
Abstract
Background:
This phase I study evaluated the safety, tolerability, maximum tolerated dose (MTD) and pharmacokinetics of two dosing schedules of oral topotecan in combination with pazopanib in patients with advanced solid tumours.
Methods:
Stage I of this study was to determine whether there was an impact of pazopanib on topotecan exposure. In stage II, the MTD and safety profile of oral topotecan given weekly on days 1, 8 and 15 in a 28-day cycle; or daily-times-five on days 1–5 in a 21-day cycle, both in combination with daily pazopanib, were explored.
Results:
In total, 67 patients were enroled. Pazopanib co-administration caused a substantial increase in exposure to total topotecan (1.7-fold) compared with topotecan alone, which is considered clinically relevant. Topotecan had no effect on pazopanib concentrations. Safety findings were consistent with the known profile of both agents. There were three drug-related deaths, liver failure, tumour haemorrhage and myelosuppression. Two patients experienced dose-limiting toxicities (DLTs; hand–foot syndrome, myelosuppression and diarrhoea) on the weekly topotecan schedule and four patients experienced DLTs (myelosuppression) on the daily-times-five topotecan schedule. When combined with pazopanib, 800 mg daily, the recommended doses for oral topotecan are: 8 mg weekly and 2.5 mg daily-times-five. Seven of eight patients with partial response had platinum-resistant ovarian cancer. In addition, 54% of patients had stable disease with 22% stable for 6 months.
Conclusions:
Total topotecan exposure is 1.7-fold higher when co-administered with pazopanib. Both schedules of administration were tolerated and would permit further evaluation, especially the weekly schedule.
Similar content being viewed by others

Main
Pazopanib (GW786034; Votrient; GlaxoSmithKline) is an oral multi-target tyrosine kinase inhibitor (TKI) targeting receptors of vascular endothelial growth factor (VEGF-1, -2 and -3), platelet-derived growth factor (α and β) and stem cell factor (c-kit; Anon, 2013b). Pazopanib is approved by FDA and EMA, and used for the treatment of advanced renal cell carcinoma (Sternberg et al, 2010) and advanced soft tissue sarcoma (STS) (Anon, 2013b). It has demonstrated activity in the preoperative setting for NSCLC (Altorki et al, 2010) and advanced epithelial ovarian cancer, fallopian tube or primary peritoneal cancer (Bois, 2014). The recommended dose of oral pazopanib is 800 mg once daily (QD). The most common side effects (>30%) included: diarrhoea, hair and skin hypopigmentation, hypertension, nausea, fatigue, anorexia, vomiting, and elevated alanine aminotransferase (ALT) and elevated aspartate aminotransferase (AST; Anon, 2013b). Peak concentrations are achieved within 2–4 h with a mean elimination half-life (t1/2) of ∼30 h in human plasma. Pazopanib absorption is increased by food and therefore was administered on an empty stomach. Pazopanib is not extensively metabolised and major route of elimination of pazopanib is excretion of parent compound in faeces (Anon, 2013b). Metabolism of pazopanib is primarily by cytochrome P-4503A4 (CYP3A). Pazopanib is a substrate with a moderate affinity for the drug efflux transporters P-glycoprotein-1 (P-gp/ABCB1) and with a high affinity for breast cancer resistance protein (BCRP/ABCG2) (Xu et al, 2010; Minocha et al, 2012; Deng et al, 2013; Anon, 2013b).
Topotecan (Hycamtin, GlaxoSmithKline), a semisynthetic analogue of camptothecin, inhibits DNA topoisomerase I in dividing cells. By binding to the cleavable complex, topotecan blocks further replication, which leads to cell death (Creemers et al, 1994; Anon, 2013a). Intravenous (i.v.) topotecan is approved for the treatment of small cell lung cancer (SCLC), cervical cancer and metastatic ovarian carcinoma, whereas oral topotecan is approved for SCLC only (Eckardt et al, 2007; Anon, 2013a). The oral formulation enables more convenient dosing than i.v. administration, especially in combination regimens with other oral anti-cancer agents (Schellens et al, 2000) and has similar in activity to i.v. topotecan, with less grade 4 neutropenia and greater convenience of administration (Von Pawel et al, 2001). In patients with platinum-resistant recurrent ovarian cancer, the 5-day schedule had better progression-free survival than the weekly schedule, but the weekly schedule had comparable overall survival and a favourable toxicity profile (Sehouli et al, 2011). However, the 5-day schedule is limited by the occurrence of haematological toxicity (Schiller et al, 1996; Stewart, 2004).
The time to reach peak plasma concentration (Tmax) for oral topotecan is 2 h. Following oral administration, ∼20% was recovered as parent (total topotecan) drug in urine and 33% of the oral dose was found to be unchanged (total topotecan) in faeces (Herben et al, 1999). The contribution of metabolism to topotecan total body clearance (CL) is limited (<10%). Topotecan undergoes reversible pH-dependent hydrolysis, yielding topotecan carboxylate. Elimination t1/2 for oral topotecan is between 4 and 6 h (Herben et al, 1999). The absorption of oral topotecan is limited largely due to BCRP- and P-gp-mediated efflux of oral topotecan in the intestinal epithelium that varies among subjects (Maliepaard et al, 1999; Schellens et al, 2000; Maliepaard et al, 2001).
