy subjects from Tongji Hospital (Wuhan, China). All patients were diagnosed upon CT and histopathological analyses, and all sera were collected at a preoperative stage following a standardised protocol. The serum of healthy controls were obtained with informed consent from volunteers who received medical examinations at Tongji Hospital.
In this study, sera from six healthy subjects, six subjects with benign tumours, nine patients with TNM=IA and four patients with TNM=IB were collected and used in the study. There were 104 serum samples from 19 healthy subjects, 25 subjects with benign tumours, 60 NSCLC patients were used in validation. Information on collected clinical samples are listed in Table 1 and Supplementary Table S2. Patients were graded using the seventh edition of TNM classification of ‘the International Association for the Study of Lung Cancer’ (IASLC).
The serum samples were prepared by collecting blood in empty tubes, and were maintained at room temperature for 30 min before centrifuging at 400 g for 15 min at 4 °C (West-Nielsen et al, 2005). After centrifugation, the sera were divided into 200 μl aliquots in cryotubes and immediately stored at −80 °C until use. Each aliquot was thawed no more than two times prior to use.
Enrichment of glycoproteins by lectin affinity chromatography
Lectins AAL/AAGL and AAL2/AANL were both purified from A. aegerita as previously described (Jiang et al, 2012; Jin et al, 2014). Lectin-coupled Sepharose 4B medium was prepared according to the manufacturer’s instructions (GE Healthcare, Piscataway, NJ, USA). The extraction of glycoproteins with lectins was performed briefly as follows: 900 μg pooled sera from patients or control individuals were diluted in 2 ml of 1 × binding buffer (20 mM Tris-Cl, pH 7.4, 0.5 M NaCl, 1 mM CaCl2, 1 mM MnCl2), were loaded onto the prepared sepharose-bound AAL/AAGL or AAL2/AANL column (1 ml coupled medium) and incubated for 30 min at 4 °C. After washing the column with 4 ml binding buffer three times to remove unbound proteins, the bound glycoproteins were eluted twice with 4 ml elution buffer (200 mM lactose in binding buffer for AAL/AAGL-column, 200 mM N-acetylglucosamine in binding buffer for AAL2/AANL column) (Yang et al, 2005b; Jiang et al, 2012) and concentrated and desalted with a 10-kDa Amicon centrifugal filter unit (Millipore Corp., Billerica, MA, USA). Twenty-five microlitres of the concentrated eluted fraction were electrophoresed by SDS–PAGE and checked by silver staining. The concentration of total protein and eluted glycoproteins were measured using a BCA protein assay kit (Thermo, Waltham, MA, USA).
Lectin blot and western blotting
Lectin blotting was performed as follows: 12 μg of proteins was subjected to 12% SDS–PAGE. After electrophoresis, the gels were transferred onto PVDF membranes (Millipore Corp.) and the membranes were blocked with 0.3% TBST (0.3% Tween-20 in 1 × TBS) containing 5% BSA at 4 °C overnight. The blot was incubated with AAL/AAGL or AAL2/AANL (0.5 μg/ml) at RT for 1 h and the membranes were washed six times with 0.3% TBST, and probed with rabbit polyclonal antibodies against AAL/AAGL or AAL2/AANL (1 : 10 000) at RT for 1 h. The rabbit polyclonal antibodies were prepared from ‘in-house’ as described in Jiang et al (2012) and Jin et al (2014). After a further six washes with 0.3% TBST, the blots were incubated with anti-rabbit IgG HRP-conjugated antibodies (1 : 10 000; Pierce, Waltham, MA, USA) for 1 h at RT, washed six times with 0.3% TBST and developed using an ECL system (Bio-rad, Hercules, CA, USA).
For western blots, the membranes were blocked in 3% BSA in 0.1% TBST and probed with mouse monoclonal antibody against AACT (1 : 5000; Protein-tech, Wuhan, China), and rabbit polyclonal antibodies against AGP1 (1 : 5000; Protein-tech), CFB (1 : 5000; Protein-tech) and HPX (1 : 5000; Protein-tech) as primary antibodies, overnight at 4 °C. The membranes were incubated with HRP-conjugated goat-anti-mouse IgG or goat-anti-rabbit IgG as secondary antibody (diluted 1 : 5000; Protein-tech) for 1 h at RT, and detected by Supersignal West Pico Chemiluminescent HRP Substrate (Thermo). The loading control sample in each gel was used as an internal standard for quantification, and densitometric analysis of each band was measured using Image J software (Ahn et al, 2014).
Protein digestion and labelling with iTRAQ reagents
To prepare proteins for iTRAQ labelling, the eluted glycoproteins were precipitated in a triple volume of buffer (50% acetone, 50% ethanol and 0.1% acetic acid) at −20 °C for 2 h. The glycoproteins were resuspended in 8 M urea, reduced in 10 mM DTT at 50 °C for 1 h, and alkylated by 40 mM iodoacetamide at RT for 30 min and finally digested with trypsin (Mass spectrometry grade; Promega, Madison, WI, USA) overnight at 37 °C. The digested peptides were desalted with C18 columns (Waters, Sep-PaK Vac C18) and evaporated using speed vacuum.
One hundred microgram peptides of each sample (Healthy, Benign, Stage IA and Stage IB) were labelled with 4-plex iTRAQ reagent kit according to the manufacturer’s protocol (SCIEX, San Diego, CA, USA) and the conditions for each labelled reagent was performed as follows: Healthy, iTRAQ reagent 114; Benign, iTRAQ reagent 115; Stage IA, iTRAQ reagent 116 and Stage IB, iTRAQ reagent 117. After labelling, the four samples were pooled and desalted using a C18 column (Waters, Sep-PaK Vac C18), then dried and stored at −80 °C prior to further analysis.
