
Abstract
Background:
Osteosarcoma (OS) is the most common bone malignancy in the paediatric population, principally affecting adolescents and young adults. Minimal advancements in patient prognosis have been made over the past two decades because of the poor understanding of disease biology. Runx2, a critical transcription factor in bone development, is frequently amplified and overexpressed in OS. However, the molecular and biological consequences of Runx2 overexpression remain unclear.
Methods:
si/shRNA and overexpression technology to alter Runx2 levels in OS cells. In vitro assessment of doxorubicin (doxo)-induced apoptosis and in vivo chemosensitivity studies. Small-molecule inhibitor of c-Myc transcriptional activity was used to assess its role.
Results:
Loss of Runx2 sensitises cells to doxo-induced apoptosis both in vitro and in vivo. Furthermore, in conjunction with chemotherapy, decreasing Runx2 protein levels activates both the intrinsic and extrinsic apoptotic pathways. Transplanted tumour studies demonstrated that loss of endogenous Runx2 protein expression enhances caspase-3 cleavage and tumour necrosis in response to chemotherapy. Finally, upon doxo-treated Runx2 knockdown OS cells there was evidence of enhanced c-Myc expression and transcriptional activity. Inhibition of c-Myc under these conditions resulted in decreased activation of apoptosis, therefore insinuating a role for c-Myc in dox-induced activation of apoptotic pathways.
Conclusions:
Therefore, we have established a novel molecular mechanism by which Runx2 provides a chemoprotective role in OS, indicating that in conjunction to standard chemotherapy, targeting Runx2 may be a new therapeutic strategy for patients with OS.
Similar content being viewed by others


Main
Osteosarcoma (OS) is the most common bone malignancy in the paediatric population, and it comprises about 3% of all paediatric tumours. The current treatment regimen for patients with OS includes neoadjuvant chemotherapy, surgical resection of the tumour with assessment of tumour necrosis, and subsequent post-operative systemic chemotherapy. Active chemotherapeutic agents used in the treatment of OS include doxorubicin (doxo), cisplatin, and high-dose methotrexate (Bueno et al, 2004; Clark et al, 2008; Geller and Gorlick, 2010; Rainusso et al, 2013). Owing to a lack of reliable predictive biomarkers of therapeutic efficacy, the response to chemotherapy is assessed histologically after tumour resection. Poor responding tumours show less than 90% necrosis, whereas a good response is defined as greater than 90% or complete necrosis. The tumour response to chemotherapy is directly correlated with long-term patient survival (Provisor et al, 1997; Kager et al, 2003; Miwa et al, 2013). With the current treatment regimen, the survival rates are 65–75% for patients with localised disease, but only about 20% for those with evidence of metastatic disease (Wang, 2005; Ta et al, 2009; Geller and Gorlick, 2010). Overall, these survival rates have not significantly improved over the past 30 years despite ongoing studies (Tang et al, 2008; Mirabello et al, 2009; Ta et al, 2009).
Although specific gene alterations have been associated with OS, the mechanisms leading to the development and progression of the disease are not well defined (Tang et al, 2008). One gene often amplified and aberrantly expressed in OS is the transcription factor Runx2 (Man et al, 2004; Nathan et al, 2009; Sadikovic et al, 2009; Won et al, 2009). Runx2 is the master regulator of skeletal development and directly regulates the cell fate decision between proliferation, apoptosis, and differentiation in osteoblasts (Karsenty and Wagner, 2002; Franceschi et al, 2007; Teplyuk et al, 2008; Ghali et al, 2010; Olfa et al, 2010). Overexpression of Runx2 has been correlated with an increase in proliferation of OS cells in vitro, poor clinical outcome, and poor response to chemotherapeutic regimens (Pereira et al, 2009; San Martin et al, 2009; Won et al, 2009; Sadikovic et al, 2010; van der Deen et al, 2012; Lucero et al, 2013; Lamoureux et al, 2014; Lee et al, 2015). Runx2 also prevents apoptosis in LNCaP cells after treatment with docetaxel, which implicates a role for Runx2 in chemoresistance in non-osseous tumours (Browne et al, 2012).
It has been suggested that Runx2 gene expression is a marker for chemotherapeutic resistance in OS (Sadikovic et al, 2010). However, before Runx2 can be considered as a viable biomarker or therapeutic target for OS, it is imperative that investigations into understanding its biological role in OS be performed before prolonged inhibition of this critical transcription factor can be considered. Here, we show that the knockdown of Runx2 sensitises OS to cell death following treatment with doxo, in vitro and in vivo. We have also provided a novel mechanism for the increased doxo-induced apoptosis in Runx2 knockdown cells by providing evidence that c-Myc has a role in the activation of pro-apoptotic pathways, and inhibition of c-Myc abolishes molecular characteristics of apoptotic activation in Runx2 knockdown cells after chemotherapy treatment. Thus, we have demonstrated a novel chemoprotective role for Runx2 in OS, which implies that inhibition of Runx2 with concurrent chemotherapy is a potential therapeutic intervention for patients with OS.