Various preclinical and clinical studies of anti-angiogenic agents in combination with chemotherapy showed mild toxicity and improved anti-tumour activity (Pasquier et al, 2010). The observed additive effect could be attributed to the anti-angiogenic drugs that act by normalising tumour vasculature, which can then lead to improved delivery of cytotoxic drugs to the tumour (Jain, 2001). Further, topotecan showed to be a potent inhibitor of hypoxia-inducible factor HIF-1α and HIF-2α subunits, leading to decreased VEGF expression and angiogenic activity and contribute to the mode of action of the pazopanib and topotecan combination. Other theories are based on timing of anti-angiogenic drugs during chemotherapy-free periods (Shaked et al, 2008; Roodhart et al, 2010). Preclinical models of pazopanib and topotecan co-administration showed significantly improved anti-tumour activity compared with the respective single agents (Kerbel and Kamen, 2004; Kumar et al, 2011). Prolonged combination therapy with low-dose topotecan and pazopanib in mouse models demonstrated sustained anti-angiogenic activity (Kumar et al, 2011). A study in patients with gynaecologic tumours showed the lack of a statistically significant drug–drug interaction between pazopanib and low-dose topotecan (Turner et al, 2013).
The aim of this study was to evaluate the safety, tolerability, maximum tolerated dose and pharmacokinetics of two topotecan dosing schedules in patients with advanced solid tumours.
Patients and methods
Patient selection
Eligible patients were those with histologically or cytologically confirmed diagnosis of a progressive advanced solid tumour that was resistant to standard therapy or for whom there was no established therapy. Other inclusion criteria were: written informed consent; ⩾18 years; Eastern Cooperative Oncology Group performance status of ⩽1; able to swallow and retain oral medications; adequate haematological (neutrophils ⩾1.5 × 109 per litre; haemoglobin ⩾6.2 mmol l−1; platelets ⩾100 × 109 per litre), hepatic (bilirubin ⩽1.5 × upper limit of normal (ULN); AST and ALT ⩽3 × ULN or ⩽5 × ULN in case of liver metastases) and renal function (Cockroft-Gault creatinine CL ⩾50 ml min−1 and urine protein creatinine ratio and partial thromboplastin time ⩽1.2 × ULN). Exclusion criteria were: less than 4 weeks since last chemo, radio or biologic therapy or surgery or less than 6 weeks since last prior nitrosurea or mitomycin C chemotherapy; administration of investigational drugs within 30 days or 5 elimination half-lives; prior treatment with pazopanib or investigational anti-angiogenic compounds; uncontrolled infection; pregnancy or lactating (all patients with child-bearing potential had to use adequate contraceptive protection); poorly controlled hypertension (systolic ⩾140 mm Hg or diastolic blood pressure ⩾90 mm Hg); prolonged QTc interval; class III or IV heart failure; vascular events within 6 months; therapeutic heparin or warfarin use; leptomeningeal or brain metastases; and any other condition that would interfere with the patient’s ability to comply with the dosing schedule and protocol-specified evaluations. The study was conducted in accordance with the guidelines for good clinical practice, and was approved by local Medical Ethics committees.
Study design and treatment administration
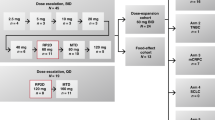
This was a two-stage, two-arm, open-label, dose-escalation phase I study (NCT00732420, www.clinicaltrials.gov). From September 2008 to September 2013 three centres participated in the study. These included the Abramson Cancer Center of the University of Pennsylvania in the USA, University Medical Center Utrecht and the Netherlands Cancer Institute Antoni van Leeuwenhoek Hospital, Amsterdam, both in the Netherlands. In the drug–drug interaction study portion (P1), the impact of pazopanib on the exposure of oral topotecan was investigated. In P2, the combination regimens were explored in a dose-escalation phase and a dose-expansion phase. There were two different combination regimens for oral topotecan: P2A: topotecan once weekly (day 1, 8 and 15) in a 28-day cycle; P2B: topotecan on days 1–5 in a 21-day cycle, whereas oral pazopanib was taken daily (QD) throughout the cycle. Patients were enroled in sequential cohorts of three to six patients and the maximal tolerated dose (MTD) was defined as the highest dose level at which not more than one out of six patients experienced a dose-limiting toxicity (DLT) after completing one treatment cycle.
Study procedures, safety and efficacy assessments
Written informed consent was obtained before study-specific assessments. Demographic data, concomitant medications and medical history were recorded. Complete physical examinations, including ECG, and clinical laboratory tests were performed at screening and at regular intervals during cycle 1, during any following cycles and at study termination. Toxicities were graded according to the National Cancer Institute Common Terminology Criteria of Adverse Events version 3.0. Radiological tumour assessments were performed at baseline and every two cycles. Tumour measurements were carried out according to RECIST 1.0. Patients remained on treatment until disease progression, unmanageable toxicity had developed or withdrawal of consent. Patients were considered to be evaluable for safety when they completed cycle 1.
Dosing
Pazopanib monohydrochloride was provided as 200 and 400 mg tablets. Oral topotecan was provided as capsules containing topotecan HCL, equivalent to 0.25 or 1.00 mg. Different dose levels of pazopanib and topotecan are summarised in Table 1. Pazopanib and topotecan were administered with water on an empty stomach either 1 h before a meal or 2 h after a meal. Dose reductions following each cycle were allowed twice. Specific guidelines were prescribed for management of hypertension and diarrhoea. Oral topotecan was administered as a flat dose, in this study, to facilitate the interpretation of the pharmacokinetic data and decrease medication errors, as there is no reduction in variability in systemic exposure to topotecan by BSA-based dosing vs flat dosing (Mathijssen et al, 2007).
In P1, the interaction portion of the study, pazopanib was dosed continuously from day 2 at 800 mg. Topotecan 4 mg was administered only on day 1 and 15 of cycle 1. This order was chosen with the aim to enable pharmacokinetic sampling of both drugs as monotherapy and in combination therapy. On completion of the P1 (Days 1–15), subjects continued pazopanib monotherapy (continuation phase) in 28-day cycles.
