
Abstract
Background:
We conducted a phase 1 trial in patients with locally advanced cervical cancer by injecting 0.5 ml of the CK2-antagonist CIGB-300 in two different sites on tumours to assess tumour uptake, safety, pharmacodynamic activity and identify the recommended dose.
Methods:
Fourteen patients were treated with intralesional injections containing 35 or 70 mg of CIGB-300 in three alternate cycles of three consecutive days each before standard chemoradiotherapy. Tumour uptake was determined using 99Tc-radiolabelled peptide. In situ B23/nucleophosmin was determined by immunohistochemistry.
Results:
Maximum tumour uptake for CIGB-300 70-mg dose was significantly higher than the one observed for 35 mg: 16.1±8.9 vs 31.3±12.9 mg (P=0.01). Both, AUC24h and biological half-life were also significantly higher using 70 mg of CIGB-300 (P<0.001). Unincorporated CIGB-300 diffused rapidly to blood and was mainly distributed towards kidneys, and marginally in liver, lungs, heart and spleen. There was no DLT and moderate allergic-like reactions were the most common systemic side effect with strong correlation between unincorporated CIGB-300 and histamine levels in blood. CIGB-300, 70 mg, downregulated B23/nucleophosmin (P=0.03) in tumour specimens.
Conclusion:
Intralesional injections of 70 mg CIGB-300 in two sites (0.5 ml per injection) and this treatment plan are recommended to be evaluated in phase 2 studies.
Similar content being viewed by others


Main
The CK2-mediated phosphorylation has a significant role in the pathogenesis and development of both solid and haematopoietic tumours (Duncan and Litchfield, 2008; Piazza et al, 2012). Of note, CK2 signalling pathways seem to reinforce biochemical cascades indispensable for tumour growth, proliferation and resistance to conventional and novel cytotoxic agents (Piazza et al, 2012). Therefore, CK2 inhibition could represent a rational therapeutic approach to treat both solid and haematological tumours. In fact, different CK2 inhibitors have already provided successful proof-of-concept for such rationality at the experimental setting (Ahmada et al, 2005; Prudent et al, 2010; Siddiqui-Jain et al, 2010) with perspectives to become a novel cancer-targeted therapeutics in future. Among the CK2 experimental inhibitors, only the CX-4945 small molecule and the CIGB-300 cell-permeable peptide have already reached the clinical stage in phase 1 trials.
The CIGB-300 treatment inhibits the CK2-mediated phosphorylation of the B23/nucleophosmin protein-inducing apoptosis in vitro and in vivo and halts tumour growth in animals (Perea et al, 2004, 2008, 2009). Interestingly, it has been observed that a B23/nucleophosmin downregulation on CIGB-300-treated tumour cells (Rodriguez-Ulloa et al, 2010) probably is a consequence of inhibiting the CK2-mediated phosphorylation in such a protein (Tawfic et al, 1995). Importantly, the CIGB-300 treatment modulates a diverse array of proteins involved in apoptosis, cell proliferation, ribosomal biogenesis, anti-cancer drug resistance and angiogenesis in vitro (Rodriguez-Ulloa et al, 2010). Likewise, evidences of synergistic effect between CIGB-300 and some standard chemotherapeutics like cisplatin and paclitaxel, have been also observed both in vitro and in vivo in cervical and lung cancer models (Perera et al, 2014). Such premises along with preclinical data demonstrating the significant antitumour activity of CIGB-300 by intratumour injections in human cervical tumours (Perea et al, 2008), encouraged the subsequent evaluation of this peptide-based drug candidate in patients with cervical malignancies using intralesional injections.
Although the systemic routes are preferred for the administration of most anti-cancer drugs, we have hypothesised that local delivery for CIGB-300 could lead to favourable tumour uptake in patients with relative easy-to-access tumours like those ones in the uterine cervix. Under this clinical hypothesis, a First-in-Human trial using CIGB-300 intralesional injections in patients with high-grade squamous intraepithelial lesions demonstrated to be safe and tolerable (Solares et al, 2009). In that study, 90% of the patients experienced a significant colposcopic response, 19% experienced full histological regression and human papillomavirus DNA was negative in 48% of the previously positive patients. Furthermore, CIGB-300 has been also used under compassionate use clauses by this route. Remarkably, a successful antitumour response was observed in a chemoradio refractory patient diagnosed with germinoma who experienced significant tumour shrinking with a sustained improvement of his quality of life after intratumour injection of CIGB-300 (Perea et al, 2011).
Finally, a phase 1 trial in patients with cervical cancer FIGO stages IB2/II using 2 ml-intralesional injections of CIGB-300 in a single site on tumours indicated a plateau in terms of tumour uptake between doses of 35 and 70 mg of the peptide (Soriano-García et al, 2013). Therefore, we hypothesised that less volume (1 ml) applied into two equidistant tumour sites would allow a greater CIGB-300 tumour uptake using the highest dose, hence maximising the therapeutic chances for this peptide.