Materials and Methods
Cell culture, transfections, and inhibitors
Osteosarcoma cell lines were cultured in Dulbecco’s modified Eagle’s medium and supplemented with 10% FBS and maintained at 37 °C with 5% CO2. The m824 and c2984 cell lines were derived from p53+/− mouse OSs in the laboratory (Ma et al, 2009). Stable c2984, DLM8, and HOS control or Runx2 knockdown cell lines were generated by transfecting Open Biosystems pLKO.1 TRC control or pLKO.1 shRunx2 vector (clone ID: TRCN0000013655) utilising the Lipofectamine LTX reagent (Invitrogen, Grand Island, NY, USA). Positively transfected clones were selected with puromycin (2 μg ml−1), and real-time PCR confirmed Runx2 knockdown. FlexiTube (Qiagen, Valencia, CA, USA, catalogue# GS12393) siRNA for Runx2 was transfected per manufacturer’s recommendation. Stable m824 cell lines overexpressing Runx2 were generated by transfecting a FLAG-Runx2 or empty vector using the Lipofectamine LTX reagent, and positively transfected clones were selected using neomycin (1 mg ml−1). Cells were cultured in the presence of DMSO (vehicle), doxo (Sigma, St Louis, MO, USA), or the c-Myc inhibitor 10058-F4 (Sigma) for the reported amount of time. Doxorubicin dosing for each mouse and human OS cells was determined through analysis of killing curves previously performed. For caspase westerns, 293 cells untreated or treated with staurosporine (Sigma), a known inducer of caspase-dependent apoptosis, was used as a positive control.
Cell proliferation assays
Growth assays were performed by plating 1000 cells per well in a 96-well dish. Cell growth was assessed daily by addition of the Cell Counting Kit-8 (CCK-8) reagent (Dojindo Laboratories, Kumamoto, Japan), according to the manufacturer’s instructions (Cell counting kit-8 (CCK-8) assay kit; Dojindo Laboratories). Each cell line was plated in triplicate, and the value presented represents the average of the samples. The P-values were calculated comparing values from control cells to shRunx2 cells on each day.
Quantitative real-time PCR
Total mRNA was extracted with the RNeasy Mini kit (Qiagen) and quantified using Bio-Rad spectrophotometer (Bio-Rad, Hercules, CA, USA). Total RNA (500 ng) were used for cDNA synthesis using the High-Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR with iQ SYBR Green Super Mix (Bio-Rad) was performed using QuantiTect gene-specific primer pairs (Qiagen) utilising the StepOnePlus real-time PCR machine (Applied Biosystems). The relative mRNA expression was calculated with the ΔΔCT method.
Western blot analysis
Cells were lysed in RIPA buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% Na-deoxycholate) supplemented with protease inhibitors (Complete mini, Roche, Indianapolis, IN, USA) and phosphatase inhibitors (Sigma, P0044 and P5726). Nuclear lysates were isolated utilising the NE-PER nuclear and cytoplasmic extraction reagents (Promega) following the manufacturer’s instructions. Lysates were boiled in NuPAGE LDS sample buffer (Invitrogen), separated on NuPAGE Novex 4%–12% Bis-Tris Gel (Invitrogen), and transferred to polyvinylidene difluoride membranes. The blots were probed overnight with antibodies to Runx2 (Cell Signaling, Danvers, MA, USA, D1H7), Caspase-9 (Cell Signaling, 9502), Cleaved Caspase-9 (Cell Signaling, 9501), Caspase-8 (Cell Signaling), Anti-Fas Antibody, clone 7C10, Rat mAB (Millipore, Darmstadt, Germany, 1C12), Caspase-3 (Cell Signaling, 9662), Fas-Associated protein with Death Domain (FADD; Santa Cruz Biotechnology, Dallas, TX, USA, H-181), TBP (Thermo Scientific, Grand Island, NY, USA, MA1–21516), tubulin (Fisher Scientific, Waltham, MA, USA, Ab-2, clone DM1A), c-Myc (Santa Cruz Biotechnology), or GAPDH (EMD Millipore, Billerica, MA, USA, AB2302). Blots were incubated with the appropriate secondary HRP-conjugated antibodies for 1 h, and signal was detected utilising Millipore Immobilon Western chemiluminescent HRP substrate (Millipore). FADD protein levels were quantified utilising the Image J software (http://imagej.nih.gov/ij), and were normalised to GAPDH.
Immunoprecipitation
C2984 cells were treated with 50 nM doxo or DMSO for 48 h. Cells were then lysed with NP-40 buffer and centrifuged to clear the lysates. The lysates were then quantified using the BCA Assay (Thermo Fisher). A measure of 500 μg of lysate wwere then incubated with Fas or negative control IgG overnight at 4°. Lysates were then incubated with Protein A or Protein G magnetic beads for 2 h at 4°. Bead pellets were collected by magnetic separation and washed with lysis buffer and re-suspended in 4 × LDS NuPAGE Sample Buffer (Invitrogen). Protein complexes were then separated on a 4–12% SDS–PAGE gel and transferred to PVDF membranes. Membranes were then incubated overnight with respective antibodies. 5% of the input lysate was used as control.
Luciferase transcription factor reporter assays
To test for c-Myc activity, the Myc Cignal reporter assay from Qiagen (CCS-012L) was used. c2984 control or shRunx2 cells were transiently transfected with the c-Myc luciferase reporter plasmid and treated with DMSO or doxo 24 h post transfection. The Dual Luciferase Assay system (Promega) was used to analyse reporter activity. The firefly luciferase signal was normalised to the renilla luciferase signal, and the data presented in the graphs is the normalised average of triplicate assays. P-values were calculated comparing control shRNA cells treated with doxo to shRunx2 cells treated with doxo.