Hybrid Quadrupole-TOF LC-MS/MS and data analysis
The iTRAQ labelled samples were analysed by LC-MS/MS using a Hybrid Quadrupole-TOF LC-MS/MS Mass Spectrometer (TripleTOF 5600; AB Sciex Instruments, Foster City, San Diego, CA, USA). In brief, the samples were subjected to liquid chromatography, the mobile phases consisted of H2O (solvent A) and acetonitrile (ACN, solvent B) where both phases contained 0.1% v/v formic acid and 3% DMSO. The flow rate was maintained at 300 nl min−1. The peptides were eluted from the C18 column by a gradient running from 5 to 80% acetonitrile over 100 min (Software Version: Analyst TF 1.6, Software Application: Eksigent AS2), followed by nanospray into the Hybrid Quadrupole-TOF mass spectrometer (TripleTOF 5600, AB Sciex). The spray voltage was set at 2.3 kVA, ion tolerance was set to 50 mDa and mass spectra were acquired in the positive mode with a m/z window of 350–1250.
Data acquisition was performed with Eksigent Software v. 3.12.1. For iTRAQ quantification, ProteinPilot 4.5 software (AB Sciex’ Software revision number: 1656) was used to analyse label-based quantitation peptides and protein identifications with the integrated Paragon search algorithm (revision number: 4.5.0.0, 1654) as described previously (Zeng et al, 2012).
Query searches were conducted against a Uniprot human reference database (version: 2013.10) using the following data analysis parameters: Sample type: iTRAQ 4-plex (peptide labelled); Cys. alkylation: iodoacetamide (IAA); Digestion: trypsin; Instrument: TripleTOF 5600; Species: Homo sapiens; Search Effort: rapid. All peptides used for the calculation of protein ratios were unique to the given protein or proteins within the group. The protein confidence threshold cutoff was set at greater than 0.05 (Unused ProtScore) and with at least one peptide with 95% confidence.
N-deglycosylation by peptide N-glycosidase F (PNGase F)
The sera of five healthy individuals were pooled, and 2 μg of pooled sera were denatured in denaturing buffer at 100 °C for 10 min and then restored to room temperature. For complete deglycosylation, the denatured protein was directly incubated with 1 μl of PNGase F (New England BioLabs, Ipswich, MA, USA) in reaction buffer at 37 °C for 2 h. The deglycosylated proteins were then separated by SDS–PAGE and analysed by immunoblotting.
Immunohistochemistry
Fresh tissues were removed from diagnosed patients' bodies by operation, and immediately fixed in formalin and rehydrated with 20% sucrose solution, before finally embedded using a standard with O.C.T embedding medium (Optional Cutting Temperature, Tissue-TeK; SAKURA, Torrance, CA, USA) for frozen tissue sections. Immunohistochemistry was performed as described previously (Zeng et al, 2012). The 8 μm tissue sections were incubated with anti-AACT (1 : 200; Protein-tech) antibody or anti-AGP1 (1 : 200; Protein-tech) overnight at 4 °C, and then incubated with 1 : 200 dilution of biotinylated secondary antibody followed by staining with Avidin/Biotinylated enzyme Complex (ABC Tissue staining; VECTASTAIN ABC systems; VECTOR, Burlingame, CA, USA) according to the manufacturer’s instructions. The tissue sections were incubated with 3,3′-diaminobenzidine (DAB; Sigma, St Louis, MO, USA) until a brown colouration was formed. The sections were then stained with haematoxylin, washed with ethanol-dimethylbenzene and sealed by neutral resins.
Analysis of AGP1 protein levels in the Lewis lung cancer mouse model
Female C57BL/6 mice were purchased from the animal centre of the epidemic prevention sector in Hubei province (permission number: SCXK 2008-0005). All mice used in this study were 6 weeks of age. The LLC cells used were purchased from the cell resource centre of Shanghai Institutes for Biological Sciences and cultured in DMEM medium (GIBCO, Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (GIBCO, Invitrogen). After the LLC cells were harvested, the C57BL/6 female mice (n=14) were inoculated with LLC cells (1 × 106) through the tail vein (Ahn et al, 2014). The sera were collected from the tail vein using a capillary tube for several days until all mice were killed and the harvested serum samples were used for western blotting (WB) analysis.
CEA ELISA and lectin-based ELISA of AACT
Protein levels of CEA were measured by ELISA. The ELISA kit for CEA was purchased from USCNK (Wuhan, China) and all samples were tested according to the manufacturers’ instructions.
Lectin-based ELISAs of AACT were performed as previously described (Liang et al, 2015). Briefly, 100 μl of 4.2 μg ml−1 AAL2/AANL in 0.015 M sodium carbonate buffer (pH 9.6) was added to each well of a 96-well plate and incubated at 4 °C overnight. The coated plate was then blocked with 3% (w/v) BSA in PBST (0.1%, v/v, Tween-20 in PBS, pH 7.4) at RT for 1 h, followed by washing with PBST. Serum samples were diluted 30-fold with PBST, and then 100 μl of sample was applied to each well. After 1 h incubation at RT, the plate was washed five times with PBST and incubated with anti-AACT monoclonal antibody (Protein-tech; 1 : 20 000) for 1 h at RT. After additional washing, 5000-fold diluted HRP-conjugated goat-anti-mouse IgG (Protein-tech; 1 : 5000) was added to the plate and incubated for 1 h at RT. After washing the plate five times, 100 μl TMB working solution was added to each well, followed by 50 μl stop solution (2 M sulfuric acid). To determine the concentration of AACT bound to a specific lectin, the absorbance of the plate was measured at 450 nm on a microplate reader (BMG FLUOstar OPTIMA, Gemarny).