This phase 1 trial was designed to investigate the tumour uptake, safety profile and pharmacodynamic activity of CIGB-300 injected in two different sites on tumours at 0.5 ml per injection.
Materials and methods
Patient selection
Patients histologically diagnosed as FIGO stage IIB cervical cancer were recruited in five gyneco-obstetric services in Havana and Matanzas. They were firstly evaluated in the Reference Center for Cervical Cancer at the ‘Ramón González Coro’ Hospital in Havana, and stage classification was finally concluded in the gynaecological service of the Center for Medical–Surgical Research, Havana. CIGB-300 administration and evaluations took place as in-patients in the second institution, except the first dose when patients were translated to the Clinical Investigation Center (Havana) for biodistribution/pharmacokinetic studies. Pre-inclusion evaluation included a detailed history and physical examination by video colposcopy. In addition, electrocardiogram, rectoscopy, cystoscopy, haematological counts, blood chemistry, coagulation and cervix microbiological studies were performed.
Patients were included if they were 18–75-years old, had clinical, imaging and histological diagnosis of stage IIB cervical epidermoid carcinoma (Benedet et al, 2003), a WHO general health index from 0 to 2, laboratory parameters within normal limits, and gave their written informed consent to participate. Exclusion criteria were chemotherapy or surgical, ablative, radiant, or immunomodulator treatment in the previous 30 days, psychiatric disease, pregnancy, breastfeeding, uncontrolled chronic diseases such as diabetes, hypertension, hepatic or renal chronic insufficiency, epilepsy or other malignant neoplasia, autoimmune or allergic disease, coagulation dysfunction, acute systemic infections, current administration of immunomodulating drugs, tumour extensive necrosis or genital sepsis that could limit the application of the product, and participation in another clinical trial in the last 8 weeks.
This trial followed the principles of Good Clinical Practices, in accordance with the Declaration of Helsinki and its amendments. The protocol was approved by the Ethic Committees of the participant institutions, and by the Cuban Regulatory Authority, the State Center for the Control of Drugs, Equipment & Medical Devices (CECMED): reference number: 05.009.09.B. Cuban Public Registry of Clinical Trials: RPCEC00000142.
Study design and treatment plan
A randomised, double-blind, uncontrolled phase 1 clinical trial was carried out. Patients were distributed according to a computer-generated random number list, to receive 35 mg (group I) or 70 mg (group II) of CIGB-300. It was expected to include 6–8 patients in each group. This sample size was inferred starting from a prefixed maximum limit of toxicity taking into account before experience. That limit was 30% of patients with grade 3 adverse events into each group, with a 95% confidence interval. This method for N calculation is considered specific to pilot studies with transitional therapies even when no adverse events are registered (Carter and Woolson, 2004).
CIGB-300 was synthesised and supplied by the Peptide Synthesis Unit at CIGB (Havana) in 35 mg vials, as a lyophilised powder. To keep masking for both groups of patients, the same number of vials was administered. One CIGB-300 and one placebo vial were used for the 35 mg group and two 35 mg vials were used for 70 mg group. Their composition differed only regarding the presence of CIGB-300. Random lists were kept at the sponsor’s product-preparation department, where vials were labelled. This group was independent from the monitors, investigators or other trial participants. All clinical and laboratory evaluations were blinded regarding the patients’ group allocation. The code was opened only in case of serious adverse events, deaths or at the end of the study, after database closure.
The whole content of the two vials was re-suspended in 1 ml of water for injection, later injected into two equidistant points (0.5 ml per point) from the lesion centre, ∼2 cm deep, for 10 min (5 min per injection). Injection of necrotic areas with craters or in the cervical channel was avoided. Patients received three cycles of CIGB-300 as single agent at weeks 1, 3 and 5. All of them were hospitalised and treated once daily during three consecutive days at each cycle. Tumour uptake, whole-body biodistribution and drug levels in blood were measured after the first drug administration with 99mTechnetium (Tc)-labelled CIGB-300. After last cycle (week 5), imaging evaluation was carried out before conventional chemoradiotherapy. This regimen consisted in cisplatin 40 mg m−2, once per week, during 6 weeks, concomitant to 5040 cGy of external beam radiotherapy given in 28 fractions, followed by 2600 cGy of intracavitary radiotherapy given in four high-dose fractions (650 cGy per day) over 2 weeks. Other interventions were indicated only for the management of adverse events, according to established clinical practices.
Tumour-uptake profile and biodistribution of CIGB-300 in organs
CIGB-300 was firstly radiolabelled with 99mTc and whole-body imaging studies were performed at 10 min, 1, 2, 4, 8, 12 and 24 h using a gamma camera (Park Medical, Quebec, QC, Canada) with a high energy collimator for 99mTc. Tumour static views were also taken at 3 and 24 h. Calculation of the percent of uptaken radioactivity in tumours and target organs was based on the Medical Internal Radiation Dose (MIRD) methodology for quantitative radiopharmaceutical biodistribution data acquisition (Siegel et al, 1999). Tumour mass was estimated from morphometric data obtained from initial magnetic resonance imaging (MRI). Biological half-life was adjusted by means of the Microcal Origin program, version 6.0 (Northampton, MA, USA), taking into account the radioactive decay. Monoexponential and biexponential functions were fitted for calculation of the area under the curve.