Allograft in vivo tumour studies
All animal experiments were conducted according to institutional animal care and use committee protocols after approval was obtained from the BCM Institutional Review Board (BCM Animal Protocols AN-336 and AN-5225). c2984 control shRNA and shRunx2 cells (1 × 106) were each injected subcutaneously into 10 NU/NU nude female mice. Tumour growth was measured weekly, and tumour volume (V) was determined by the formula V=(4/3) × p × [(L+W)/4]3, L being the longer cross-section and W being the shorter. Half of the control or shRunx2 mice were treated with either doxo (4 mg kg−1 per dose) or PBS at the first sign of palpable tumour, and injections were performed every 7 days for 4 weeks. Mice were weighed and tumour volume was documented two times per week. Tumour tissue was isolated at the time of killing the mice, lysed in RIPA lysis buffer using PT100 homogenizer (Polytron), and lysates were subjected to western blot analysis.
Immunohistochemistry
Tumours were fixed in 10% formalin at the time of killing the mice, paraffin-processed, sectioned, and stained with haematoxylin and eosin. Slides were scanned in the Department of Veterinary Medicine and Surgery at the MD Anderson Cancer Center. Stained tumour sections were scanned, and the percentage of necrosis in each tumour section was quantified utilising the Aperio ImageScope Viewer (Leica Biosystems, Buffalo Grove, IL, USA). Percentage necrosis was quantified by the following formula: ((area of necrosis in the tumour section/total area of the tumour section) × 100). The percentage necrosis represented in the graph represents the average of the tumours in each untreated and doxo-treated group. Statistical difference between the groups was determined by the two-sample Wilcoxon rank-sum (Mann–Whitney) test.
Apoptosis detection with Annexin V staining
c2984, DLM8, and HOS control or shRunx2, or m824 cmv10 or Runx2 cells were plated overnight. Media containing doxo was added to the cells for the time listed. Cells were then trypsinised, washed with 1 × PBS, and resuspended in 1 × Annexin V binding buffer (FITC Annexin V Apoptosis Detection Kit I, BD Pharmingen, San Jose, CA, USA). FITC Annexin V (AV) and propidium iodide were added to the cells, and incubated for 15 min at RT in the dark. Cells were analysed by flow cytometry within 1 h. Percent apoptosis represents the sum of the AV and AV/propidium iodide-positive cell populations. For apoptosis studies, the P-values were calculated comparing the control cells treated with doxo to shRunx2 cells treated with doxo.
Cytochrome c release determination
c2984 and HOS control or shRunx2 cells were plated overnight. Media containing doxo was added to the cells for the time listed. Cytochrome c release was quantified using the Flowcellect Cytochrome c kit (Millipore) following the manufacturer’s instructions. Cells were analysed by flow cytometry, and % cytochrome c release represents FITC-negative population. For cytochrome c release, the P-value was calculated comparing the control cells treated with doxo to shRunx2 cells treated with doxo.
Statistical analysis
The significance of differences between control and shRunx2 samples under different conditions was determined by Student’s t-test. A P-value ⩽0.05 was considered significant. A two-sided Student’s t-test (paired) was used to calculate the level of statistical significance for the relative tumour volumes (treated/untreated) in control mice vs in shRunx2 mice on each day observed. P values less than 0.05 were considered statistically significant. The relative tumour volume reduction after dox treatment in control vs shRunx2 cohorts was subjected to analysis of covariance. Briefly, the data were fitted into a multi-linear regression model that contains linear terms of control, shRunx2, and control-shRunx2 (interaction term) with relative tumour volume as the response variable. The statistical analysis was performed by using R software (https://www.r-project.org/).
Results
Knockdown of Runx2 sensitises OS cells to treatment with doxo in vitro via intrinsic and extrinsic apoptotic pathways
The efficacy of chemotherapeutics, including doxo, depends on the ability of these agents to trigger apoptosis in cancer cells. RUNX2 mRNA is increased in OS tumours that show a poor response to chemotherapy, and consequently, we investigated whether the expression of Runx2 confers mechanisms of chemoresistance to OS cells. We treated control and stably transduced shRunx2 OS cells with doxo, and noticed that the Runx2 knockdown cells consistently displayed enhanced chemosensitivity to lower doses of doxo than the control cells (Figure 1A). In addition, compared with control cells treated with doxo, the Runx2 knockdown cells showed a significant decrease in cell proliferation (Figure 1B and C, and Supplementary Figure 1A). These results indicate that the expression of Runx2 is integral to OS cell survival in response to doxo.

Runx2 knockdown enhances doxorubicin (doxo) sensitivity in OS. (A) Cell viability assay for c2984 control and shRunx2 mouse OS cell lines treated with the indicated doses of doxo for 24 h. Each value is the average of triplicate samples. P-values were determined comparing control and shRunx2 values at each dose of doxo. *P⩽0.05, ***P⩽0.001. (B and C) Cell survival assay for mouse c2984 (B) and human HOS (C) control and shRunx2 OS cells treated with the indicated doses of doxo for 72 h. Each value is the average of triplicate samples, and represents the absorbance levels normalised to appropriate untreated cell lines. P-values were determined comparing control and shRunx2 values at each dose of doxo. *P⩽0.05, ***P⩽0.001. Validation of Runx2 protein knockdown is shown by western blot next to survival graphs.
