
Abstract
Background:
Constitutive Wnt activation is essential for colorectal cancer (CRC) initiation but also underlies the cancer stem cell phenotype, metastasis and chemosensitivity. Importantly Wnt activity is still modulated as evidenced by higher Wnt activity at the invasive front of clonal tumours termed the β-catenin paradox. SMAD4 and p53 mutation status and the bone morphogenetic protein (BMP) pathway are known to affect Wnt activity. The combination of SMAD4 loss, p53 mutations and BMP signalling may integrate to influence Wnt signalling and explain the β-catenin paradox.
Methods:
We analysed the expression patterns of SMAD4, p53 and β-catenin at the invasive front of CRCs using immunohistochemistry. We activated BMP signalling in CRC cells in vitro and measured BMP/Wnt activity using luciferase reporters. MTT assays were performed to study the effect of BMP signalling on CRC chemosensitivity.
Results:
Eighty-four percent of CRCs with high nuclear β-catenin staining are SMAD4 negative and/or p53 aberrant. BMP signalling inhibits Wnt signalling in CRC only when p53 and SMAD4 are unaffected. In the absence of SMAD4, BMP signalling activates Wnt signalling. When p53 is lost or mutated, BMP signalling no longer influences Wnt signalling. The cytotoxic effects of 5-FU are influenced in a similar manner.
Conclusions:
The BMP signalling pathway differentially modulates Wnt signalling dependent on the SMAD4 and p53 status. The use of BMPs in cancer therapy, as has been proposed by previous studies, should be targeted to individual cancers based on the mutational status of p53 and SMAD4.
Similar content being viewed by others


Main
Mutations in APC (adenomatous polyposis coli) or CTNNB1 (β-catenin) result in constitutive activation of the Wnt pathway, an essential first step in the molecular sequence of events underlying the adenoma–carcinoma sequence. However, while APC/CTNNB1 mutations are identical throughout a clonal tumour, immunohistochemical analysis reveals heterogeneous expression of nuclear β-catenin. Nuclear β-catenin accumulation, indicative of high levels of Wnt pathway activity, is found in tumour cells at the invasive front with lower nuclear β-catenin in the centre of the tumour (Brabletz et al, 2001). This indicates that the ‘constitutively active’ Wnt signalling caused by APC/CTNNB1 mutations is actually still modulated by tumour cell intrinsic and/or extrinsic factors (Fodde and Brabletz, 2007). This results in high levels of Wnt signalling specifically in cells at the invasive front, which underlies the cancer stem cell properties and metastatic potential of these cells (Vermeulen et al, 2010). What these Wnt modulating factors are remains unknown but these nuclear β-catenin expressing cells at the invasive front only arise after an adenoma becomes a carcinoma and in the proximity of the stroma and are thought to be the result of an interaction between tumour cell intrinsic and extrinsic factors. It is likely that tumour cell intrinsic factors are, therefore, mutations occurring at this point in the adenoma to carcinoma sequence, the commonest of which are SMAD4 and p53 mutations (Cho and Vogelstein, 1992; Sjoblom et al, 2006). SMAD4 and p53 have both already been shown to influence Wnt signalling in vitro (Kim et al, 2011; Freeman et al, 2012). How these two molecules influence the Wnt pathway is unknown, but it is likely to involve modulation of a Wnt interacting pathway, as neither is directly involved in Wnt signalling. The main Wnt-antagonising pathway in the intestine is the BMP pathway (Wakefield and Hill, 2013). Tumour cell extrinsic, stromal factors that have been proposed from indirect evidence to influence tumour cell Wnt activity include the Forkhead transcription factors acting via, among others, the BMP pathway, HGF, PDGF and COX2 (Fodde and Brabletz, 2007). However, recent research in several tumour types has identified BMP pathway components as stromally produced factors influencing tumour progression (Sneddon et al, 2006; McLean et al, 2011; Karagiannis et al, 2013). Although the BMP pathway is classically thought to be tumour suppressive in nature (Hardwick et al, 2004; Beck et al, 2006; Auclair et al, 2007), our previous work in pancreatic cancer would suggest that BMPs can promote invasion in the context of SMAD4 loss (Voorneveld et al, 2013), and others have shown that related molecules (TGFβ) can do the same in the context of aberrant p53 (Adorno et al, 2009). As high levels of Wnt signalling promote invasion, we hypothesised that this may be due to these mutations altering the influence of stromally produced BMPs on Wnt signalling.