Pharmacokinetics of CIGB-300
Blood samples were drawn by venipuncture immediately, 5, 15, 30 min and 1, 2, 4, 8, 12 and 24 h after the first CIGB-300 administration. Subsequently, plasma from each sample was obtained and CIGB-300 was quantified by an in-house validated immune competition-based enzyme immunoassay (EIA). Drug disposition analysis was performed individually by a non-compartmental method with a combined linear/log–linear trapezoidal rule approach. All the pharmacokinetic parameters were determined by using the WinNonlin professional software (version 5.1, Pharsight Inc., 2005, Princetone, NJ, USA).
Safety evaluation and histamine levels in plasma
Local and systemic adverse events, including routine laboratory parameters, were carefully screened up. Systemic toxicity was evaluated during 24 h after each CIGB-300 administration, including cardiovascular monitoring during the injections and vital signs measurements (temperature, heart beats, respiration per min and blood pressure). After chemoradiotherapy, patients were 1-year followed for safety. The medical terminology for adverse events and their severity classification (in five grades) was applied according to the Common Terminology Criteria for Adverse Events (CTCAE) (2010). The causal relationship was classified as very probable (definitive), probable, possible or remote (doubtful) (Naranjo et al, 1998).
The CIGB-300-induced histamine levels were measured in plasma by a commercially available EIA kit (Labor Diagnostika, GmBH, Oberursel, Germany) before, immediately, 5, 15, 30 and 60 min after the first CIGB-300 administration.
Pharmacodynamic activity of CIGB-300 and 1-year clinical follow-up
Expression of B23/NPM in tumours biopsies was analysed by immunohistochemistry in paraffin-embedded tumour biopsies taken before and after CIGB-300 treatment using a commercial immunostaining kit (Histostain-SP, Zymed Laboratories, San Francisco, CA, USA) with a specific human B23/NPM Mab as primary antibody (Invitrogen, Camarillo, CA, USA). Deparaffined and further processed biopsies were finally stained with peroxidase stain and further contrastained with hematoxylin.
Therapeutic response was defined by MRI (Siemens AG, Erlangen, Germany), according to the RECIST criteria for solid tumours (Therasse et al, 2000). Images were taken before and after CIGB-300 treatment and quarterly during a 1-year follow-up after CIGB-300+chemoradiotherapy. Computed tomography (CT) scans (Siemens AG) and abdominal and gynaecological ultrasonographies (Toshiba, Tokyo, Japan) were used to detect proximal and distant metastasis.
In vivo studies
Female 6–8-week-old NIH nu/nu nude mice were subcutaneously inoculated with 5 × 106 of human cervical cancer SiHa cells into inguinal zone of the right flank. When tumours reached 150 mm3, 100 μg of 99m Tc-radiolabelled CIGB-300 were intratumour injected in 0.025, 0.05, 0.2 or 0.5 ml of PBS. Whole-body images were collected in living animals immediately after injection, at 0.25, 1, 4, 24 and 48 h using a planar gamma camera (Mediso Nucline TH-22, Budapest, Hungary). The region of interest associated to tumour was selected using the software ImageJ (1.48r, National Institute of Health, Bethesda, MD, USA) and results are expressed as percent of total initial image.
The antitumour activity of different CIGB-300 regimens was explored using a factorial design that combines both number of cycles of peptide administration (1, 3 or 5 cycles) and the time interval between each cycle (3, 5 or 8 days). Each cycle consisted of three consecutive intratumoural injections of 200 μg of CIGB-300. Tumours were measured with a caliper and the respective volumes were calculated using the formula: volume=width2 (mm) × length (mm)/2. Animals were maintained in pathogen-free conditions and procedures were performed in accordance with recommendations for the proper use and care of laboratory animals.
Statistical analysis
Continuous variables were expressed as mean±s.d. or median±quartile range and minimum and maximum values (range). With these variables, analyses of normality (Shapiro–Wilk’s test) and homogeneity of variance (Levene’s test) were carried out. Categorical variables were given as absolute values and percentages. Biodistribution and pharmacokinetic parameters as well as those variables related to the therapeutic effect were tested by the estimation of the 95% confidence intervals for the differences between groups as well as the calculation of the probability that the difference was >0. Tolerability was treated through the estimation of severe toxicity in both groups. For both groups the probability to surpass maximum level of toxicity (30%) was estimated. Spearman rank correlation analyses between histamine levels and CIGB-300 concentration and between histamine and severity of the allergic reaction were done. This test was also used to determine the presence of direct or inverse correlation accordingly. Immunohistochemical data were treated using paired analysis (Student’s t-test or Wilcoxon’s test) depending on the normality assumption.