After demonstrating this phenotype associated with dysregulated Runx2 expression, we were interested in the molecular mechanism(s) by which Runx2 confers chemoresistance to OS cells. We investigated if the enhanced sensitivity in the Runx2 knockdown cells was caused by an increase in apoptosis by performing FACS analysis of Annexin V staining after doxo treatment. We observed that the downregulation of Runx2 in mouse and human OS cells resulted in a significant increase in the percentage of apoptotic cells compared with the control cells (Figure 2A and B, and Supplementary Figure 1B). Importantly, to further prove that Runx2-dependent mechanisms are directly contributing to chemosensitivity, we performed the reciprocal experiment using the murine M824 OS cell line with relatively low Runx2 expression (Supplementary Figure 1C). We observed that overexpression of Runx2 protected OS cells from doxo-induced apoptosis compared with control cells. These experiments demonstrate that the expression of Runx2 is integral to OS cell survival in response to chemotherapy.

Induction of apoptosis and cytochrome c release in doxo-treated Runx2 knockdown cells in vitro. (A and B) Annexin V FACS analysis of c2984 (A) and HOS (B) control and shRunx2 cells treated with the indicated dose of doxo for 48 h (A) or 24 h (B) revealed a significant increase in apoptotic cells in the shRunx2 samples compared with control cells. Values represent the average of triplicate samples. (C and D) FACS analysis of cytochrome c release for c2984 (C) or HOS (D) control and shRunx2 cells treated with the indicated doses of doxo for 48 h (C) or 24 h (D). % cyt. c release represents FITC-negative subpopulation. Values represent the average of triplicate samples, *P⩽0.05, ***P⩽0.001.
The initiation of cell death can be achieved through the extrinsic or intrinsic apoptotic pathways. Doxorubicin induces apoptosis in a caspase-dependent manner in U2OS cells (Yuan et al, 2007). Therefore, we assessed whether the extrinsic or intrinsic pathways were activated in our Runx2 knockdown cell lines after doxo treatment. The intrinsic pathway is mediated by cytochrome c release before caspase cleavage (Wang, 2005) noticed a significant increase in the percentage of cytochrome c release after doxo treatment in mouse and human Runx2 knockdown OS cells (Figure 2C and D). Furthermore, we noticed that higher expression of the extrinsic ligand, Fas, and adaptor protein, FADD, in our mouse and human Runx2 knockdown cell lines when compared with control cell lines (Figure 3A and B, and Supplementary Figure 2A). Activation of the extrinsic pathway results in the recruitment of FADD and the formation of the death-inducing signalling complex (DISC), and this potentiates the activation of downstream caspases and subsequent apoptosis (Kavurma and Khachigian, 2003). To investigate DISC formation, we performed co-immunoprecipitation of DISC components Fas and FADD within OS upon doxo treatment in control and Runx2 knockdown cells. Upon doxo-treatment, we observed enhanced Fas and FADD interaction (Supplementary Figure 2B), thus insinuating increased DISC formation in Runx2 knockdown cells. These results demonstrate that Runx2 can influence the activation of both the intrinsic mitochondrial-mediated and extrinsic death receptor apoptotic pathways after treatment with doxo.

Activation of the extrinsic apoptotic pathways and cleavage of cell-death caspases in doxo-treated Runx2 knockdown cells in vitro. (A and B) FADD expression is significantly induced in mouse c2984 (A) and human HOS (B) shRunx2 cells after treatment with 50 nM (A) or 100 nM (B) doxo for 24 h, when compared with control cells. qPCR was performed to assess the expression levels of FADD. P-value was determined using the Students t-test comparing control and shRunx2 cells treated with doxo, and **P⩽0.01,***P⩽0.001. Western blot analysis of FADD expression in cell lysates isolated after doxo treatment in c2984 and HOS control and shRunx2 cell lines are shown in adjacent blots. GAPDH and tubulin were utilised as internal loading controls. (C) Western blot analysis of activated caspase-8 in total cell lysates isolated from c2984 control or shRunx2 cells treated with doxo (50 nM) for 48 h. Ctl=c2984 control shRNA OS cells, shRunx2=c2984 shRunx2 OS cells. Tubulin was used for loading control. (D) Western blot analysis of activated caspase-3 in c2984 control or shRunx2 cells treated with 50 nM doxo for 48 h. Ctl=c2984 control shRNA OS cells, shRunx2=c2984 shRunx2 OS cells. TBP was used as internal loading control. For control lanes, (−) and (+) represent 293 cells untreated or treated with staurosporine, respectively.
Activation of the extrinsic and intrinsic apoptotic pathways culminates in the cleavage of cellular caspases that induce cell death. We next investigated the cleavage of caspases in doxo-treated Runx2 knockdown cells. We noticed an increase in the cleavage of caspase 8, an initiator caspase involved in necrosis and apoptosis induced by the extrinsic pathway, in the Runx2 knockdown cells after doxo treatment (Figure 3C; McIlwain et al, 2013). We also observed cleavage of both caspase-3 and caspase-9, which are activated by the intrinsic pathway, in the doxo-treated Runx2 knockdown cells (Figure 3D and Supplementary Figure 2C; Porter and Janicke, 1999). Taken together, these data demonstrate that Runx2 expression has a critical role in the activation of apoptotic pathways and the cleavage of caspases following chemotherapy exposure.
Decreased expression of Runx2 enhances chemosensitivity and tumour necrosis in vivo
As we noticed an increase in doxo sensitivity in vitro, we next tested whether knockdown of Runx2 could sensitise tumours to treatment with doxo in vivo. We injected control or shRunx2-expressing OS cells subcutaneously into nude mice, allowed for palpable tumour growth, and began weekly doxo (4 mg kg−1 per dose) intraperitoneal treatments. We observed that the tumours expressing the Runx2 shRNA were more sensitive to doxo, as shown by the noticeable decrease in tumour volume after treatment (Figure 4A). Furthermore, we observed an increase in detectable cleaved caspase-3 only in the Runx2 knockdown tumours after doxo treatment (Figure 4B), which corroborated our in vitro data. A statistical significance between tumour volumes was noted at day 27 post treatment (P<0.05) and an ANOVA revealed a coefficient of −0.0078 for control and Runx2 knockdown, but it did not have statistical significance (P>0.1).