The critical interaction between BMP and Wnt signalling in the intestine is exemplified by their roles in crypt–villus homeostasis. In normal intestine, the BMP and Wnt pathways interact to control cell fate (Radtke and Clevers, 2005). BMPs that induce differentiation are produced at the top of the villus and antagonise Wnts, which are responsible for a progenitor phenotype, and are produced in the crypt. BMP antagonists from stromal myofibroblasts near the crypt base further ensure that stem cells are not exposed to BMPs, thus both epithelial and stromal cells contribute to BMP signalling (Kosinski et al, 2007). Experiments in transgenic mice show the importance of the BMP pathway in intestinal neoplasia and suggest that this occurs through the influence of the BMP pathway on the Wnt pathway (He et al, 2004).
In summary, we hypothesised that a combination of SMAD4 loss, p53 mutations and BMP signalling may explain the high levels of Wnt signalling seen in CRC cells at the invasive front that is thought to be responsible for the stem-like phenotype, metastatic potential and chemoresistance of these cells. We therefore set out to investigate the influence of BMP signalling on Wnt signalling in CRC, how two of the most common mutations occurring late in the adenoma–carcinoma sequence influence this and how this influences CRC chemosensitivity.
Materials and Methods
Patient information (stage I/II CRC)
Formalin-fixed, paraffin-embedded tissues from 94 patients with stage I/II CRC who received surgery between 1980 and 2001 were used for the study. We studied stage I/II cancers because these cancers are at a stage before metastasising. In these cancers, one might expect to find both tumours with a low metastatic potential as tumours with a high metastatic potential. Using cancers with more advanced stage is likely to select for the more aggressive invasive/metastatic tumours where there will be less difference in metastatic potential. Blocks were selected from the archives of the Pathology Department at the Leiden University Medical Center, Leiden, The Netherlands. All the samples were handled in a coded fashion, according to National ethical guidelines (‘Code for Proper Secondary Use of Human Tissue’, Dutch Federation of Medical Scientific Societies).
Immunohistochemistry
SMAD4 and β-catenin
Sections were deparaffinized, immersed in 0.3% hydrogen peroxide in methanol for 30 min to block for endogenous peroxidase activity and antigen retrieval was performed in 1 × Tris/EDTA, pH 9.0, for 30 min at 97 °C. Nonspecific binding sites were blocked with 10% normal goat serum for 10 min. SMAD4 primary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA; sc-7966, B-8, mouse monoclonal) were diluted in PBS/1%BSA/0.1%Triton (1 : 400) and incubated for 1 h at room temperature. β-catenin primary antibodies (BD Transduction Laboratories, Breda, The Netherlands; 610154, mouse monoclonal) were diluted in PBS/1%BSA/0.1%Triton (1 : 400) and incubated overnight at 4 °C. Poly-HRP-Goat-α-Mouse (Immunologic, Duiven, The Netherlands) antibodies were used as secondary antibodies. Peroxidase activity was detected with fastDAB (Sigma-Aldrich, St Louis, MO, USA). Every staining included a negative control where we used the same protocol but without the primary antibody.