Results
Patient characteristics and dosing history
Fourteen patients, seven per dose level, fulfilled the selection criteria to be included in the trial. Demographic and baseline data are shown in Table 1. The groups were similar regarding each variable. Most of the patients were white, their ages ranged from 25 to 61-years old (mean around 40 years), weighed 50–80 kg and were 152–177 cm tall. Mean BMI was around 26 kg m−2. All the patients were classified within stage IIB cervical cancer, and no gynaecological or general pathological antecedent stood out particularly (data not shown).
Most of the patients completed the three cycles of CIGB-300 treatment, except three of them who discontinued treatment during the third cycle because of grade 2 allergic syndrome (two cases, one from each group) and acute febrile disease (one case, group II). All the patients received at least seven doses of CIGB-300 (median: nine doses). Similarly, all the patients fulfilled the chemoradiotherapy schedule, except patient no. 2 (group I) who died because of disease progression.
CIGB-300 tumour-uptake profile and biodistribution
The time-course analysis of CIGB-300 tumour uptake is shown in Figure 1. There was a maximum CIGB-300 tumour uptake at 10 min after intratumoural administration that gradually decreased until 24 h in all of the patients. Interestingly, mean value of the maximum CIGB-300 tumour uptake was near two-fold higher after injecting 70 mg compared with 35 mg of CIGB-300 (31.3 vs 16.1 mg; P=0.01) (Table 2). After normalising the CIGB-300 tumour uptake respect to tumour mass volume estimated by MRI, there was roughly 35% increase of this parameter for the group II respect to group I (1.1±0.8 vs 0.7±0.6 mg g−1), although such difference was not statistically significant (P=0.24) (Table 2). Importantly, the AUC24 was significantly higher in the group II respect to group I (385 vs 132 mg h; P<0.001) and the CIGB-300 biological half-life was also significantly longer in patients from group II (6.2±1.9 vs 3.9±0.9 h; P<0.001) (Table 2).

Time course of CIGB-300 tumour uptake. In the first day of cycle 1, 35 or 70 mg of 99mTc-radiolabelled CIGB-300 was injected directly into tumours and whole-body gammagraphic images were taken during 24 h after administration. CIGB-300 tumour uptake is expressed as mg of CIGB-300 retained in tumours. S.e.m. is represented. Inset corresponds to a illustrative anterior whole-body image from one patient at 10 min after injecting 70 mg (group II) of radiolabelled peptide.
Immediately after the CIGB-300 intratumour injection, the unincorporated peptide escaped to blood stream and it was mainly distributed towards kidneys and liver although some radiolabelled peptide was also detected in lungs, spleen and heart at a lesser extent (Supplementary Figure 1). Interestingly, in some patients, CIGB-300 was localised in locoregional nodules and bladder (data not shown).
Pharmacokinetics of CIGB-300
The pharmacokinetic parameters of the unincorporated CIGB-300 after intralesional injection were estimated from the respective clearance curves in plasma. The highest CIGB-300 levels in blood were observed only during the first hour after injection and it was near undetectable after 8 h for both groups. No significant differences on most of the pharmacokinetic parameters were detected between both groups, except for Tmax and t1/2 where they were higher for the group II (Supplementary Table 2). Interestingly, an inverse correlation between the CIGB-300 Cmax in blood and the maximum CIGB-300 tumour uptake was observed (P=0.035, r2=−0.587).
Safety and tolerability
Table 3 lists the local and systemic toxicities observed during CIGB-300 at both dose levels. Overall, the type of events were similar in both groups and the allergic-like reactions were the most common systemic side effect, in particular hot flashes (100%) and localised itching and rashes (85.7%). A total of 39 types of adverse events were recorded, 81.7% of the 378 reports corresponded to systemic events and 58.5% occurred in the group II. Otherwise, only three types of local side effects were observed being the pain in lower abdomen the most frequent one (85.7%) in both groups. Only four grade 3 adverse events were observed in the whole trial irrespective of the CIGB-300 dose level (Table 3). They were distributed as follows: one allergic reaction, one anaemia and one tumour bleeding in group I, whereas one allergic reaction was found in group II. The severe allergic reactions were produced in the last cycle of treatment, otherwise, anaemia and tumour bleeding was observed after the first cycle. After red cell transfusion, patient with anaemia recovered normal haemoglobin values and continued the CIGB-300 treatment considering the favourable risk–benefit balance. In all the cases, the allergic reaction events lasted about 15 min with further spontaneous resolution. Cardiovascular events were mostly mild and had a rapid and spontaneous resolution. The pain in lower abdomen was successfully treated with analgesics. There were no differences between baseline and CIGB-300 post-treatment blood counts and serum chemistry values.