Knockdown of Runx2 sensitises OS cells to doxorubicin treatment in vivo. (A) Graph represents tumour volume at multiple time points over the course of doxo treatment compared with the tumour volume at the start of treatment. Values depicted in the graph represent the average volume of each mouse in the doxo-treated groups normalised to the matched average volumes of vehicle-treated groups from two independent experiments. Statistical difference between groups was determined using an unpaired Student’s t-test and was noticed at 27 days post treatment, *P<0.05. (B) Western blot analysis of activated caspase-3 in lysates isolated from c2984 control or shRunx2 tumours treated with PBS or doxo after killing the mice. Ctl=c2984 control shRNA OS cells, shRunx2=c2984 shRunx2 OS cells. GAPDH was used as internal loading control.
As the clinical efficacy of the chemotherapy regimen is assessed by percent tumour necrosis upon surgical resection, we investigated whether there was an increase in necrosis in our Runx2 knockdown tumours treated with doxo. We thoroughly analysed haematoxylin and eosin-stained tumour sections, and quantified the percentage of necrosis in multiple tumour sections (Figure 5A). We did not notice a statistically significant difference in necrosis in the untreated control shRNA and Runx2 knockdown tumours, except for one apparent outlier in our Runx2 knockdown cohort. However, when treated with doxo, we noticed a statistically significant increase in the percentage of tumour necrosis in the Runx2 knockdown tumours when compared with control shRNA tumours (Figure 5B). This suggests that the decreased Runx2 levels leads to enhanced chemosensitivity, and tumour necrosis, and contribute towards decreased tumour volume in our Runx2 knockdown tumours. Collectively, these data indicate that enhanced expression of Runx2 confers resistance to doxo in OS and the knockdown of Runx2 can potentiate chemotherapeutic responsiveness.

Knockdown of Runx2 results in an increase in tumour necrosis after doxo treatment. (A) Tumour sections from vehicle- and doxo-treated mice were isolated at the time the mice were killed. Haematoxylin and eosin (H&E) staining was performed, representative images from two mice in each group are shown. Circles enclose areas of necrosis in the tumour section. (B) Graph depicts calculated areas of necrosis per tumour section in each vehicle- or doxo-treated group. Statistical difference between the groups was determined by the Two-sample Wilcoxon rank-sum (Mann–Whitney) test.
c-Myc influences doxo-induced apoptosis in Runx2 knockdown cells
We have demonstrated that the loss of Runx2 expression augments doxo-induced FADD expression, caspase cleavage, and cell death. We next sought to elucidate the mechanisms by which Runx2 protects cells from doxo-induced apoptosis. Although c-Myc has been best characterised as having significant oncogenic functions, it can have pleiotropic roles in tumour cell biology including the regulation of apoptotic genes. Specifically, although it has previously been reported to have a cooperative role with Runx2 during lymphoma development, c-Myc can have an active role as a pro-apoptotic factor via its regulation of caspase-3 cleavage in response to doxo and FADD activation during apoptosis (Kangas et al, 1998; Prendergast, 1999; Blyth et al, 2001, 2006; Klefstrom et al, 2002; Grassilli et al, 2004; Scionti et al, 2008; Baniwal et al, 2010).
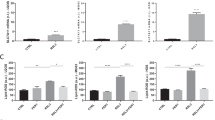
Thus, we investigated whether the transcription factor c-Myc could be actively involved in the chemotherapy-induced apoptotic pathways within OS. We assessed the relative expression of c-Myc and noticed an increase in c-Myc protein levels, but not mRNA levels (data not shown), in our shRunx2 cells treated with doxo (Supplementary Figure 3A). The exact post-transcriptional mechanisms of regulation require additional investigations. Moreover, using a c-Myc luciferase reporter and analysis of c-Myc transcriptional target genes revealed a statistically significant increase in c-Myc transcriptional activity in our Runx2 knockdown cells after doxo treatment when compared with control cells (Supplementary Figure 3B). This implies that enhanced intratumoral c-Myc transcriptional activity may contribute to the chemosensitivity seen in our shRunx2 OS cells.
Subsequently, we evaluated the effects of c-Myc inhibition on essential downstream apoptotic events. Cleavage of caspases is a biochemical event that occurs during c-Myc apoptosis and is also enhanced in our Runx2 knockdown cells after doxo treatment. Therefore, we investigated whether the activation of caspases could be diminished by inhibition of c-Myc using 10058-F4, a selective Myc-Max inhibitor (Huang et al, 2006). We treated c2984 OS cells with doxo, with or without 10058-F4, and isolated protein lysates after doxo treatment. Western blot analysis revealed that inhibition of c-Myc prevented the cleavage of caspases normally seen in shRunx2 cells post doxo treatment (Figure 6A and B). Furthermore, inhibition of c-Myc-Max interaction prevented the upregulation of FADD gene and protein expression after doxo treatment in our Runx2 knockdown cells (Figure 6C–E). Our data present a novel molecular role for c-Myc in the Runx2-mediated chemoresistance pathways in OS that could be therapeutically exploited.