All measurements were performed in triplicate.
Bioinformatics and statistical analysis
The glycan array analysis was performed as described previously (Blixt et al, 2004; Fouquaert et al, 2008; Jiang et al, 2012) and the data of glycan array (Version 5.0) were downloaded from CFG (Consortium for Functional Glycomics, http://www.functionalglycomics.org/). The heatmap of AAL/AAGL and AAL2/AANL for binding specificity was analysed by R code (V i386 3.0.1). The identified proteins showing changes were annotated by GO terms using BGI WEGO (http://wego.genomics.org.cn/cgi-bin/wego/index.pl). The protein–protein interaction networks were constructed using STRING v9.1 (http://string-db.org/) and analysed by Cytoscape (V 3.0.2). Analysis of N- or O-linked glycoforms in the identified protein list were from Uniprot database (http://www.uniprot.org/).
For WB, the densitometry data were analysed using the non-parametric unpaired t-test in GraphPad Prism (version 5.0). For all statistical comparisons, P<0.05 was considered statistically significant. The constructed ROC curves and the AUC were analysed using IBM SPSS statistics (version 19.0). Combination diagnosis was performed using binary logistic regression by calculating the new probability, and acquiring the new ROC and AUC values for combined biomarkers. To find the optimum efficiency in early diagnosis of NSCLC, Youden’s index (J) was determined and calculated as J=sensitivity+specificity−1 (Youden, 1950).
Results
Enrichment of glycoproteins from human serum by lectin affinity chromatography
The two lectins from edible mushroom A. aegerita are first used for lectin affinity chromatography for identifying glycoprotein biomarkers of lung cancer. The binding affinity of AAL/AAGL and AAL2/AANL to 611 glycan candidates was tested based on the glycan array analysis as shown in Figure 1A and Supplementary Table S1. The glycan-binding profile presented the affinity of AAL/AAGL and AAL2/AANL at different concentrations and the results indicate that AAL/AAGL was a galectin with specific affinity for β-galactosides (Yang et al, 2005a) and selective binding for Gal-terminated glycans (Bian et al, 2011). AAL2/AANL showed high binding selectivity to glycans with non-reduced GlcNAc termini. It suggested that using these two lectins to extract Gal- and GlcNAc-terminated glycoproteins based on their specificity to identify effectively early biomarkers from the serum of NSCLC patients is reasonable and available.

Glycan array analysis and lectin overlay analysis of glycoproteins enriched by AAL/AAGL and/or AAL2/AANL. (A) The heat map shows the affinity of AAL/AAGL and AAL2/AANL for different glycans at different concentrations and is based on the glycan array. The log10(RFU) values is represented on the bar at the bottom. Colours increasing in red intensity indicate higher binding affinity. (B) Workflow showing the integrated strategy for the identification and validation of candidate biomarkers in NSCLC. (C) SDS–PAGE of whole protein serum samples. Twenty micrograms of each serum sample were separated by SDS electrophoresis and visualised by Coomassie Brilliant Blue staining. From top to bottom the band size of markers are 116.0, 66.2, 45.0, 35.0, 25.0 and 18.4 kDa. (D) Twelve micrograms of proteins from four pooled sera samples (each of 3 μg) were assessed by lectin blotting. EF represents pooled sera from individuals who were healthy or had benign lung diseases, Stage IA and Stage IB patients were enriched for glycoproteins by AAL/AAGL or AAL2/AANL coupled Sepharose 4B chromatography. Twenty-five microlitres of eluted proteins were detected by SDS–PAGE and lectin blotting. M, marker; H, healthy controls; B, benign samples; IA, Stage IA; IB, Stage IB. A full colour version of this figure is available at the British Journal of Cancer journal online.
For biomarker discovery and verification, we employed the following procedural workflow as summarised in Figure 1B. Twenty-five serum samples used for discovery were separated by SDS–PAGE, and all the detected serum samples were homogeneously loaded showing no significant differences between individuals (Figure 1C). Of which, four pooled sera samples were also analysed by lectin blotting and no visible differences were found between samples (Figure 1D). Whole-sera samples were purified by lectin affinity chromatography, the fractions were separated by SDS–PAGE and silver stained. Several proteins were found to be enriched in the eluted fractions (Figure 1E and F, left) with no visible differences observed between the four subject groups. However, from the eluted fractions, large differences in protein levels were found between healthy controls and patients in lectin blots, indicating glycoprotein content changed in patients (Figure 1E and F, right).
Protein identification and quantitative proteomics of lectin-enriched samples
Our elution fraction from lectin affinity chromatography was analysed further by mass spectrometry. The enriched glycoproteins of the four groups were digested and labelled with four different iTRAQ reagents to allow for quantitation and identification by LC-MS/MS. Detailed information of the proteins identified by MS are shown in Supplementary Table 3. A total of 107 proteins were identified with two lectins, among of which 96 proteins were enriched by AAL/AAGL and 49 proteins were identified by AAL2/AANL (Figure 2A and Supplementary Table S3A and C). The protein list was filtered on the basis of significant changes in expression. After filtering, a total of 53 proteins were differentially expressed (27 proteins were up-regulated and 26 proteins were down-regulated) from our MS detection results, of which 45 proteins were enriched by AAL/AAGL and 20 proteins were enriched by AAL2/AANL (Figure 2A and Supplementary Table S3B and D) with 12 proteins enriched by both two lectins (Figure 2A). The 53 differentially expressing proteins (named DEPs) are listed in Table 2. Seventy-two percent of the identified proteins were N-linked glycoforms with terminal GlcNAc, and is consistent with reports that GlcNAc-terminal proteins are accumulated in lung cancer tissues (Satomaa et al, 2009).