In the P2A of the dose-escalation component, patients started with continuous pazopanib monotherapy at day −14, which was before administration of topotecan once weekly on days 1, 8 and 15 in 28-day cycles. The presence of steady-state levels of pazopanib ensured accurate determination of cycle 1 DLTs during the oral topotecan dosing.
In the P2A dose expansion, continuous pazopanib dosing started on day 2 and topotecan was dosed on days 1, 8 and 15 of a 28-day cycle.
In the P2B dose-escalation, patients started with continuous pazopanib monotherapy at day −14, which was before administration of topotecan on a daily-times 5-consecutive days (on days 1, 2, 3, 4 and 5) of a 21-day cycle.
In the P2B dose expansion, oral topotecan was given continuous on days 1–5 every cycle of 21 days (daily-times-five). Dosing with pazopanib began on day 6 of cycle 1.
Dose-limiting toxicities
A DLT was defined as: any grade 3 or 4 clinically significant non-haematological toxicity (excluding grade ⩾3 nausea and vomiting without maximal anti-emetic prophylaxis); grade 4 neutropenia with fever or infection or grade 4 neutropenia ⩾5 days or grade ⩾3 neutropenia requiring delay in the next cycle; grade 4 thrombocytopenia (<25 000 mm−3/<25.0 × 109 per litre; inadequately controlled grade 3 hypertension in spite of maximal two antihypertensive drugs; grade 4 hypertension; grade 3 proteinuria during uncontrolled hypertension and/or renal impairment or lack of improvement to grade ⩽2 upon interruption of pazopanib; grade 4 proteinuria; delay of next cycle of ⩽2 weeks due to unresolved toxicity; grade 2 non-haematological toxicity beyond cycle 1 and any grade ⩽2 toxicity that was considered a DLT.
Pharmacokinetic sampling and analysis
Blood samples for the determination of pazopanib in P1 and P2A dose expansion, were obtained on days 14 and 15 at baseline and at 1, 2, 3, 4, 6, 8, 10–12 and 24 h following administration of pazopanib. Pharmacokinetic sampling was also performed for total topotecan at day 1 and day 15 at baseline and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10–12 and 24 h following administration of topotecan.
In P2B dose expansion, the pharmacokinetic sampling was performed according the same schedules but on day 5 of cycle 1 for topotecan alone, day 21 of cycle 1 for pazopanib alone and day 5 of cycle 2 (day 26) for both topotecan and pazopanib.
At each collection point, for pazopanib, 2 ml of whole blood was withdrawn into a tube containing potassium ethylenediaminetetra acetic acid. For total topotecan, 3 ml of whole blood was withdrawn into lithium heparinised collection tube. After separation, the plasma was stored frozen at −30 °C. Plasma concentrations of pazopanib and total topotecan (both carboxylate and lactone form) were quantified using a validated high-performance liquid chromatography method (Rosing H et al, 1995, Hurwitz et al, 2009). Total topotecan and pazopanib concentrations and actual sample collections times were used to carry out non-compartmental analysis using WinNonLin (v 6.2). The following pharmacokinetic parameters were determined: area under the concentration–time curve extrapolated to infinity (AUC0–∞), terminal half-life (t1/2) for total topotecan and only AUC(0–24) for pazopanib. The maximum observed plasma concentration (Cmax), time to maximum observed plasma concentration (tmax) and concentration at 24 h (C24) was directly obtained from the plasma concentration data.
Pharmacogenetics (Pg)
Pg and biomarker analyses in blood and on archive tumour samples were collected. Separate written informed consent was required for Pg sampling. Several genes involved in safety and efficacy of the study drugs were planned to be investigated for single-nucleotide polymorphisms (SNP). These included genes coding for drug targets such as UDP-glucoronyltransferase (UGT1A1) and drug transporters such as P-gp. Additional analysis was included for polymorphisms in the hemochromatosis (HFE) gene, which was recently shown to be associated with ALT elevation in renal cell cancer patients treated with pazopanib (Xu et al, 2010).
Statistical methods
Safety and preliminary anti-tumour activity data, as well as calculated pharmacokinetic parameters were summarised and tabulated using descriptive statistics. The effect of pazopanib on total topotecan and of total topotecan on pazopanib was assessed. Pharmacokinetic parameters, including AUC0–∞, Cmax and t1/2 for total topotecan, and AUC(0-24), Cmax and C24 for pazopanib, were analysed using a mixed effects model on log-transformed data with treatment as a fixed effect and subject as a random effect. The geometric last squares mean ratio and associated 90% confidence interval (CI) were calculated after transforming the log-transformed results back to the original scale. For paired data, tmax was compared between the groups using the Wilcoxon-matched pairs method (Steinijans and Diletti, 1983). With point estimates and 90% CIs for median differenced were calculated.
Results
In total, nine patients with mean age 59 (range 37–78) were treated in the interaction study (P1). In dose-escalation and -expansion study (P2), 58 patients with mean age of 52.3 (range 18–72) years were treated. The patient characteristics are listed in Table 2.
Dose escalation
Planned dose escalation is summarised in Table 1. The weekly dose of 10 mg topotecan and continuous 800 mg pazopanib (P2A) was determined to be above the maximal tolerated dose as two out of six patients experienced a DLT (one grade 3 diarrhoea and one grade 3 neutropenia). The next lower-dose level (8 mg topotecan and 800 mg pazopanib) was taken forward into the expansion cohort. The expansion cohort consisted of additional 11 patients who did not experience any DLT, as well as the initial three patients during the dose-escalation part.