c-Myc activity is required for efficient doxorubicin (doxo)-induced apoptosis in Runx2 knockdown OS cells. Inhibition of c-Myc prevents caspase-3 (A) and caspase-8 (B) cleavage in Runx2 knockdown cells. c2984 control or shRunx2 cells were treated with 50 nM doxo, with or without 10058-F4 (40 μM), and western blot analysis was performed on total cell lysates isolated 48 h post treatment utilising a caspase-3- (A) and caspase-8- (B) specific antibodies. Ctl=c2984 control shRNA mouse OS cells, shR=c2984 shRunx2 mouse OS cells. GAPDH and tubulin were used as loading controls. (+) represents 293 cells treated with staurosporine. (C and D) Doxo-induced FADD expression is attenuated by c-Myc inhibition in mouse (C) and human (D) OS. Control or shRunx2 cells were treated with doxo, with or without 10058-F4 (40 μM). Total RNA was isolated 24 h (D) or 48 h (C) post treatment. qPCR was performed to assess the expression levels of FADD. P-value was determined using the Student’s t-test comparing shRunx2 cells treated with doxo, or doxo plus 10058-F4, and **P⩽0.01, ***P⩽0.001. (E) Inhibition of c-Myc prevents FADD protein induction in Runx2 knockdown cells. HOS control or shRunx2 cells were treated with doxo, with or without 10058-F4 (40 μM), and western blot analysis was performed on total cell lysates isolated 24 h post treatment utilising a FADD-specific antibody. Ctl=HOS control shRNA mouse OS cells, shR=HOS shRunx2 mouse OS cells. GAPDH was used as a loading control.
Discussion
Runx2 is a vital transcription factor that directly regulates multiple cell fate decisions, including apoptosis in response to TNF-α, during normal osteoblast differentiation (Ghali et al, 2010; Olfa et al, 2010). Moreover, it has been implicated as a putative oncogene and marker for chemoresistance in OS (Nathan et al, 2009; Pereira et al, 2009; San Martin et al, 2009; Sadikovic et al, 2010; Lamoureux et al, 2014; Lee et al, 2015). The biological consequences of Runx2 overexpression and its molecular role in chemoprotective pathways in OS have not been clearly defined. We have identified a novel mechanism for Runx2 in antagonising chemotherapy-induced apoptosis in vitro, and more importantly, provide significant evidence of its chemoprotective role in OS in vivo. Our study also provides molecular evidence that Runx2 contributes towards inhibiting apoptosis after doxo treatment by repressing the activation of caspase-3 in vitro and in vivo, although this mechanism needs to be further investigated as its direct effects on caspase-3 have not been reported.
We have identified a novel role for c-Myc in chemotherapy-induced apoptosis of OS. We have shown that the inhibition of c-Myc prevents caspase cleavage and induction of FADD expression in the doxo-treated Runx2 knockdown OS cells. c-Myc and Runx2 cooperate to induce T-cell lymphoma, whereby Runx2 functions to prevent the apoptosis normally induced by aberrant c-Myc expression by an unknown mechanism (Blyth et al, 2006). Our data suggest that loss of Runx2 allows for increased c-Myc-dependent apoptosis in OS cells after doxo treatment. Studies into how Runx2 functions to prevent c-Myc induced apoptosis after doxo treatment is an exciting area of future research, as the cooperation between these two oncogenes has been implicated in the pathogenesis of other human tumours (Blyth et al, 2006).
In conclusion, we have shown a synergistic effect between Runx2 downregulation and doxo treatment in vitro and in vivo, compared with treatment with doxo alone. In addition, our data suggest that Runx2 functions to prevent c-Myc-mediated apoptosis after doxo treatment. Currently, there are no clinically useful Runx2 inhibitors. We provide strong evidence to support the claim that Runx2 serves as a biomarker for chemoresistance in OS, and thus targeting Runx2 with small-molecule inhibitors in combination with standard chemotherapy regimens can represent a novel effective therapeutic approach for patients afflicted with this deadly disease.
Change history
03 November 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Baniwal SK, Khalid O, Gabet Y, Shah RR, Purcell DJ, Mav D, Kohn-Gabet AE, Shi Y, Coetzee GA, Frenkel B (2010) Runx2 transcriptome of prostate cancer cells: insights into invasiveness and bone metastasis. Mol Cancer 9: 258.
Blyth K, Terry A, Mackay N, Vaillant F, Bell M, Cameron ER, Neil JC, Stewart M (2001) Runx2: a novel oncogenic effector revealed by in vivo complementation and retroviral tagging. Oncogene 20 (3): 295–302.
Blyth K, Vaillant F, Hanlon L, Mackay N, Bell M, Jenkins A, Neil JC, Cameron ER (2006) Runx2 and MYC collaborate in lymphoma development by suppressing apoptotic and growth arrest pathways in vivo. Cancer Res 66 (4): 2195–2201.
Browne G, Nesbitt H, Ming L, Stein GS, Lian JB, McKeown SR, Worthington J (2012) Bicalutamide-induced hypoxia potentiates RUNX2-mediated Bcl-2 expression resulting in apoptosis resistance. Br J Cancer 107 (10): 1714–1721.
Bueno M, Fillat MF, Strasser RJ, Maldonado-Rodriguez R, Marina N, Smienk H, Gomez-Moreno C, Barja F (2004) Effects of lindane on the photosynthetic apparatus of the cyanobacterium Anabaena: fluorescence induction studies and immunolocalization of ferredoxin-NADP+ reductase. Environ Sci Pollut Res Int 11 (2): 98–106.