P53, BMP2, BMP4, BMP6 and BMP9
The sections were deparaffinized and immersed in 0.3% hydrogen peroxide in methanol for 30 min. Antigen retrieval was achieved by heating for 10 min to 97 °C in sodium citrate, pH 6.0, and nonspecific binding sides were blocked by incubation with TENG-T (10 mmol l−1 Tris, 5 mmol l−1 EDTA, 0.15 mol l−1 NaCl, 0.25% gelatin, 0.05% Tween 20, pH 8.0) for 10 min. P53 (sc-126, DO-1, mouse monoclonal), BMP2 (sc-6895, N-4, goat polyclonal), BMP4 (sc-393329, D-6, mouse monoclonal) and BMP6 (sc-27409, D-19, goat polyclonal) primary antibodies were obtained from Santa Cruz Biotechnology. BMP9 antibodies (ab35088, rabbit polyclonal) were obtained from Abcam (Cambridge, UK). All primary antibodies were diluted in PBS/1%BSA/0.1%Triton (1 : 100). Incubation took place overnight at 4 °C and the Dako detection system LSAB+ System-HRP (Dako, Glostrup, Denmark) was used. Peroxidase activity was detected with fastDAB (Sigma-Aldrich). Every staining included a negative control where we used the same protocol but without the primary antibody. Antibody specificity was tested using western blot analysis on colorectal cancer cell lines and colorectal cancer samples (Supplementary Figure 1).
Tissue analysis
The analysis was performed in a blinded fashion by two investigators independently. The scoring was done according to the scoring systems supplied in the Supplementary Methods.
Cell lines
HCT116, DLD-1, SW480, RKO, LS174T, HT-29 and HEK-293 cells were obtained from the ATCC. HCT116 SMAD4−/−, HCT116 p53 R248, HCT116 p53−/− and DLD-1 SIL/+ cells were the kind gift of Dr B Vogelstein (Johns Hopkins University, Baltimore, MD, USA). A description of the generation of the cell lines was previously published (Zhou et al, 1998; Sur et al, 2009). All cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Paisley, Scotland) with 4.5 g l−1 glucose and with 580 mg l−1 L-glutamine. This was supplemented with penicillin (50 U ml−1) and streptomycin (50 μg ml−1) and with 10% fetal calf serum (FCS) (Gibco) unless stated otherwise. All in vitro experiments were performed on cells growing exponentially.
Reagents
Stock solutions of recombinant human BMP2 ligands (R&D systems, Minneapolis, MN, USA) were prepared in phosphate-buffered saline (PBS) and subsequently dissolved in culture medium (100 ng ml−1) containing 0.5% FCS. Stock solutions of LDN-193189 (AxonMedchem BV, Groningen, The Netherlands) were prepared in dimethyl sulphoxide (DMSO) and subsequently dissolved in a culture medium containing 10% FCS (5 nM).
BMPR2 transfection
Cells were transiently transfected with either a pcDNA4/TO*BMPR2 plasmid or pcDNA4/TO control vector (Invitrogen, Breda, The Netherlands). pcDNA4/TO*BMPR2 plasmid was constructed by digesting a previously developed pcDNA3.1*BMPR2 construct (Rosenzweig et al, 1995) using HindIII and BamHI restriction enzymes and subsequently placing the BMPR2 sequence into a pcDNA4/TO plasmid. The efficiency of transfection was evaluated by co-transfection with pmaxGFP control vector (from Amaxa GmbH, Cologne, Germany). Efficiency of transfection was determined by the measurement of GFP-positive cells and was at least 70%. All the experiments where BMPR2 transfection was used, were done in normal culture conditions, which includes 10% FCS.
Luciferase reporter assays
Transcriptional activity of canonical BMP, TGF-β and Wnt signalling was measured by transfection of BRE-Luc, CAGA-Luc or WRE-luc/MRE-luc, respectively (Dennler et al, 1998; Korchynskyi and Ten, 2002; van et al, 2011). Transfection efficiency was corrected by co-transfection of a CMV promoter-driven Renilla luciferase vector (Promega, Leiden, The Netherlands). Transfections were performed using Lipofectamine 2000 (Invitrogen). Luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega) on a Luminometer (Berthold Technologies, Bad Wildbad, Germany).