A second dosing schedule (P2B) was explored based on daily-times-five oral topotecan dose every 3 weeks. Dosing started at 1.75 mg topotecan daily-times-five in combination with 400 mg pazopanib continuously. The combination of 3 mg topotecan daily-times-five with 800 mg pazopanib continuously resulted in a cohort of three patients that all experienced at least one DLT: grade 4 neutropenia lasting ⩾5 days; grade 3 thrombocytopenia, anaemia, leucocytopenia and bleeding; and grade 4 thrombocytopenia in combination with grade 3 neutropenia. The next lower-dose level, 2.5 mg topotecan daily-times-five with 800 mg pazopanib daily in 21-day cycle was expanded, where one out of six enroled patients experienced DLT (grade 4 neutropenia lasting >5 days in combination with grade 4 thrombocytopenia). Therefore, this dose was taken forward into the expansion cohort. Eight additional patients were enroled in the expansion cohort, in which no further DLT was observed.
Safety
The summary of drug-related adverse events (grades 3, 4 and 5) in dose-escalation study (P2) is listed in Table 3. Two patients enroled in P2 (1a and 2a) received monotherapy but were included in the safety assessment. The most frequently occurring treatment-related haematological toxicities grades ⩾3 were neutropenia (15 and 25.9%), thrombocytopenia (12 and 20.6%), leucocytopenia (6 and 10%) and anaemia (3 and 5%). The daily-times-five topotecan regimen had higher rate of haematologic toxicities than the weekly schedule. In both the drug combination regimens, the most frequently occurring treatment-related non-haematological toxicities grades ⩾3 were fatigue (5 and 8%) and hypertension (4 and 7%). Gastrointestinal side effects such as nausea, vomiting and diarrhoea, all grades, occurred with comparable percentage in both schedules. Deaths related to study drug occurred in three patients. One patient with lung adenocarcinoma treated in P1 developed a fatal pulmonary haemorrhage while on pazopanib, 3 days following the last topotecan dose. The second death occurred also in P1 in a patient with synovial sarcoma, without history of hepatic disease, who developed hepatic failure while on pazopanib, 21 days following the last topotecan dose. At autopsy there was extensive liver necrosis and congestion, ascites and sub-acute heart congestion. Drug concentration values were consistent with therapeutic exposure. Several other causes, sub-clinical heart failure and paracetamol toxicity, were evaluated for their contribution to the observed hepatic toxicity, but none were confirmed; therefore, this event was classified as a pazopanib-related liver failure. One patient in the 3 mg topotecan daily-times-five and daily 800 mg pazopanib experienced grade 3 neutropenia and grade 4 thrombocytopenia on day 22 and treatment was interrupted. However, the patient died on 35th day of drug-related pancytopenia resulting in pneumonia and septic shock. This side effect has been reported in literature (O’Brien et al, 2006). Non-fatal reversible treatment-related liver toxicity (elevated ALT and AST) occurred in 13 patients out of 67 patients from P1 and P2 (19%) during the whole study. The only other grade 4 elevated ALT and AST (1 out of 67, 1.5% each) was also observed in the P1 while on pazopanib, 15 days post last topotecan dose. This patient had colorectal carcinoma with liver metastases and experienced partial recovery after discontinuation of study medication and administration of oral steroids. Also, for this patient plasma concentration obtained at time of the liver event was not elevated (32.6 μg ml−1). Therefore, this hepatic toxicity event was considered to be only partially due to pazopanib.
Pharmacokinetics
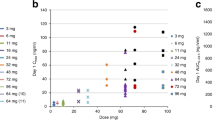
The pharmacokinetic population included data from patients who received all doses in P1 and received all MTD doses in P2 (Table 4). Figure 1 shows the mean plasma concentration–time curves of pazopanib (A) and total topotecan (B) of patients treated in the drug–drug interaction study (P1) and individual total topotecan concentration–time curves in the MTD part of the study (P2) for weekly (C) and daily-times-five (D) schedules. Because of the safety reasons and one of the drugs dose delay/interruption, only 50% of patients from P2 expansion phase had paired available Pharmacokinetics (PK) measurements. No differences in pazopanib plasma concentration can be seen following pazopanib dose alone vs co-administration with topotecan (Figure 1A). However, a marked increase in mean total topotecan exposure was observed on the sampling day when topotecan was co-administered with pazopanib, compared with dosing alone (Figure 1B–D). Pazopanib exposure on both schedules was similar with or without co-administration of topotecan (figures not shown). Detailed pharmacokinetic data are summarised in Table 4 and Table 5. Total topotecan exposure was increased when co-administered with pazopanib. Total topotecan Cmax increased ∼1.9-fold in P1 and both schedules in P2, whereas AUC(0–∞) increased between 1.5- and 1.7-fold depending on study part and schedule. Individual increases in ratios ranged from 0.58 to 3.5 and 0.66 to 2.9 for Cmax and AUC(0–∞), respectively, for the topotecan 4 mg single dose in P1, from 1.0 to 3.0 and 1.5 to 2.1 for Cmax and AUC(0–∞), respectively, for 8.0 mg on weekly schedule and from 1.0 to 3.6 and 1.1 to 1.9 for Cmax and AUC(0–∞), respectively, for the topotecan 2.5 mg daily-times-five schedule. The minor differences between the three doses and schedules were likely due to the small sample sizes. Total topotecan mean t½ values were ∼4–5 h and were similar whether topotecan was dosed alone or with pazopanib. PK analysis of the two patients with severe liver toxicity did not suggest any significant difference in plasma drug exposure to other patients.

Mean (s.d.) plasma concentration–time curves of pazopanib (A) and total topotecan (B) during part 1 (P1) following single administration of pazopanib (day 14, n =7), single administration of topotecan (day 1, n =6) and concomitant administration of pazopanib and topotecan (day 15); (C) Plasma concentration–time curves per patient treated with 8 mg topotecan weekly and 800 mg pazopanib daily in P2A and (D) treated with 2.5 mg topotecan daily-times-five and 800 mg pazopanib daily in P2B.