Clark JC, Dass CR, Choong PF (2008) A review of clinical and molecular prognostic factors in osteosarcoma. J Cancer Res Clin Oncol 134 (3): 281–297.
Franceschi RT, Ge C, Xiao G, Roca H, Jiang D (2007) Transcriptional regulation of osteoblasts. Ann N Y Acad Sci 1116: 196–207.
Geller DS, Gorlick R (2010) Osteosarcoma: a review of diagnosis, management, and treatment strategies. Clin Adv Hematol Oncol 8 (10): 705–718.
Ghali O, Chauveau C, Hardouin P, Broux O, Devedjian JC (2010) TNF-alpha's effects on proliferation and apoptosis in human mesenchymal stem cells depend on RUNX2 expression. J Bone Miner Res 25 (7): 1616–1626.
Grassilli E, Ballabeni A, Maellaro E, Del Bello B, Helin K (2004) Loss of MYC confers resistance to doxorubicin-induced apoptosis by preventing the activation of multiple serine protease- and caspase-mediated pathways. J Biol Chem 279 (20): 21318–21326.
Huang MJ, Cheng YC, Liu CR, Lin S, Liu HE (2006) A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp Hematol 34 (11): 1480–1489.
Kager L, Zoubek A, Potschger U, Kastner U, Flege S, Kempf-Bielack B, Branscheid D, Kotz R, Salzer-Kuntschik M, Winkelmann W, Jundt G, Kabisch H, Reichardt P, Jurgens H, Gadner H, Bielack SS (2003) Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol 21 (10): 2011–2018.
Kangas A, Nicholson DW, Holtta E (1998) Involvement of CPP32/Caspase-3 in c-Myc-induced apoptosis. Oncogene 16 (3): 387–398.
Karsenty G, Wagner EF (2002) Reaching a genetic and molecular understanding of skeletal development. Dev Cell 2 (4): 389–406.
Kavurma MM, Khachigian LM (2003) Signaling and transcriptional control of Fas ligand gene expression. Cell Death Differ 10 (1): 36–44.
Klefstrom J, Verschuren EW, Evan G (2002) c-Myc augments the apoptotic activity of cytosolic death receptor signaling proteins by engaging the mitochondrial apoptotic pathway. J Biol Chem 277 (45): 43224–43232.
Lamoureux F, Baud'huin M, Rodriguez Calleja L, Jacques C, Berreur M, Redini F, Lecanda F, Bradner JE, Heymann D, Ory B (2014) Selective inhibition of BET bromodomain epigenetic signalling interferes with the bone-associated tumour vicious cycle. Nat Commun 5: 3511.
Lee DH, Qi J, Bradner JE, Said JW, Doan NB, Forscher C, Yang H, Koeffler HP (2015) Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. Int J Cancer 136 (9): 2055–2064.
Lucero CM, Vega OA, Osorio MM, Tapia JC, Antonelli M, Stein GS, van Wijnen AJ, Galindo MA (2013) The cancer-related transcription factor Runx2 modulates cell proliferation in human osteosarcoma cell lines. J Cell Physiol 228 (4): 714–723.
Ma O, Cai WW, Zender L, Dayaram T, Shen J, Herron AJ, Lowe SW, Man TK, Lau CC, Donehower LA (2009) MMP13, Birc2 (cIAP1), and Birc3 (cIAP2), amplified on chromosome 9, collaborate with p53 deficiency in mouse osteosarcoma progression. Cancer Res 69 (6): 2559–2567.
Man TK, Lu XY, Jaeweon K, Perlaky L, Harris CP, Shah S, Ladanyi M, Gorlick R, Lau CC, Rao PH (2004) Genome-wide array comparative genomic hybridization analysis reveals distinct amplifications in osteosarcoma. BMC Cancer 4: 45.
McIlwain DR, Berger T, Mak TW (2013) Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 5 (4): a008656.
Mirabello L, Troisi RJ, Savage SA (2009) Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer 115 (7): 1531–1543.
Miwa S, Takeuchi A, Shirai T, Taki J, Yamamoto N, Nishida H, Hayashi K, Tanzawa Y, Kimura H, Igarashi K, Ooi A, Tsuchiya H (2013) Prognostic value of radiological response to chemotherapy in patients with osteosarcoma. PLoS One 8 (7): e70015.
Nathan SS, Pereira BP, Zhou YF, Gupta A, Dombrowski C, Soong R, Pho RW, Stein GS, Salto-Tellez M, Cool SM, van Wijnen AJ (2009) Elevated expression of Runx2 as a key parameter in the etiology of osteosarcoma. Mol Biol Rep 36 (1): 153–158.
Olfa G, Christophe C, Philippe L, Romain S, Khaled H, Pierre H, Odile B, Jean-Christophe D (2010) RUNX2 regulates the effects of TNFalpha on proliferation and apoptosis in SaOs-2 cells. Bone 46 (4): 901–910.
Pereira BP, Zhou Y, Gupta A, Leong DT, Aung KZ, Ling L, Pho RW, Galindo M, Salto-Tellez M, Stein GS, Cool SM, van Wijnen AJ, Nathan SS (2009) Runx2, p53, and pRB status as diagnostic parameters for deregulation of osteoblast growth and differentiation in a new pre-chemotherapeutic osteosarcoma cell line (OS1). J Cell Physiol 221 (3): 778–788.