Stable knockdown of SMAD4
Lentiviral constructs expressing shRNAs targeting SMAD4 (TRCN0000040028) and a non-targeting control construct (SHC002) were obtained from the Sigma MISSION shRNA library (Sigma-Aldrich). Production of lentiviruses by transfection into 293T cells has been described earlier (Carlotti et al, 2004). Cells were selected using puromycin. The shSMAD4 transduced CRC cell lines were constructed and used previously (Voorneveld et al, 2014).
Real time PCR
Total RNA was isolated using Trizol (Invitrogen) according to the manufacturer’s instructions. cDNA was synthesised from 1 μg of total RNA using Random primers (Promega) and MMLV-reverse transcriptase (Invitrogen). PCR was performed using the iCycler Thermal Cycler and iQ5 Multicolour Real Time PCR Detection System (Bio-Rad). GAPDH expression was used to normalise for variance. Primer sequences and protocols can be provided upon request.
Wnt specific RT–PCR array
HCT116 and HCT116 SMAD4−/− cells were transfected with a pcDNA4/TO*BMPR2 plasmid or pcDNA4/TO control vector and after 24 h, the cells were lysed and RNA was isolated using RNeasy (Qiagen, Venlo, The Netherlands). The Human WNT Signalling Targets RT2 Profiler PCR Array was purchased from SABIOsciences (Qiagen) and used according to the manufacturer’s instructions.
Immunofluorescence
Cells were allowed to adhere to poly-l-lysine (Sigma-Aldrich)-coated coverslips, fixed in 4% paraformaldehyde and stained in permeabilisation buffer (PBS containing 0.05% Triton X-100) using mouse monoclonal β-catenin (1 : 100) and goat-anti-mouse 594 nm (1 : 200) antibodies. Slides were embedded in SlowFade Gold (Invitrogen). Images were obtained using a Leica TCS SP2 confocal system (Leica, Mannheim, Germany) and processed using ImageJ software.
Chemosensitivity
Cells were transfected with either a pcDNA4/TO*BMPR2 plasmid or the pcDNA4/TO control vector 48 h before the treatment with different concentrations of 5-fluorouracil (5-FU). After 24 h of 5-FU treatment, cell viability was measured by adding 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution for 2 h at 37 °C (0.5 mg ml−1) after which the absorbance of the samples was measured at 562 nm. In another experimental set-up, cells were treated with 5 nM LDN-198189 or control (DMSO) for 4 days before the treatment with different concentrations of 5-FU.
Statistical analysis
In vitro experiments were analysed using a two-tailed Student’s t-test. Significant differences are presented as following: *P<0.05; **P<0.01 ***P<0.001. All the experiments were done with a minimum of three independent experiments. For the tissue analysis, the Fisher’s exact test was used as appropriate and performed using SPSS Statistics version 20 (IBM, New York, NY, USA). A P-value of <0.05 was considered significant. The hierarchical cluster analysis was performed using JMP 10 (Cary, NC, USA).
Results
BMPs are expressed abundantly in both the tumour and stroma at the invasive front
To explore to what extent the BMP signalling pathway can potentially be activated by BMP ligands, we assessed the levels of BMP ligand expression at the invasive front. We stained and scored BMP2, BMP4, BMP6 and BMP9 in the invasive front of 94 colorectal cancers using immunohistochemistry. BMP2, BMP4, BMP 6 and BMP9 ligands were expressed in the tumour in 45.2%, 10.7%, 50.0% and 90.5%, respectively (Supplementary Figure 2). In the surrounding stroma, we detected one or more BMP ligands in 56.0% of the cases. Overall, in all of the cancer specimens, there were one or more BMPs expressed in the tumour, stroma or both. From this, we concluded that there are sufficient BMP ligands present at the invasive front to potentially activate BMP signalling.