As the transient allergic reactions after CIGB-300 administration have been associated to the transient histamine induction (Soriano-García et al, 2013), we also verified such biochemical event in this clinical trial. Figure 2 shows a very similar induction pattern of plasmatic histamine after the first CIGB-300 injection in both groups that is characterised by a rapid increase and returning to normal levels (1 ng ml−1) after 15 min. Interestingly, a significant correlation was detected between the histamine levels and the unincorporated CIGB-300 in blood (P<0.0001) (Supplementary Table 3). Although there was no significant correlation between the severity of ‘allergic-like’ events and histamine levels, the overall histamine median was higher when grade 2 events occurred, different from grade 1 events: 3.5 vs. 0.7 ng ml−1.

Plasmatic histamine levels after the first CIGB-300 intratumour injection. Data correspond to the histamine mean values from patients who received 35 mg (solid line, N=7) or 70 mg (dashed line, N=7) of CIGB-300. Standard deviations are not shown for simplifying the illustration.
Pharmacodynamic activity and therapeutic effect
To explore pharmacodynamic activity in the CIGB-300 treatment plan assessed here, we studied the in situ levels of B23/nucleophosmin in paraffin-embedded tumour biopsies before and after completing the CIGB-300 therapeutic regimen. Interestingly, CIGB-300 treatment reduced nucleolar B23/NPM expression in 10 out of the 12 evaluated patients and such suppressive effect was statistically significant in group II (P=0.03) (Table 4). Otherwise, B23/nucleophosmin expression at the nucleoplasm was not considerably modified.
Data from long-term clinical response are shown in Table 5. Importantly, during 1-year follow-up after chemoradiotherapy, a better and faster clinical response was observed in those patients from group II according to the RECIST criteria. Complete response was achieved in 6 out of 7 patients by the third month in this group, whereas only 2 out of 6 was observed in group I. Interestingly, by the sixth month and thereafter, 100% of the patients exhibited complete response in group II whereas in group I, three patients did not respond completely, two of them died (Table 5). Overall, there were four deaths in this clinical trial because of disease progression, but three of them corresponded to the group I. There were not new adverse events during 1-year follow-up. Longer follow-up is currently ongoing and no other deaths are recorded.
In vivo preclinical studies
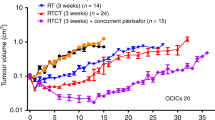
To verify how much the injection volume could influence the tumour uptake, we injected a fixed 99mTc-radiolabelled CIGB-300 dose in 0.025, 0.05, 0.2 or 0.5 ml of PBS in cervical cancer tumours xenografted in nude mice. The injections in lowest volumes (0.025 and 0.05) exhibited the highest CIGB-300 tumour uptake, both immediately after application and thereafter (Figure 3A). Otherwise, higher injection volume exhibited worse tumour uptake.

In vivo preclinical studies. (A) CIGB-300 tumour uptake with different injection volumes. Intratumour injections of 100 μg of 99Tc-radiolabelled CIGB-300 in 0.025, 0.05, 0.2 or 0.5 ml of PBS were performed in SiHa tumour-bearing nude mice. Whole-body images were collected in living animals immediately after injection and at 0.25, 1, 4, 24 and 48 h. Data are expressed as percent of total initial image in different times. Inset corresponds to an example of the initial uptake on each injection volume tested. (B) Antitumour activity of CIGB-300 in animal model. The antitumour activity of different CIGB-300 regimens was explored using a factorial design. CIGB-300 (200 μg) was intratumor injected during 3-, 5- or 8-consecutive days. Concurrently, 1, 3 or 5 cycles of 3-consecutive day injections were investigated. Tumour mass volume is plotted at different times. Insert corresponds to the tumour mass volume at day 42.
We also investigated the number of CIGB-300 cycles required to achieve a significant antitumour effect, as well as the time interval required between each cycle, we performed a factorial designed testing 1, 3 or 5 cycles of peptide administration with drug holidays of 3, 5 or 8 days between each cycle. Data indicated a comparable or ‘saturable’ antitumour effect for 3 and 5 cycles of CIGB-300 irrespectively of the timing among each cycle (Figure 3B). Therefore, three cycles consisting of three consecutive daily injections seem to be the minimal frequency required to reduce tumour growth by CIGB-300 administration (Figure 3B).
Discussion
Data from this clinical trial delineate a new optimised protocol for the CIGB-300 intralesional administration in women with cervical cancer stage IIB before standard chemoradiotherapy. The injection of CIGB-300 70 mg (in 1 ml) in two different sites equidistantly from the tumour centre (0.5 ml per injection) guarantees the best tumour-uptake profile. It means, better tumour uptake (31.3±12.9 mg), AUC24 (385±167 mg h; P<0.001) and t1/2 (6.2±1.9 h) compared with injection of CIGB-300 35 mg. However, the variable baseline tumour size along with relative low number of patients in each group could influence the limited increase of the CIGB-300 tumour uptake for 70 mg respect to 35 mg when correcting it by the estimated tumour mass.