Schematic workflow on the discovery of serum biomarkers in NSCLC patients by iTRAQ labelling and comparative data analysis. (A) Data analysis of identified proteins. (B) Quantitative analysis of identified proteins. Proteins were selected for further analysis which produced intensity ratios showing greater than 1.5 or less than 0.67 changes. Ratio changes were based on iTRAQ reporter ions in the disease state compared with healthy controls. (C) GO annotation analysis of total identified DEPs. The three main categories (biological process, cellular component and molecular function) are described. (D) The protein–protein interactions networks of the 53 identified proteins showing changes. Ellipses in red represent up-regulated proteins and blue represents down-regulated proteins in patients compared with healthy control sera. The four proteins indicated in green were selected for further verification. A full colour version of this figure is available at the British Journal of Cancer journal online.
The comparative analyses of DEPs between different patient groups are summarised in Figure 2B. Compared with healthy controls, cancer patients at stages IA and IB produced 10 and 8 proteins that were up-regulated (≥1.5-fold), and 14 and 16 proteins were down-regulated (≤0.67-fold) among the 96 proteins enriched by AAL/AAGL. Seven and five were up-regulated (≥1.5-fold) and six and nine down-regulated (≤0.67-fold) of 49 proteins with AAL2/AANL enrichment, respectively.
GO annotation and protein–protein interaction networks of differentially altered lectin-binding proteins
The 53 DEPs were further annotated by Gene Ontology (GO) analysis. All DEPs were classified according to their biological process, cellular component and molecular function (Figure 2C and Supplementary Table S3E), and most of these proteins were found to be involved in biological regulation, stimuli response, metabolism, immunity and localisation. The most common molecular functional annotations were in transporter activity and protein binding. Detailed information of GO annotation of the DEPs are listed in Supplementary Table S3E. The DEPs protein–protein interactions networks were also analysed using STRING v9.1 and Cytoscape software (Figure 2D and Supplementary Table S3F). Overall there were 21 up-regulated proteins (in red) and 22 down-regulated proteins (in blue). The remaining 10 DEPs showed no interactions with other proteins after querying in STRING v9.1; however, six proteins were shown to be up-regulated and four proteins were down-regulated.
Quantitation with LC-MS/MS and N-linked glycosylation analysis of the selected four proteins
To narrow down the candidate biomarkers, based on being involved in a cancer progression and commercially available antibodies, four DEPs (AACT, AGP1, CFB and HPX) were selected for further screening in an antibody-based immunoassay. MS/MS spectra of AACT, AGP, CFB and HPX showed expressional changes (Figure 3A–D and Supplementary Table S5), with the three proteins (AACT, AGP1, CFB) down-regulated and the HPX protein up-regulated.

Quantitative LC-MS/MS and N-linked glycosylation analysis of AACT, AGP1, CFB, HPX proteins. (A–D) Left, the amino-acid sequences ADLSGITGAR, NWGLSVYADKPETTK, AVISPGFDVFAK and DYFWPCPGR were used to identify AACT, AGP1, CFB and HPX, respectively; Right, the released iTRAQ reporter ions were used to provide the relative quantitation of each protein (Healthy, 114 isobaric tag; Benign, 115 isobaric tag; Stage IA, 116 isobaric tag; Stage IB, 117 isobaric tag). (E–H) Five pooled healthy serum samples were treated with or without PNGase F and analysed by immunoblotting for anti-AACT, anti-AGP1, anti-CFB and anti-HPX antibodies. −, PNGase F-untreated crude samples; +, PNGase F-treated crude samples. A full colour version of this figure is available at the British Journal of Cancer journal online.
To confirm whether the AACT, AGP1, CFB and HPX proteins were glycosylated in our samples, we treated our sera samples with PNGase F and immunoblotted the proteins. The results showed the 65-kDa AACT band was downshifted to 48 kDa after PNGase F treatment, indicating that the 65-kDa band was an N-linked glycosylated form of AACT (Figure 3E). The 44-kDa AGP1 band was downshifted to 24 kDa (Figure 3F); proteins CFB and HPX were also reduced in size after PNGase F treatment (Figure 3G and H). These results demonstrate that the four proteins are N-linked glycoproteins and suggest that glycosylation modification or protein expression levels are potentially altered during tumour development.
Validation of dysregulated glycoproteins in clinical serum samples of lung tumours
To verify the expressional changes of selected proteins were consistent with the MS results, the four glycoproteins were subjected to WB across all 66 serum samples (Supplementary Figure S1). For every WB analysis, we used the same normalisation control (H1). Densitometric analysis was used to measure band intensities and the resulting values were normalised to the control value. The serum levels of AACT, AGP1 and CFB were reduced significantly in cancer patients (Figure 4A–C), with significant decreases in very early Stage IA, whereas HPX were shown to be at slightly higher levels in tumour samples compared with those in benign diseases (Figure 4D). Our immunoblot results are in agreement and is consistent with our MS detection data.