Pazopanib mean C24h plasma concentrations were >25 μg ml−1 in P1 and >35 μg ml−1 in P2 both schedules.
Pharmacogenetic analysis
Pharmacogenetic analysis was performed in only two patients with severe hepatotoxicity in the P1. Both the patient with fatal hepatotoxicity and the patient with grade 4 ALT and AST elevation were found to be heterozygous for the ABCB1 gene (*6 and *7, rs1045642 and rs2032582), which has been associated with decreased P-gp activity (Bosch et al, 2006). The latter patient also had one copy of the UGT1A6*3A allele (S7A, rs6759892), which has been associated with lower UGT1A6 expression and possibly higher likelihood of paracetamol-induced hepatotoxicity (Shrestha et al, 2011).
Preliminary anti-tumour activity
Eight out of 50 patients evaluable in combination therapy (P2) showed partial tumour response (PR; 8 out of 50, 16%). Seven patients with PR had platinum-resistant ovarian cancer (47%, 7 out of 15 evaluable ovarian cancer) and one cervical cancer. Five patients with PR were observed in P2A and 3 in P2B. Duration of response (PR) was median 24 weeks (range 16–63). Twenty-seven patients showed stable disease (SD) as best response (54%). Seventeen were enroled in P2A and 10 in P2B. SD >6 months were observed in 11 (22%) patients, in patients with ovarian (3) and colon–rectum cancer (2) and other tumour types (6). The most frequent tumours that showed SD were STS and ovarian. Early progression of disease was seen in 30% of the patients.
Discussion
Pharmacokinetics and tolerance of the combination of oral topotecan and pazopanib treatment were studied in this phase I study. MTD of weekly oral topotecan was 8 mg on days 1, 8 and 15 in combination with 800 mg pazopanib daily in a 28-day cycle. For the daily-times-five regimen, MTD is 2.5 mg oral topotecan on days 1 through 5 with 800 mg pazopanib daily in the 21-day cycle. Both schedules could be administered for longer periods and showed initial signs of anti-tumour activity.
Pazopanib substantially increased exposure of total topotecan by 1.8-fold for Cmax and 1.7-fold for AUC(0–∞), which is considered clinically relevant, but did not increase t½ values, when compared with topotecan alone. This suggests that the effects of pazopanib on topotecan pharmacokinetics were pre-systemic (increase in oral bioavailability) and not related to changes in elimination. These findings can be compared with an effect of concomitant administration of oral topotecan and elacridar, the known potent inhibitor of BCRP and P-gp that resulted in a 2.8- and 2.4-fold increase in total topotecan Cmax and AUC, respectively, and confirmed the effects of transporter modulation on the pharmacokinetics of topotecan. In contrast, there was only a 10% decrease in topotecan CL after i.v. administration and co-administration with elacridar (Kruijtzer et al, 2002). These results indicate that the effects of elacridar on orally administered topotecan pharmacokinetics were also primarily pre-systemic and likely due to inhibition of transporters in the gut. These efflux transporters are located in the intestine where they act to limit drug absorption from the lumen (Maliepaard et al, 1999, 2001). BCRP and P-gp are also expressed in the liver and the kidney where they promote drug excretion into bile and urine, respectively (Maliepaard et al, 1999; Schellens et al, 2000; Maliepaard et al, 2001). However, topotecan is primarily renally excreted and undergoes little metabolism (Herben et al, 1999). Although both topotecan and pazopanib are found to be high-affinity substrates for BCRP, topotecan is a weak substrate for P-gp and pazopanib moderate (Maliepaard et al, 1999; Minocha et al, 2012; Anon, 2013a, 2013b), which means that pazopanib binds has a higher affinity for P-gp.
Previously published total topotecan exposure when 14 mg topotecan was dosed weekly without pazopanib (published data, Von Gruenigen et al, 2012) was similar to total topotecan plasma exposure following a weekly 8 mg oral topotecan dose in combination with daily pazopanib 800 mg (Von Gruenigen et al, 2012). This is not unexpected as pazopanib increased AUC (0–∞) ∼1.7-fold and the difference between 8 and 14 mg dose is ∼1.7.
The previous study with low-dose oral topotecan and pazopanib indicated there was no statistically significant drug–drug interaction, which might be because the model did not adequately describe the data, the study was not powered to see an effect, or the low-dose topotecan shows different results than standard doses. Their finding was based on a population pharmacokinetic analysis that included oral clearance (CL/F), central volume (Vc/F), absorption rate constant (ka) and lag time. The CL/F estimate with 95% CIs was 11.5 l h−1 (5.9–17.1). But this value is greater than two-fold lower the median post hoc estimation quoted in their study (26.7 l h−1) and the absolute CL for total topotecan 24.8 (l h−1; Turner et al, 2013).
Pazopanib plasma concentrations did not differ between two regimens and were above (>20.6 μg ml−1) levels previously associated with longer PFS in renal cancer (Suttle et al, 2010). The severe haematological toxicities (grade ⩾3) were not more frequently reported in the present study with topotecan and pazopanib co-administration, than in the study with single weekly topotecan administration (Von Gruenigen et al, 2012). This is likely due to similar total topotecan concentrations in both studies.
In present study, the daily-times-five regimen resulted in more severe haematologic toxicity in comparison with the weekly topotecan regimen, whereas the dose density of the recommended topotecan dose in the daily-times-five regimen was lower (4.16 mg per week vs 6 mg per week).