Opposite to these findings, former data injecting 2 ml of CIGB-300 at a single tumour site showed lower and not different maximum tumour-uptake levels between 35 and 70 mg of CIGB-300 (14.9±6.4 vs 10.4±8.3 mg) (Soriano-García et al, 2013). Therefore, besides number of applications, the injection volume itself could negatively influence on the tumour-uptake profile. Otherwise, lower volume could favour higher intratumoural CIGB-300 concentration with minor escape to the vascular and lymphatic vessels. This hypothesis was concurrently verified here in preclinical assays where an inverse relation between injection volume and the CIGB-300 tumour uptake was observed in cervical cancer tumours xenografted in nude mice. Furthermore, the injection in two equidistant tumour sites could decrease the risk of delivery into a necrosis area and permit homogeneous tumour uptake.
Consistently, after intralesional injection, the unincorporated CIGB-300 escaped rapidly into blood stream and distributed in kidneys and liver although some accumulation was also detected in lungs, heart and spleen in some patients. Interestingly, an inverse correlation between Cmax of unincorporated CIGB-300 and the maximum CIGB-300 tumour uptake was observed. It means, patients who exhibited higher CIGB-300 tumour uptake had lower plasmatic concentrations of this peptide, and vice versa.
The rationale of using the CIGB-300 regimen investigated in this clinical trial was encouraged and supported by data from a factorial design performed in preclinical studies which indicated that three cycles of three consecutive daily injections was quite enough to halt tumour growth in cervical–uterine tumour-bearing nude mice. Importantly, such CIGB-300 regimen was safe and well tolerated in patients with locally advanced cervical cancer as evidenced in this clinical trial. In fact, it was not MTD as grade 3 adverse events did not surpass the limit of toxicity (30% of patients in any group). However, CIGB-300 70 mg had been reported as MTD in a previous clinical study when intralesional injections were applied in a different administration regimen (Soriano-García et al, 2013). In this clinical trial only two grade 3 systemic ‘allergic-like’ (one in each group) events were recorded during treatment and the onset of allergic reactions paralleled with the CIGB-300 Cmax in blood which in turn, correlated with plasmatic histamine. Altogether, the CIGB-300 regimen based in three cycles of three consecutive day injections seemed to be the minimal intervention required to induce antitumour effect in vivo, it was safe in patients and therefore, merits to be assayed in phase 2 clinical trials.
In vitro studies have revealed that CIGB-300 impairs the CK2-mediated phosphorylation on B23/NPM with subsequent induction of nucleolar disassembly and apoptosis (Perera et al, 2009); and degradation of B23/NPM on CIGB-300-treated cells has been documented in non-small cell lung cancer cells (Rodriguez-Ulloa et al, 2010). Likewise, the B23/NPM downregulation in patients with cervical cancer treated with CIGB-300 has been previously observed (Soriano-García et al, 2013). Data from this clinical trial confirm that B23/NPM levels can be modulated by CIGB-300 intralesional injections as observed in 10 out of the 12 evaluated patients. Interestingly, there was a dose-dependent reduction on nucleolar B23/NPM expression in tumour biopsies that was significantly higher in the group treated with CIGB-300 70 mg. Such a finding agrees with the higher CIGB-300 tumour uptake observed in the group of patients treated with CIGB-300 70 mg. Therefore, the in situ B23/NPM expression meets potentialities as a feasible response biomarker for CIGB-300 and merits being further validated in clinical trials designed to explore efficacy of CIGB-300.
Besides the CIGB-300 tumour-uptake profile, safety and pharmacodynamics, this phase 1 clinical study also permitted to explore efficacy signs after combining CIGB-300 with chemoradiotherapy. Interestingly, during 1-year follow-up, a sustained clinical response was mainly observed in those patients that initially received 70 mg of CIGB-300 as determined by the number of patients with complete response according to the RECIST criteria.
In conclusion, this clinical trial provides a safe and feasible protocol for the intralesional administration of CIGB-300 where 70 mg is the dose with better risk–benefit balance alone and in combination with chemoradiotherapy. The favourable toxicity profile along with the efficacy signs could support further phase 2–3 clinical trials in high-risk cervical cancer, particularly in stage IIB–IVA patients with positive nodes and/or those with poor tolerance/response to radiotherapy.
Change history
12 May 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ahmad KA, Wang G, Slaton J, Ungera G, Ahmed K (2005) Targeting CK2 for cancer therapy. Anticancer Drugs 16 (10): 1037–1043.
FIGO. Staging classification and clinical practice guidelines of gynecologic cancer Benedet JL, Hacker NF, Ngan HY (2003). Committee on Gynecologic Oncology, IGCS Guidelines Committee 2nd edn pp 36–40. http://www.figo.org/publications.
Common Terminology Criteria for Adverse Events (CTCAE) (2010) Version 4.0, Published: May 28, 2009 (v4.03: June 14, 2010), DCTD, NCI, NIH, DHHS. http://ctep.cancer.gov.
Carter RE, Woolson RF (2004) Statistical design considerations for pilot studies transitioning therapies from the bench to the bedside. J Transl Med 2 (1): 37–39.