Porter AG, Janicke RU (1999) Emerging roles of caspase-3 in apoptosis. Cell Death Differ 6 (2): 99–104.
Prendergast GC (1999) Mechanisms of apoptosis by c-Myc. Oncogene 18 (19): 2967–2987.
Provisor AJ, Ettinger LJ, Nachman JB, Krailo MD, Makley JT, Yunis EJ, Huvos AG, Betcher DL, Baum ES, Kisker CT, Miser JS (1997) Treatment of nonmetastatic osteosarcoma of the extremity with preoperative and postoperative chemotherapy: a report from the Children's Cancer Group. J Clin Oncol 15 (1): 76–84.
Rainusso N, Wang LL, Yustein JT (2013) The adolescent and young adult with cancer: state of the art — bone tumors. Curr Oncol Rep 15 (4): 296–307.
Sadikovic B, Thorner P, Chilton-Macneill S, Martin JW, Cervigne NK, Squire J, Zielenska M (2010) Expression analysis of genes associated with human osteosarcoma tumors shows correlation of RUNX2 overexpression with poor response to chemotherapy. BMC Cancer 10: 202.
Sadikovic B, Yoshimoto M, Chilton-MacNeill S, Thorner P, Squire JA, Zielenska M (2009) Identification of interactive networks of gene expression associated with osteosarcoma oncogenesis by integrated molecular profiling. Hum Mol Genet 18 (11): 1962–1975.
San Martin IA, Varela N, Gaete M, Villegas K, Osorio M, Tapia JC, Antonelli M, Mancilla EE, Pereira BP, Nathan SS, Lian JB, Stein JL, Stein GS, van Wijnen AJ, Galindo M (2009) Impaired cell cycle regulation of the osteoblast-related heterodimeric transcription factor Runx2-Cbfbeta in osteosarcoma cells. J Cell Physiol 221 (3): 560–571.
Scionti I, Michelacci F, Pasello M, Hattinger CM, Alberghini M, Manara MC, Bacci G, Ferrari S, Scotlandi K, Picci P, Serra M (2008) Clinical impact of the methotrexate resistance-associated genes C-MYC and dihydrofolate reductase (DHFR) in high-grade osteosarcoma. Ann Oncol 19 (8): 1500–1508.
Ta HT, Dass CR, Choong PF, Dunstan DE (2009) Osteosarcoma treatment: state of the art. Cancer Metastasis Rev 28 (1-2): 247–263.
Tang N, Song WX, Luo J, Haydon RC, He TC (2008) Osteosarcoma development and stem cell differentiation. Clin Orthop Relat Res 466 (9): 2114–2130.
Teplyuk NM, Galindo M, Teplyuk VI, Pratap J, Young DW, Lapointe D, Javed A, Stein JL, Lian JB, Stein GS, van Wijnen AJ (2008) Runx2 regulates G protein-coupled signaling pathways to control growth of osteoblast progenitors. J Biol Chem 283 (41): 27585–27597.
van der Deen M, Akech J, Lapointe D, Gupta S, Young DW, Montecino MA, Galindo M, Lian JB, Stein JL, Stein GS, van Wijnen AJ (2012) Genomic promoter occupancy of runt-related transcription factor RUNX2 in Osteosarcoma cells identifies genes involved in cell adhesion and motility. J Biol Chem 287 (7): 4503–4517.
Wang LL (2005) Biology of osteogenic sarcoma. Cancer J 11 (4): 294–305.
Won KY, Park HR, Park YK (2009) Prognostic implication of immunohistochemical Runx2 expression in osteosarcoma. Tumori 95 (3): 311–316.
Yuan XW, Zhu XF, Huang XF, Sheng PY, He AS, Yang ZB, Deng R, Feng GK, Liao WM (2007) Interferon-alpha enhances sensitivity of human osteosarcoma U2OS cells to doxorubicin by p53-dependent apoptosis. Acta Pharmacol Sin 28 (11): 1835–1841.
Acknowledgements
We thank James LeRoux in the Department of Veterinary Medicine and Surgery at the MD Anderson Cancer Center for his assistance with scanning slides. This project was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574) and the expert assistance of Joel M Sederstrom, and by the Department of Defense Predoctoral Fellowship (W81XWH-11-1-0114) awarded to Alison Roos, Cancer Prevention and Research Institute of Texas Grant RP101335 to Lawrence A Donehower and Jason T Yustein, Sarcoma Foundation of America to Lawrence A Donehower, and by the WWW.W Foundation, Inc. (QuadW; Jason T Yustein), the St Baldrick’s Foundation (Jason T Yustein).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Roos, A., Satterfield, L., Zhao, S. et al. Loss of Runx2 sensitises osteosarcoma to chemotherapy-induced apoptosis. Br J Cancer 113, 1289–1297 (2015). https://doi.org/10.1038/bjc.2015.305
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.305
Keywords
This article is cited by
-
Proteomic analysis reveals key differences between squamous cell carcinomas and adenocarcinomas across multiple tissues
Nature Communications (2022)
-
Transcription factor Runx2 changes the expression of some matricellular proteins in metastatic breast cancer cells
Molecular Biology Reports (2022)
-
Runt related transcription factor-1 plays a central role in vessel co-option of colorectal cancer liver metastases
Communications Biology (2021)
-
Impact of RUNX2 on drug-resistant human pancreatic cancer cells with p53 mutations
BMC Cancer (2018)
-
RUNX2 Promotes Malignant Progression in Glioma
Neurochemical Research (2018)

{kind=link}
{kind=link}
{kind=link}