Verification of selected protein candidate biomarkers by western blotting, ROC curve analysis, immunohistochemistry and animal model studies. (A–D) Densitometry analysis of western blots of 66 individual sera. The densitometric ratios were calculated as the value for an individual sample divided by the normalisation control value (positive control samples: H1). (A) AACT, (B) AGP1, (C) CFB, (D) HPX. *P<0.05; **P<0.01; ***P<0.001. (E, F) ROC curves for differentiating Stage IA (E) or Stage (IA+IB) (F) from HB samples. (G) The performance of candidate proteins in distinguishing early NSCLC are listed. Grey bold indicates the improved performance of individual protein or combined proteins. (H) Immunohistochemistry analysis of AACT and AGP1 in Stage IB AD patients compared with normal lung tissue specimens. Upper, AACT (no haematoxylin staining), original magnification, × 40; under, AGP1 (haematoxylin staining), original magnification, × 200. (I) Serum AGP1 protein levels were reduced in the sera of LLC mice (n=14).
ROC analysis of the four protein biomarkers in the early detection of NSCLC
Dot plot analysis was performed to compare the serum proteins levels of four candidates in the NSCLC samples with controls. To detect the diagnostic performance of selected candidates, ROC curves were constructed to test their performance. The down-regulation of proteins AACT, AGP1 and CFB showed good performance in very early Stage IA, and the AUC values were 0.826, 0.823 and 0.816, respectively (Supplementary Table S6). Combinational diagnosis was able to improve the diagnostic performance in Stage IA or Stage (IA+IB) from healthy controls (Supplementary Table S6), which is helpful for clinical diagnosis and yield a higher AUC value with better sensitivity or specificity than a single biomarker. Assessing the combination of AACT and AGP1 significantly enhanced the performance in distinguishing Stage IA from healthy samples with 94.4% sensitivity (AUC=0.837) and discriminate Stage (IA+IB) from healthy controls achieving a sensitivity of 94.1% (AUC=0.849).
A good biomarker for early diagnosis would show significant differences between healthy and early stage tumour groups but also between benign disease and early stage tumour groups. AACT is the most promising biomarker of the four proteins studied in our analysis as it was decreased significantly in very early-stage tumour development. The AUC for AACT between the HB (healthy and benign samples) and Stage IA samples was 0.759 at a sensitivity of 83.3%. Likewise, AGP1 was able to differentiate Stage IA from HB samples with 94.4% sensitivity (Figure 4E and G). AACT could distinguish Stage (IA+IB) from HB samples with an AUC of 0.774 and 94.1% sensitivity (Figure 4F and G). In addition, the combinational measurements of AACT and AGP1 proteins as biomarkers were able to differentiate Stage IA or Stage (IA+IB) from HB with above 94% sensitivity (Figure 4). The diagnostic performance of other two biomarkers (CFB and HPX) is also shown in Figure 4.
Validation of the proteins AACT and AGP1 as biomarkers in human lung tissues and mice models
To confirm whether the protein biomarkers AACT and AGP1 can be applied in a clinical setting, the protein levels of these biomarkers were assessed in human lung tissues and a mouse model. The expression levels of AACT and AGP1 in human lung tissue specimens were detected by immunohistochemistry (Figure 4H) with AACT and AGP1 protein levels shown to be significantly lower in Stage I AD than in healthy tissues. This down-regulation in expression is in agreement with our mass spectrometry and western blot findings.
As above, both the expression levels of AACT and AGP1 in human lung tissue specimens were detected by immunohistochemistry, and we also want to detect the changes of their expression levels with lung cancer development using the LLC orthotropic mouse model. However, there is no commercial antibody against mouse AACT, so only the AGP1 protein expression levels were examined in the LLC mouse model. After mice were inoculated with LLC cells (n=14), the sera were collected at 0, 1, 3, 5, 8, 11, 14 and 19 days from the tail veil (Figure 4I, upper). Thirty-eight days after LLC inoculation, all mice were killed and the mouse lungs were examined for cancer growth. The LLC-injected mice developed tumour nodules in the lung (Figure 4I, middle). WB analysis indicated that AGP1 serum levels were significantly down-regulated between days 5 and 19 with tumour growth (Figure 4I, bottom). Our results confirm that AGP1 serum levels were reduced in early stage lung cancer development in the mouse LLC model, which is consistent with our MS result.
AACT glycosylation changes detected by lectin-based ELISA and GlcNAcylated AACT can significantly improve the diagnostic efficiency of CEA for NSCLC
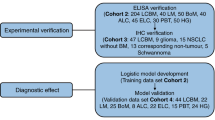
The glycosylation modifications of glycoproteins play an important role in cancer progression, so we detected the glycoform of serum AACT from 100 samples using lectin-based ELISA. The images are shown in Figure 5A. The GlcNAcylated AACT can significantly decrease in Stage I, but will be recovered in late stage, which were consistent with the results of western blot (Supplementary Figure S2). The GlcNAcylated AACT can effectively differentiate Stage I from benign samples with an AUC of 0.932 and 90.9% sensitivity at a specificity of 93.8%, and significantly differ Stage I from HB samples with an AUC of 0.908 and 90.9% sensitivity at a specificity of 86.2% (Table 3), which has better clinical application than the total AACT protein level.

Lecin-based ELISA of AACT and ROC curves analysis of combined GlcNAcylated AACT with CEA. (A) Dot plot showing the results of Lecin-based ELISA of AACT. (B) Analysis of ELISA for CEA. *P<0.05; ***P<0.001. (C) ROC curves analysis of combined GlcNAcylated AACT with CEA (the red line and the blue line was overlapped in comparison Stage I to Benign samples). A full colour version of this figure is available at the British Journal of Cancer journal online.