The frequency of the gastrointestinal side effects (nausea/vomiting and diarrhoea) showed an additive effect in the combination treatment, when compared with safety information of single-agent treatment with both pazopanib and topotecan from the literature (Matrana et al, 2013). No clinically diagnosed congestive heart failure (CHF) was reported in our study, despite the fact that the recently published review with VEGFR-TKIs showed all-grade pazopanib-related CHF in 6.1% of patients (Qi et al, 2014).
Elevated transaminases were seen in 22% of the patients that corresponds to previously reported frequencies for single-agent pazopanib (0%–35%) (Hurwitz et al, 2009; Altorki et al, 2010; Sternberg et al, 2010; Matrana et al, 2013). Grades 3 and 4 liver enzyme increase were seen in <1% of patients treated with pazopanib (Anon, 2013b). Rare but potentially severe and fatal hepatotoxicity has been observed with pazopanib treatment. There was one occurrence of fatal liver necrosis and another patient with liver metastases who developed grade 4 toxic hepatitis after pazopanib exposure. The liver necrosis could have been affected by the ABCB gene polymorphism, concomitant administration of topotecan and pazopanib and concomitant paracetamol treatment. Hepatotoxicity is not likely to be related to topotecan therapy based on experience from previous phase I–III trials (Anon, 2013a).
In the present study, stomatitis of all grades was observed two times more frequently (22 vs 11%) than in the pazopanib literature (Anon, 2013b). The reason for this could be underreporting of oral adverse events secondary to TKIs, as they more closely resemble aphthous stomatitis than oral mucositis or stomatitis caused by conventional agents (Boers-Doets et al, 2012).
In total, 8 patients out of evaluable 50 (16%) had PR and 27 patients (54%) had SD. Seven out of 15 (47%) heavily pretreated patients with platinum-resistant ovarian cancer evaluable for anti-tumour activity showed PR as their best response. In the literature, topotecan alone in patients with recurrent ovarian cancer, in the daily-times-five oral regimen yielded objective response ranging from 13 to 16.3% when administered as second line and/or later lines of therapy (Creemers et al, 1996; Ten Bokkel Huinink et al, 1997; Bookman et al, 1998; Bodnar et al, 2009), and 30% (6 out of 20) as a salvage i.v. topotecan single therapy (Bodnar et al, 2009). Seven of the total number of patients had STS and all of them had SD, as their best response with mean duration of 15.5 weeks (s.d.=4.9).
Conclusion
Pazopanib substantially increased exposure of total topotecan by 1.8-fold for Cmax and 1.7-fold for AUC(0–∞), which is considered clinically relevant, but did not increase t1/2 values, whereas topotecan had no effect on the exposure of pazopanib. The combination of both oral topotecan chemotherapy and angiogenesis inhibitor pazopanib was found to be active and tolerated by patients with solid tumours in two administration regimens: oral topotecan with 8 mg weekly in a 28-day cycle and 2.5 mg five times weekly in a 21-day cycle, both in combination with pazopanib 800 mg daily permitting further evaluation especially the weekly schedule.
Preliminary anti-tumour activity was observed in patients with advanced platinum-resistant ovarian cancer, who had all been pretreated with carboplatin.
Change history
01 September 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Altorki N, Lane ME, Bauer T, Lee PC, Guarino MJ, Pass H, Felip E, Peylan-Ramu N, Gurpide A, Grannis FW, Mitchell JD, Tachdjian S, Swann RS, Huff A, Roychowdhury DF, Reeves A, Ottesen LH, Yankelevitz DF (2010) Phase II proof-of-concept study of pazopanib monotherapy in treatment-naive patients with stage I/II resectable non-small-cell lung cancer. J Clin Oncol 28: 3131–3137.
Anon (2013a) Hycamptin SmPC/Topotecan. Summary of product characteristics. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000123/WC500051542.pdf.
Anon (2013b) Pazopanib SmPC/Votrient 200 mg and 400 mg film coated tablets Summary of Product Characteristics. The electronic Medicines Compendium (eMC). Available at http://www.medicines.org.uk/emc/medicine/23148/spc.
Bodnar L, Wcislo G, Nasilowska A, Szarlej-Wcislo K, Gasowska-Bodnar A, Smoter M, Szczylik C (2009) Salvage therapy with topotecan in heavily pretreated ovarian cancer patients. J Cancer Res Clin Oncol 135: 815–821.
Boers-Doets CB, Epstein JB, Raber-Durlacher JE, Ouwerkerk J, Logan RM, Brakenhoff JA, Lacouture ME, Gelderblom H (2012) Oral adverse events associated with tyrosine kinase and mammalian target of rapamycin inhibitors in renal cell carcinoma: a structured literature review. Oncologist 17: 135–144.
Bois ADu (2014) Randomized, double-blind, phase III trial of pazopanib versus placebo in women who have not progressed after first-line chemotherapy for advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (AEOC): Results of an international Intergro. ASCO Abstract LBA5503. Available at http://meetinglibrary.asco.org/content/115898-132.
Bookman MA, Malmström H, Bolis G, Gordon A, Lissoni A, Krebs JB, Fields SZ (1998) Topotecan for the treatment of advanced epithelial ovarian cancer: an open-label phase II study in patients treated after prior chemotherapy that contained cisplatin or carboplatin and paclitaxel. J Clin Oncol 16: 3345–3352.
Bosch TM, Meijerman I, Beijnen JH, Schellens JH (2006) Genetic polymorphisms of drug-metabolising enzymes and drug transporters in the chemotherapeutic treatment of cancer. Clin Pharmacokinet 45: 253–285.
Creemers GJ, Lund B, Verweij J (1994) Topoisomerase I inhibitors: topotecan and irenotecan. Cancer Treat Rev 20: 73–96.