Association between expression of β-catenin, SMAD4 and p53 at the invasive front in human colorectal cancer tissue
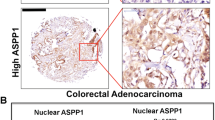
We analysed the expression patterns of SMAD4, p53 and β-catenin at the invasive front of 94 CRCs using immunohistochemistry. We have made use of immunohistochemical analysis methodology that others have shown to correlate well with the mutation status p53 (see Supplementary Methods) (Curtin et al, 2004; Yemelyanova et al, 2011). Allelic loss of 18q and SMAD4 mutation also correlates with SMAD4 expression, but SMAD4 expression can also be reduced without the presence of mutations or allelic imbalance (Alazzouzi et al, 2005). A total of 59.6% of the cancers have a high expression of nuclear β-catenin, 55.3% have aberrant p53 expression and 40.4% have loss of expression of SMAD4 (Table 1). There is no association between age, gender, stage and immunohistochemical scoring for p53, nuclear SMAD4 and nuclear β-catenin, except that patients with SMAD4-positive cancers are younger (P=0.04). Left-sided tumours have significantly higher levels of nuclear β-catenin (P=0.001) and SMAD4-negative cancers are associated with Microsatellite stability (P=0.008) and left-sided tumours (P=0.04; Supplementary Table 1). There is a strong association between loss of SMAD4 expression and high expression of nuclear β-catenin (P=0.001), and there is an association between aberrant p53 expression and high expression of nuclear β-catenin (P=0.04) (Table 1). To graphically visualise the relationship between the expression profiles of these three molecules, we used a hierarchical cluster analysis (Figure 1A). This shows that the majority of cancers with high nuclear β-catenin staining (n=56) are either SMAD4 negative and/or p53 aberrant (n=46/56, 82%). Figure 1B and Supplementary Figure 3 show examples of the four clusters based on these expression patterns. When comparing patient characteristics, cluster 1 and 2 cancers are more often left-sided compared with cluster 3 and 4 cancers (Supplementary Table 2).

The majority (82%, 46/56) of colorectal cancers exhibiting high levels of nuclear β -catenin at the invasive front have abnormal expression of SMAD4 or p53 or both. (A) Hierarchical cluster analysis of the expression patterns of β-catenin combined with SMAD4 and p53. The invasive front of 94 stage I/II cancers was stained and scored for nuclear SMAD4, p53 and β-catenin. (B) Examples of cluster 2 and 3 expression patterns. ( × 400).
BMP/Wnt activity in colorectal cancer cell lines
We investigated the effects of BMP signalling on Wnt signalling activity in CRC cell lines. We measured the BMP and Wnt signalling activity in a panel of CRC cell lines and a control cell line HEK-293 (embryonic kidney cells) using luciferase reporter assays and found an inverse correlation between the activity of the two pathways (Figure 2): Low BMP signalling activity is associated with a high Wnt signalling activity and vice versa. Mathematically, BMP and Wnt signalling show a log correlation following the equation WRE-luc=10^(−0.564 × log(BRE-luc)+3.907) with a correlation coefficient of R2=0.94

Graph to show the relationship between basal levels of BMP and Wnt signalling in CRC cell lines. CRC cell lines HCT116, RKO, HT-29, SW480, DLD-1 and LS174T and the embryonic kidney cell line HEK-293 (used as non-cancer cell line) were transfected with either BRE-luc (BMP activity) or WRE-luc (Wnt activity). MRE-luc was used to control for transcriptional changes not associated with Wnt signalling. A cmv-Renilla plasmid was co-transfected to control for the transfection efficiency. The graph shows the BMP activity plotted against the Wnt activity in each cell line. Regression analysis reveals a Log regression with a correlation of R2=0.94. (The experiment was performed three times. Grey areas depict the s.d. of each cell line. Dotted lines show the 95% confidence interval of the nonlinear curve fit).