In a clinical setting, CEA is a widely used serum biomarker for the diagnosis and prognosis of many kinds of cancers including lung cancer. In our study we show CEA was detected in all collected sera samples. The efficiency of CEA for correct diagnosis of early patients was low (Figure 5B and Supplementary Table S6); however we found that combined measurements of GlcNAcylated AACT and CEA show an excellent efficiency in differing Stage I from HB samples (AUC=0.914) with 93.1% specificity, which is much higher than the specificity of CEA (27.6%) (Figure 5C and Table 3). Moreover, combined GlcNAcylated AACT and CEA resulted in a better clinical diagnostic efficacy in distinguishing cancer (NSCLC) from HB samples than CEA or GlcNAcylated AACT used alone, and yielded an AUC of 0.817 with 93.1% specificity (Figure 5C and Table 3). These results imply that GlcNAcylated AACT may be a more effective early biomarker for NSCLC in clinic.
Discussion
The discovery of cancer biomarkers in the serum of patients is a promising research area. Lectins extracted from serum glycoproteins as biomarkers are a reasonable and viable form of diagnosis in early lung cancer. In this study, the two lectins (AAL/AAGL and AAL2/AANL) were first used to enrich Gal- and GlcNAc-terminated glycoproteins from human serum, and the glycosylation of serum proteins significantly increased in patients compared with healthy controls (Figure 1). In total, 107 proteins were identified, of which 62.6% proteins were GlcNAc-terminated glycosylated (Table 2 and Supplementary Table S4), which suggested that our lectins possess the high specificity to GlcNAc-terminal residues and can effectively enrich these glycoforms. As previously reported, higher GlcNAc-terminal glycans were accumulated in lung cancer tissue (Satomaa et al, 2009). Overall these studies show that altered GlcNAcylation of proteins have the potential to make promising targets for the discovery of NSCLC biomarkers, and merits further research in the search for novel biomarkers involving lectins.
In this study, we selected four proteins (AACT, AGP1, CFB and HPX) for further biomarker validation and show these to be associated with cellular processes involved in tumour development and malignant cancer progression. The abnormal expression of AACT has been shown in various tumour types, such as lung adenocarcinoma cell lines (Higashiyama et al, 1995) and down-regulated in human HCC (Santamaria et al, 2013). It has been reported that AACT is indicated as a putative urinary biomarker for the diagnosis of NSCLC and the higher AACT was also detected from urine samples of NSCLC at stage IV compared with healthy individuals (Zhang et al, 2010). Our results showed both the AACT protein levels and the GlcNAcylated AACT levels measured from serum samples of NSCLC at Stage I were reduced, subsequently recovered and even slightly increased in the late stage, which is probably due to the complexity of regulation in the development of tumour. AGP1 is an acute phase protein in human and mice, and shown to have anti-inflammatory properties. The fucosylation and sialylation of AACT and AGP1 are altered in inflammation and cancers (Peracaula et al, 2010). CFB is a biomarker and has been shown to be involved in the development of cancers such as colorectal cancer (Choi et al, 2013) and pancreatic ductal adenocarcinoma (Lee et al, 2014). HPX is a glycoprotein to bind and transport of free haeme in peripheral blood. The fucosylated form of haemopexin (Fuc-Hpx) has been reported as a good serum marker for HCC, which has an AUC of 0.952 at an optimal sensitivity of 92% and a specificity of 92% (Comunale et al, 2009). The overall protein levels of our identified four biomarkers were altered after lectin enrichment, and the glycosylation of the four proteins and their specific structure of oligosaccharides would merit further investigation of their involvement in NSCLC.
From our results we show AACT to possess better diagnostic performance with statistically significant difference and the higher value of AUC with effective sensitivity (Figure 4). Moreover, GlcNAcylated AACT can significantly differ Stage I from Benign with an AUC of 0.932 and 90.9% sensitivity at a specificity of 93.8% and significantly differ Stage I from HB samples with an AUC of 0.908 and 90.9% sensitivity at a specificity of 86.2% (Table 3), which has better advantage in clinical application than the total AACT protein level. It is also shown that combination application of GlcNAcylated AACT and CEA acquires a significantly higher specificity and clinical diagnostic efficacy in NSCLC than CEA or GlcNAcylated AACT used alone (Figure 5C). Our data suggest that the measurement of GlcNAcylated AACT levels can serve as an effective diagnostic biomarker for early NSCLC during medical examinations and could potentially be used as a first-line screen to benefit high-risk patients.
Smoking frequently can easily develop lung diseases and the high-risk populations are easy to cause the occurrence of lung cancer, so it is necessary to analyse the smoking factor in the diagnosis of lung cancer. Each group (healthy, benign and tumour) includes the smoking cases (Table 1) and about 50% samples in the collected sera are smoking cases. The smoking data of AACT and GlcNAcylated AACT were analysed (data not shown), and there are no obvious difference between smokers and non-smokers no matter AACT or GlcNAcylated AACT used in early diagnosis of NSCLC in our small-scale samples.
In conclusion, our findings show that serum AACT especially GlcNAcylated AACT can serve as a good early biomarker for NSCLC detection and improve the clinical diagnostic efficiency of CEA. To further confirm our data analysis, a larger patient cohort for clinical application is required, especially if this is to be implemented for medical examination in the early screening of NSCLC.
Change history
01 March 2016
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ahn JM, Sung HJ, Yoon YH, Kim BG, Yang WS, Lee C, Park HM, Kim BJ, Lee SY, An HJ, Cho JY (2014) Integrated glycoproteomics demonstrates fucosylated serum paraoxonase 1 alterations in small cell lung cancer. Mol Cell Proteomics 13: 30–48.