Duncan JS, Litchfield DW (2008) Too much of a good thing: the role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim Biophys Acta 1784 (1): 33–47.
Naranjo CA, Shear NH, Busto U (1998) Adverse drug reactions. In: Principles of Medical Pharmacology, Kalant H, Roschlau WHE (eds) 6th edn pp 791–800. Oxford University Press: New York.
Perea SE, Reyes O, Puchades Y, Mendoza O, Vispo NS, Torrens I, Santos A, Silva R, Acevedo B, López E, Falcón V, Alonso DF (2004) Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the Casein Kinase 2 (CK2). Cancer Res 64 (19): 7127–7129.
Perea SE, Reyes O, Baladron I, Perera Y, Farina H, Gil J, Rodriguez A, Bacardi D, Marcelo JL, Cosme K, Cruz M, Valenzuela C, Lopez-Saura PA, Puchades Y, Serrano JM, Mendoza O, Castellanos L, Sanchez A, Betancourt L, Besada V, Silva R, Lopez E, Falcon V, Hernandez I, Solares M, Santana A, Dıaz A, Ramos T, Lopez C, Ariosa J, Gonzalez LJ, Garay H, Gomez D, Gomez R, Alonso DF, Sigman H, Herrera L, Acevedo B (2008) CIGB-300, a novel proapoptotic peptide that impairs the CK2 phosphorylation and exhibits anticancer properties both in vitro and in vivo. Mol Cell Biochem 316 (1–2): 163–167.
Perea SE, Baladron I, Garcia Y, Perera Y, Lopez A, Soriano JL, Batista N, Palau A, Hernandez I, Farina H, Garcia I, Gonzalez L, Gil J, Rodriguez A, Solares M, Santana A, Cruz M, Lopez M, Valenzuela C, Reyes O, Lopez-Saura PA, Gonzalez CA, Diaz A, Castellanos L, Sanchez A, Betancourt L, Besada V, Gonzalez LJ, Garay H, Gomez R, Gomez DE, Alonso DF, Perrin P, Renualt J-Y, Sigman H, Herrera L, Acevedo B (2011) CIGB-300, a synthetic peptide-based drug that targets the CK2 phosphoaceptor domain. Translational and clinical Research. Mol Cell Biochem 356 (1–2): 45–50.
Perera Y, Farina HG, Hernández I, Mendoza O, Serrano JM, Reyes O, Gómez DE, Gómez RE, Acevedo BE, Alonso DF, Perea SE (2008) Systemic administration of a peptide that impairs the protein kinase (CK2) phosphorylation reduces solid tumor growth in mice. Int J Cancer 122 (1): 57–62.
Perera Y, Farina HG, Gil J, Rodríguez A, Castellanos L, Gómez RE, Alonso DF, Acevedo BE, Perea SE (2009) Anticancer peptide CIGB-300 binds to nucleophosmin/B23, impairs its CK2-mediated phosphorylation, and leads to apoptosis through its nucleolar disassembly activity. Mol Cancer Ther 8 (5): 1189–1196.
Perera Y, del Toro N, Gorovaya L, Fernandez-de-Cossio F, Farina HG, Perea SE (2014) Synergistic interactions of the anticasein kinase 2 CIGB-300 peptide and chemotherapeutic agents in lung and cervical preclinical cancer models. Mol Clin Oncol 2 (6): 935–944.
Piazza F, Manni S, Ruzzene M, Pinna LA, Gurrieri C, Semenzato G (2012) Protein kinase CK2 in hematologic malignancies: reliance on a pivotal cell survival regulator by oncogenic signaling pathways. Leukemia 26 (6): 1174–1179.
Prudent R, Moucadel V, Nguyen C-H, Barette C, Schmidt F, Florent J-C, Lafanech L, Sautel CF, Duchemin-Pelletier E, Spreux E, Filhol O, Reiser J-B, Cochet C (2010) Antitumor Activity of pyridocarbazole and benzopyridoindole derivatives that inhibit protein kinase CK2. Cancer Res 70 (23): 9865–9874.
Rodriguez-Ulloa A, Ramos Y, Gil J, Perera Y, Castellanos-Serra L, Garcia Y, Betancourt L, Besada V, Gonzalez LJ, Fernandez-de-Cossio J, Sanchez A, Serrano JM, Farina H, Alonso DF, Acevedo BE, Padron G, Musacchio A, Perea SE (2010) Proteomic profile regulated by the anticancer peptide CIGB-300 in non-small cell lung cancer (NSCLC) cells. J Proteome Res 9 (10): 5473–5483.
Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O’Brien SE, Bliesath J, Omori M, Huser N, Ho C, Proffitt C, Schwaebe MK, Ryckman DM, Rice WG, Anderes K (2010) CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits pro- survival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res 70 (24): 10288–10298.
Siegel JA, Thomas SR, Stubbs JB, Stabin MG, Hays MT, Koral KF, Robertson JS, Howell RW, Wessels BW, Fisher DR, Weber DA, Brill AB (1999) MIRD pamphlet no. 16: techniques for quantitative radiopharmaceutical biodistribution data acquisition and analysis for use in human radiation dose estimates. J Nucl Med 40 (2): 37S–61S.