Creemers GJ, Bolis G, Gore M, Scarfone G, Lacave AJ, Guastalla JP, Despax R, Favalli G, Kreinberg R, Van Belle S, Hudson I, Verweij J, Ten Bokkel Huinink WW (1996) Topotecan, an active drug in the second-line treatment of epithelial ovarian cancer: results of a large European phase II study. J Clin Oncol 14: 3056–3061.
Deng Y, Sychterz C, Suttle AB, Dar MM, Bershas D, Negash K, Qian Y, Chen EP, Gorycki PD, Ho MY (2013) Bioavailability, metabolism and disposition of oral pazopanib in patients with advanced cancer. Xenobiotica 43: 443–453.
Eckardt JR, von Pawel J, Pujol JL, Papai Z, Quoix E, Ardizzoni A, Poulin R, Preston AJ, Dane G, Ross G (2007) Phase III study of oral compared with intravenous topotecan as second-line therapy in small-cell lung cancer. J Clin Oncol 25: 2086–2092.
Herben VM, Rosing H, ten Bokkel Huinink WW, van Zomeren DM, Batchelor D, Doyle E, Beusenberg FD, Beijnen JH, Schellens JH (1999) Oral topotecan: bioavailablity and effect of food co-administration. Br J Cancer 80: 1380–1386.
Hurwitz HI, Dowlati A, Saini S, Savage S, Suttle AB, Gibson DM, Hodge JP, Merkle EM, Pandite L (2009) Phase I trial of pazopanib in patients with advanced cancer. Clin Cancer Res 15: 4220–4227.
Jain RK (2001) Normalising tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med 7: 987–989.
Kerbel RS, Kamen BA (2004) The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer 4: 423–436.
Kruijtzer CM, Beijnen JH, Rosing H, ten Bokkel Huinink WW, Schot M, Jewell RC, Paul EM, Schellens JH (2002) Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J Clin Oncol 20: 2943–2950.
Kumar S, Mokhtari RB, Sheikh R, Wu B, Zhang L, Xu P, Man S, Oliveira ID, Yeger H, Kerbel RS, Baruchel S (2011) Metronomic oral topotecan with pazopanib is an active antiangiogenic regimen in mouse models of aggressive pediatric solid tumor. Clin Cancer Res 17: 5656–5667.
Maliepaard M, Scheffer GL, Faneyte IF, van Gastelen MA, Pijnenborg AC, Schinkel AH, van De Vijver MJ, Scheper RJ, Schellens JH (2001) Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res 61: 3458–3464.
Maliepaard M, van Gastelen MA, de Jong LA, Pluim D, van Waardenburg RC, Ruevekamp-Helmers MC, Floot BG, Schellens JH (1999) Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. Cancer Res 59: 4559–4563.
Mathijssen RHJ, de Jong FA, Loos WJ, van der Bol JM, Verweij J, Sparreboom A (2007) Flat-fixed dosing versus body surface area based dosing of anticancer drugs in adults: does it make a difference? Oncologist 12: 913–923.
Matrana MR, Duran C, Shetty A, Xiao L, Atkinson BJ, Corn P, Pagliaro LC, Millikan RE, Charnsangave C, Jonasch E, Tannir NM (2013) Outcomes of patients with metastatic clear-cell renal cell carcinoma treated with pazopanib after disease progression with other targeted therapies. Eur J Cancer 49: 3169–3175.
Minocha M, Khurana V, Qin B, Pal D, Mitra AK (2012) Enhanced brain accumulation of pazopanib by modulating P-gp and Bcrp1 mediated efflux with canertinib or erlotinib. Int J Pharm 436: 127–134.
O’Brien ME, Ciuleanu TE, Tsekov H, Shparyk Y, Cuceviá B, Juhasz G, Thatcher N, Ross GA, Dane GC, Crofts T (2006) Phase III trial comparing supportive care alone with supportive care with oral topotecan in patients with relapsed small-cell lung cancer. J Clin Oncol 24: 5441–5447.
Pasquier E, Kavallaris M, Andre N (2010) Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol 7: 455–465.
Qi WX, Shen Z, Tang LN, Yao Y (2014) Congestive heart failure risk in cancer patients treated with VEGFR-TKIs: a systematic review and meta-analysis of 36 clinical trials. Br J Clin Pharmacol 78 (4): 748–762.
Roodhart JM, Langenberg MH, Vermaat JS, Lolkema MP, Baars A, Giles RH, Witteveen EO, Voest EE (2010) Late release of circulating endothelial cells and endothelial progenitor cells after chemotherapy predicts response and survival in cancer patients. Neoplasia 12: 87–94.
Rosing H, Doyle E, Davies BE, Beijnen JH (1995) High-performance liquid chromatographic determination of the novel antitumour drug topotecan and topotecan as the total of the lactone plus carboxylate forms, in human plasma. J Chromatogr B Biomed Appl 668 (1): 107–115.
Schellens JH, Malingre MM, Kruijtzer CM, Bardelmeijer HA, van Tellingen O, Schinkel AH, Beijnen JH (2000) Modulation of oral bioavailability of anticancer drugs: from mouse to man. Eur J Pharm Sci 12: 103–110.
Schellens JH, Maliepaard M, Scheper RJ, Scheffer GL, Jonker JW, Smit JW, Beijnen JH, Schinkel AH (2000) Transport of topoisomerase I inhibitors by the breast cancer resistance protein. Potential clinical implications. Ann N Y Acad Sci 922: 188–194.
Schiller JH, Kim K, Hutson P, DeVore R, Glick J, Stewart J, Johnson D (1996) Phase II study of topotecan in patients with extensive-stage small-cell carcinoma of the lung: an Eastern Cooperative Oncology Group Trial. J Clin Oncol 14: 2345–2352.