Ahn YH, Shin PM, Oh NR, Park GW, Kim H, Yoo JS (2012) A lectin-coupled, targeted proteomic mass spectrometry (MRM MS) platform for identification of multiple liver cancer biomarkers in human plasma. J Proteomics 75: 5507–5515.
Aoyagi Y, Saitoh A, Suzuki Y, Igarashi K, Oguro M, Yokota T, Mori S, Suda T, Isemura M, Asakura H (1993) Fucosylation index of alpha-fetoprotein, a possible aid in the early recognition of hepatocellular carcinoma in patients with cirrhosis. Hepatology 17: 50–52.
Aoyagi Y, Suzuki Y, Isemura M, Nomoto M, Sekine C, Igarashi K, Ichida F (1988) The fucosylation index of alpha-fetoprotein and its usefulness in the early diagnosis of hepatocellular carcinoma. Cancer 61: 769–774.
Bian CF, Zhang Y, Sun H, Li DF, Wang DC (2011) Structural basis for distinct binding properties of the human galectins to Thomsen-Friedenreich antigen. PLoS One 6: e25007.
Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J, Razi N, Stevens DJ, Skehel JJ, van Die I, Burton DR, Wilson IA, Cummings R, Bovin N, Wong CH, Paulson JC (2004) Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc Natl Acad Sci USA 101: 17033–17038.
Choi JW, Liu H, Shin DH, Yu GI, Hwang JS, Kim ES, Yun JW (2013) Proteomic and cytokine plasma biomarkers for predicting progression from colorectal adenoma to carcinoma in human patients. Proteomics 13: 2361–2374.
Comunale MA, Wang M, Hafner J, Krakover J, Rodemich L, Kopenhaver B, Long RE, Junaidi O, Bisceglie AM, Block TM, Mehta AS (2009) Identification and development of fucosylated glycoproteins as biomarkers of primary hepatocellular carcinoma. J Proteome Res 8: 595–602.
Dennis JW, Granovsky M, Warren CE (1999) Glycoprotein glycosylation and cancer progression. Biochim Biophys Acta 1473: 21–34.
Drake PM, Schilling B, Niles RK, Prakobphol A, Li B, Jung K, Cho W, Braten M, Inerowicz HD, Williams K, Albertolle M, Held JM, Iacovides D, Sorensen DJ, Griffith OL, Johansen E, Zawadzka AM, Cusack MP, Allen S, Gormley M, Hall SC, Witkowska HE, Gray JW, Regnier F, Gibson BW, Fisher SJ (2012) Lectin chromatography/mass spectrometry discovery workflow identifies putative biomarkers of aggressive breast cancers. J Proteome Res 11: 2508–2520.
Ferrigno D, Buccheri G, Biggi A (1994) Serum tumour markers in lung cancer: history, biology and clinical applications. Eur Respir J 7: 186–197.
Fouquaert E, Peumans WJ, Smith DF, Proost P, Savvides SN, Van Damme EJ (2008) The "old" Euonymus europaeus agglutinin represents a novel family of ubiquitous plant proteins. Plant Physiol 147: 1316–1324.
Hassanein M, Callison JC, Callaway-Lane C, Aldrich MC, Grogan EL, Massion PP (2012) The state of molecular biomarkers for the early detection of lung cancer. Cancer Prev Res (Phila) 5: 992–1006.
Higashiyama M, Doi O, Yokouchi H, Kodama K, Nakamori S, Tateishi R (1995) Alpha-1-antichymotrypsin expression in lung adenocarcinoma and its possible association with tumor progression. Cancer 76: 1368–1376.
Hoffman PC, Mauer AM, Vokes EE (2000) Lung cancer. Lancet 355: 479–485.
Indovina P, Marcelli E, Pentimalli F, Tanganelli P, Tarro G, Giordano A (2013) Mass spectrometry-based proteomics: the road to lung cancer biomarker discovery. Mass Spectrom Rev 32: 129–142.
Jiang S, Chen Y, Wang M, Yin Y, Pan Y, Gu B, Yu G, Li Y, Wong BH, Liang Y, Sun H (2012) A novel lectin from Agrocybe aegerita shows high binding selectivity for terminal N-acetylglucosamine. Biochem J 443: 369–378.
Jin Y, Che T, Yin Y, Yu G, Yang Q, Liu W, Ye X, Yu W, Alok S, Chen Y, Wong BH, Cheng P, Sun H (2014) Lethal protein in mass consumption edible mushroom Agrocybe aegerita linked to strong hepatic toxicity. Toxicon 90C: 273–285.
Lee MJ, Na K, Jeong SK, Lim JS, Kim SA, Lee MJ, Song SY, Kim H, Hancock WS, Paik YK (2014) Identification of human complement factor B as a novel biomarker candidate for pancreatic ductal adenocarcinoma. J Proteome Res 13: 4878–4888.
Liang Y, Ma T, Thakur A, Yu H, Gao L, Shi P, Li X, Ren H, Jia L, Zhang S, Li Z, Chen M (2015) Differentially expressed glycosylated patterns of alpha-1-antitrypsin as serum biomarkers for the diagnosis of lung cancer. Glycobiology 25: 331–340.
Molina R, Filella X, Aug JM, Fuentes R, Bover I, Rifa J, Moreno V, Canals E, Viñolas N, Marquez A, Barreiro E, Borras J, Viladiu P (2003) Tumor markers (CEA, CA 125, CYFRA 21-1, SCC and NSE) in patients with non-small cell lung cancer as an aid in histological diagnosis and prognosis. Tumor Biol 24: 209–218.
Mulshine JL, Sullivan DC (2005) Clinical practice. Lung cancer screening. N Engl J Med 352: 2714–2720.