Solares AM, Santana A, Baladrón I, Valenzuela C, Diaz J, Ariosa JM, González CA, Diaz A, Castillo D, Gómez R, Alonso DF, Herrera L, Perea SE, Acevedo BE, López-Saura P (2009) Safety and preliminary efficacy data of a novel casein kinase 2 (CK2) peptide inhibitor administered intralesionally at four dose levels in patients with cervical malignancies. BMC Cancer 9: 146.
Soriano-García JL, López-Díaz A, Solares-Asteasuainzarra M, Baladrón-Castrillo I, Batista-Albuerne N, García-García I, González-Méndez L, Perera-Negrín Y, Valenzuela-Silva CM, Pedro AP, Quevedo-Sotolongo LS, Hernández-González I, Silveira-Pablos JM, Chong-López A, Alonso DF, Gómez RE, Renault JY, Perrin P, Sigman H, Gold S, Perea-Rodríguez SE, Acevedo-Castro BE, Herrera-Martínez L, López-Saura PA (2013) Pharmacological and safety evaluation of CIGB-300, a casein kinase 2 inhibitor peptide, administered intralesionally to patients with cervical cancer stage IB2/II. J Cancer Res Ther 1 (6): 163–173.
Tawfic S, Olson MO, Ahmed KJ (1995) Role of protein phosphorylation in post-translational regulation of protein B23 during programmed cell death in the prostate gland. Biol Chem 270 (36): 21009–21015.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92 (3): 205–216.
Acknowledgements
We thank the doctors Margot Martínez, Joel Frígola, Javier Rivero, Virginia Hernández, Carlos Sabatier, Batista Cuellar, Dagnelia Castillo, the nurses María de los Ángeles Viamontes, Susana Romero, the imagenologists Alexei Nerey, Ricardo Téllez, Juan González, Fiodor Martínez, and Lic. María Elena Echevarría and Lisset Seijo for their participation in the clinical work. We also thank Yunia Delgado, Antonieta Herrera, Grettel Melo, Ketty Cruz, María Adela Delgado, Elizeth García, Marisol Cruz, Lourdes Hernández, Yusleidy Brea, for their assistance and Matilde López for their contribution in the drug formulation. This work was supported by HeberBiotec S.A, Havana (product, insurance, reagents), by Biorec, and by the Ministry of Public Health of Cuba (hospital facilities and general medical care of the patients).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
IG, CMV, IB, VR, YP, JAA, LG, BEA, PALS and SEP are employees of the Center for Genetic Engineering and Biotechnology (CIGB), Havana network, where CIGB-300 is produced. DFA and RG are employed by Biorec. SEP is the main author of the patent WO 03/054002 and US patent # 7374767 that sustain this project. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Appendix
Appendix
The CERVIFARM-300-II Study Group are: Josué de la Torre-Pupo, Mauricio Catalá-Ferrer, Manuel Rodríguez-Molina, Emi Hernández-Fernández, Belkis Herrera-Páez, Anay Miranda-Félix, Yolanda Cruz-Gómez, Ivón Howland-Álvarez, Katia Aroche-Estalella (Center for Medical-Surgical Research), Elsa M Masquida-García, Elvia Sánchez-Mendoza, Marilín G Pérez-Valdés (Clinical Investigation Center), Zaida Barbón-Gassó (‘Ramón González Coro’ Hospital), Luis S Quevedo-Sotolongo (Central Clinic ‘Cira García’, Havana), Juan M Silveira-Pablos (National Institute of Oncology, Havana), José R Mendoza-Montpelier (‘Julio R Alfonso’ Hospital, Matanzas), Ileana Gutiérrez (‘Enrique Cabrera’ Hospital, Havana), Alieski Cruz-Pereira, José Suárez-Alba, Yaleisys Rosales-Pantoja, Ivette Raíces-Cruz, Leovaldo Álvarez-Falcón, Cimara Bermúdez-Badell (CIGB).
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Sarduy, M., García, I., Coca, M. et al. Optimizing CIGB-300 intralesional delivery in locally advanced cervical cancer. Br J Cancer 112, 1636–1643 (2015). https://doi.org/10.1038/bjc.2015.137
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.137
Keywords
This article is cited by
-
Protein kinase CK2 – diverse roles in cancer cell biology and therapeutic promise
Molecular and Cellular Biochemistry (2023)
-
Evaluation of Cell-Penetrating Peptides as Mucosal Immune Enhancers for Nasal Vaccination
International Journal of Peptide Research and Therapeutics (2021)
-
Upregulation of IGF2R evades lysosomal dysfunction-induced apoptosis of cervical cancer cells via transport of cathepsins
Cell Death & Disease (2019)
-
Therapeutic targeting of CK2 in acute and chronic leukemias
Leukemia (2018)

{kind=link}