Sehouli J, Stengel D, Harter P, Kurzeder C, Belau A, Bogenrieder T, Markmann S, Mahner S, Mueller L, Lorenz R, Nugent A, Wilke J, Kuznik A, Doering G, Wischnik A, Sommer H, Meerpohl HG, Schroeder W, Lichtenegger W, Oskay-Oezcelik G (2011) Topotecan weekly versus conventional 5-day schedule in patients with platinum-resistant ovarian cancer: a randomized multicenter phase II trial of the North-Eastern German Society of Gynecological Oncology Ovarian Cancer Study Group. J Clin Oncol 29: 242–248.
Shaked Y, Henke E, Roodhart JM, Mancuso P, Langenberg MH, Colleoni M, Daenen LG, Man S, Xu P, Emmenegger U, Tang T, Zhu Z, Witte L, Strieter RM, Bertolini F, Voest EE, Benezra R, Kerbel RS (2008) Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell 14: 263–273.
Shrestha B, Reed JM, Starks PT, Kaufman GE, Goldstone JV, Roelke ME, O'Brien SJ, Koepfli KP, Frank LG, Court MH (2011) Evolution of a major drug metabolizing enzyme defect in the domestic cat and other felidae: phylogenetic timing and the role of hypercarnivory. PLoS One 6: e18046.
Steinijans VW, Diletti E (1983) Statistical analysis of bioavailability studies: parametric and nonparametric confidence intervals. Eur J Clin Pharmacol 24: 127–136.
Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, Barrios CH, Salman P, Gladkov OA, Kavina A, Zarbá JJ, Chen M, McCann L, Pandite L, Roychowdhury DF, Hawkins RE (2010) Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 28: 1061–1068.
Stewart DJ (2004) Topotecan in the first-line treatment of small cell lung cancer. Oncologist 9 (Suppl 6): 33–42.
Suttle B, Ball HA, Molimard M et al (2010) Relationship between exposure to pazopanib (P) and efficacy in patients (pts) with advanced renal cell carcinoma (mRCC). J Clin Oncol 28 (Suppl 15): 3048.
ten Bokkel Huinink W, Gore M, Carmichael J, Gordon A, Malfetano J, Hudson I, Broom C, Scarabelli C, Davidson N, Spanczynski M, Bolis G, Malmström H, Coleman R, Fields SC, Heron JF (1997) Topotecan versus paclitaxel for the treatment of recurrent epithelial ovarian cancer. J Clin Oncol 15: 2183–2193.
Turner DC, Tillmanns TD, Harstead KE, Throm SL, Stewart CF (2013) Combination metronomic oral topotecan and pazopanib: a pharmacokinetic study in patients with gynecological cancer. Anticancer Res 33: 3823–3829.
Von Gruenigen VE, Frasure HE, Smith DA, Fusco NL, Eaton SM, DeBernardo RL, Heugel AM, Waggoner SE (2012) Toxicity of weekly oral topotecan in relation to dosage for gynecologic malignancies: a phase I study. Anticancer Drugs 23: 724–730.
Von Pawel J, Gatzemeier U, Pujol JL, Moreau L, Bildat S, Ranson M, Richardson G, Steppert C, Rivière A, Camlett I, Lane S, Ross G (2001) Phase ii comparator study of oral versus intravenous topotecan in patients with chemosensitive small-cell lung cancer. J Clin Oncol 19: 1743–1749.
Xu CF, Reck BH, Xue Z, Huang L, Baker KL, Chen M, Chen EP, Ellens HE, Mooser VE, Cardon LR, Spraggs CF, Pandite L (2010) Pazopanib-induced hyperbilirubinemia is associated with Gilbert’s syndrome UGT1A1 polymorphism. Br J Cancer 102: 1371–1377.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
This work was supported by GSK. JS, SL, PL, PW, DAS are employed by GSK. The remaining authors declare no conflicts of interest.
Additional information
STATEMENT OF TRANSLATIONAL RELEVANCE
Combining oral topotecan and pazopanib showed an acceptable safety profile in this phase I study in 67 enroled patients and was consistent with the known profile of both single agents. The daily-times-five regimen resulted in more severe haematologic toxicity in comparison with the weekly topotecan regimen. Pazopanib co-administration caused a substantial increase in exposure of total topotecan (1.7-fold) compared with topotecan alone, which is considered clinically relevant. Topotecan had no effect on pazopanib concentrations. Both regimens could be administered for longer periods and showed initial signs of anti-tumour activity. PR showed 8 out of 50 patients (16%), with a median duration of response of 24 weeks. Seven of them had platinum-resistant ovarian cancer. In addition, 54% of patients had SD as the best response with 22% stable for 6 months.
Our results suggested that the effects of pazopanib on topotecan pharmacokinetics were pre-systemic and not related to changes in elimination. Administration of oral topotecan in combination with pazopanib is found to be active and moderate to well tolerated by patients with solid tumours in two administration regimens.
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Kerklaan, B., Lolkema, M., Devriese, L. et al. Phase I and pharmacological study of pazopanib in combination with oral topotecan in patients with advanced solid tumours. Br J Cancer 113, 706–715 (2015). https://doi.org/10.1038/bjc.2015.257
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.257
Keywords
This article is cited by
-
Phase II study of pazopanib with oral topotecan in patients with metastatic and non-resectable soft tissue and bone sarcomas
British Journal of Cancer (2021)
-
Targeted therapies in gynecological cancers: a comprehensive review of clinical evidence
Signal Transduction and Targeted Therapy (2020)
-
Revisiting the role of ABC transporters in multidrug-resistant cancer
Nature Reviews Cancer (2018)
-
Pharmacokinetics of metronomic chemotherapy: a neglected but crucial aspect
Nature Reviews Clinical Oncology (2016)