Narayanasamy A, Ahn JM, Sung HJ, Kong DH, Ha KS, Lee SY, Cho JY (2011) Fucosylated glycoproteomic approach to identify a complement component 9 associated with squamous cell lung cancer (SQLC). J Proteomics 74: 2948–2958.
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55: 74–108.
Peracaula R, Sarrats A, Rudd PM (2010) Liver proteins as sensor of human malignancies and inflammation. Proteomics Clin Appl 4: 426–431.
Plebani M, Basso D, Navaglia F, De Paoli M, Tommasini A, Cipriani A (1995) Clinical evaluation of seven tumour markers in lung cancer diagnosis: can any combination improve the results? Br J Cancer 72: 170–173.
Santamaria M, Pardo-Saganta A, Alvarez-Asiain L, Di Scala M, Qian C, Prieto J, Avila MA (2013) Nuclear alpha1-antichymotrypsin promotes chromatin condensation and inhibits proliferation of human hepatocellular carcinoma cells. Gastroenterology 144: 818–828 e814.
Satomaa T, Heiskanen A, Leonardsson I, Angstrom J, Olonen A, Blomqvist M, Salovuori N, Haglund C, Teneberg S, Natunen J, Carpen O, Saarinen J (2009) Analysis of the human cancer glycome identifies a novel group of tumor-associated N-acetylglucosamine glycan antigens. Cancer Res 69: 5811–5819.
Srivastava S, Srivastava RG (2005) Proteomics in the forefront of cancer biomarker discovery. J Proteome Res 4: 1098–1103.
Sung HJ, Ahn JM, Yoon YH, Rhim TY, Park CS, Park JY, Lee SY, Kim JW, Cho JY (2011) Identification and validation of SAA as a potential lung cancer biomarker and its involvement in metastatic pathogenesis of lung cancer. J Proteome Res 10: 1383–1395.
Tarro G, Perna A, Esposito C (2005) Early diagnosis of lung cancer by detection of tumor liberated protein. J Cell Physiol 203: 1–5.
Wallace WA (2009) The challenge of classifying poorly differentiated tumours in the lung. Histopathology 54: 28–42.
Wen CL, Chen KY, Chen CT, Chuang JG, Yang PC, Chow LP (2012) Development of an AlphaLISA assay to quantify serum core-fucosylated E-cadherin as a metastatic lung adenocarcinoma biomarker. J Proteomics 75: 3963–3976.
West-Nielsen M, Hogdall EV, Marchiori E, Hogdall CK, Schou C, Heegaard NH (2005) Sample handling for mass spectrometric proteomic investigations of human sera. Anal Chem 77: 5114–5123.
Yang L, Parkin DM, Li LD, Chen YD, Bray F (2004) Estimation and projection of the national profile of cancer mortality in China: 1991-2005. Br J Cancer 90: 2157–2166.
Yang N, Tong X, Xiang Y, Zhang Y, Liang Y, Sun H, Wang DC (2005a) Molecular character of the recombinant antitumor lectin from the edible mushroom Agrocybe aegerita. J Biochem 138: 145–150.
Yang N, Tong X, Xiang Y, Zhang Y, Sun H, Wang DC (2005b) Crystallization and preliminary crystallographic studies of the recombinant antitumour lectin from the edible mushroom Agrocybe aegerita. Biochim Biophys Acta 1751: 209–212.
Youden WJ (1950) Index for rating diagnostic tests. Cancer 3: 32–35.
Zeng GQ, Zhang PF, Deng X, Yu FL, Li C, Xu Y, Yi H, Li MY, Hu R, Zuo JH, Li XH, Wan XX, Qu JQ, He QY, Li JH, Ye X, Chen Y, Li JY, Xiao ZQ (2012) Identification of candidate biomarkers for early detection of human lung squamous cell cancer by quantitative proteomics. Mol Cell Proteomics 11: M11103946.
Zhang Y, Li Y, Qiu F, Qiu Z (2010) Comparative analysis of the human urinary proteome by 1D SDS-PAGE and chip-HPLC-MS/MS identification of the AACT putative urinary biomarker. J Chromatogr B 878: 3395–3401.
Zhao C, Sun H, Tong X, Qi Y (2003) An antitumour lectin from the edible mushroom Agrocybe aegerita. Biochem J 374: 321–327.
Acknowledgements
We would like to thank Professor Xiangdong Fu (University of California, San Diego, USA) for his helpful suggestions on the study. We thank Haolong Zeng for iTRAQ labelling and Chunjie Yu for the animal experiments. The present research project was funded by the Natural Science Foundation of China (NSFC) program (31370849, 31301426), Research Fund for the Doctoral Program of Higher Education of China (20130146120019), the National Key Technologies R&D Program (2009ZX09103–676, 2012ZX10001-006), the Chinese 111 project (B06018) and Wuhan university project (No. 204-274103).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Jin, Y., Wang, J., Ye, X. et al. Identification of GlcNAcylated alpha-1-antichymotrypsin as an early biomarker in human non-small-cell lung cancer by quantitative proteomic analysis with two lectins. Br J Cancer 114, 532–544 (2016). https://doi.org/10.1038/bjc.2015.348
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.348
Keywords
This article is cited by
-
Identification a novel clinical biomarker in early diagnosis of human non-small cell lung cancer
Glycoconjugate Journal (2019)
-
Compound kushen injection suppresses human acute myeloid leukaemia by regulating the Prdxs/ROS/Trx1 signalling pathway
Journal of Experimental & Clinical Cancer Research (2018)

{kind=link}
{kind=link}