BMP activation results in paradoxical Wnt activation in a subset of CRC cell lines
It is known that BMP signalling can inhibit Wnt signalling in normal intestinal epithelium, but it is not known whether the major mutations found in CRC influence this pathway interaction. We, therefore, activated BMP signalling in a panel of CRC cell lines and HEK-293 using a pcDNA4/TO plasmid expressing WT-BMPR2 and measured Wnt signalling activity using WRE/MRE-luciferase. We have previously shown that transfection of BMPR2 results in a reliable and robust increase in BMP signalling activity (BRE-luc) (Kodach et al, 2008) and it does not lead to the activation of TGF-β signalling (CAGA-luc) (Supplementary Figure 4). The activation of BMP signalling results in a reduction of Wnt signalling only in HCT116 and LS174T cells and the control cell line HEK-293. In HT-29, RKO and SW480 cells, the activation of BMP signalling results in an increase in Wnt signalling (Figure 3A). To elucidate whether mutations might affect the BMP–Wnt interaction, we looked at the known mutation profile of these cell lines to see if this suggested a pattern (Figure 3B). Interestingly, the two cell lines in which BMP signalling has a negative effect on Wnt signalling (HCT116 and LS174T) are both SMAD4 positive and p53 WT. This would suggest that if either SMAD4 is lost and/or p53 is mutant, the BMP–Wnt interaction is either reversed or abolished.

Activation of BMP signalling differentially modulates Wnt signalling dependent on the SMAD4 and p53 status. (A) CRC cell lines HCT116, LS174T, HT-29, SW480, RKO and DLD-1 and the embryonic kidney cell line HEK-293 (used as non-cancer cell line) were transfected with WRE-luc and MRE-luc. The next day, the cells were transfected with either BMPR2 (to activate BMP signalling) or the empty control vector pcDNA4/TO. After 24 h, the cells were lysed and luciferase activity was measured. The control (pcDNA) condition was set to 1. (B) An overview of APC/β-catenin, KRAS/BRAF, p53 and SMAD4 mutation status in the cell lines. (C) HCT116, HCT116 SMAD4−/−, HCT116 R248 (p53 mutated) and HCT116 p53−/− cells were transfected with WRE-luc and MRE-luc. The next day, the cells were transfected with either BMPR2 (to activate BMP signalling) or the empty control vector pcDNA4/TO. After 24 h, the cells were lysed and luciferase activity was measured. (D) DLD-1 (harbouring a p53 mutation) and DLD-1 SIL/+ (one WT copy p53) were transfected with WRE-luc/MRE-luc. The next day, the cells were treated with either 100 ng ml−1 BMP2 or the control vehicle (both diluted in medium with 0,5% FCS). After 24 h, the cells were lysed and luciferase activity was measured. (E) SMAD4 was stably knocked down in LS174T cell using lentiviral shRNA transduction. The empty shRNA SHC002 was used to create a control cell line. The cells were transfected with WRE-luc/MRE-luc. The next day the cells were treated with either 100 ng ml−1 BMP2 or the control vehicle (both diluted in medium with 1% FCS). After 24 h, the cells were lysed and luciferase activity was measured. (All the experiments were performed three times. Mean±s.e.m. is shown, *P<0.05, **P<0.01, ***P<0.001)
The effect of BMP signalling on Wnt signalling is dependent on the SMAD4 and p53 status
To investigate the influence of SMAD4 and p53 status on the BMP–Wnt signalling interaction, we first compared several cell lines in which the p53 and SMAD4 status was manipulated. We activated BMP signalling in colorectal cancer cells that have intact p53 expression (HCT116) and compared this with p53 null cells (HCT116 p53−/−) or p53 mutant cells (HCT116 p53 R248). In p53 expressing cells, BMP signalling reduces Wnt signalling activity, whereas a mutation in p53 or the absence of p53 abolishes the inhibiting effect of BMP signalling (Figure 3C). To further confirm the role of p53 in influencing the BMP–Wnt interaction, we treated DLD-1 cells, that naturally contain one mutated and one wild-type p53 allele, with 100 ng ml−1 BMP2 and compared these with DLD-1 p53 SIL/+ cells, where the mutant p53 allele has been silenced (Figure 3D). Activating BMP signalling does not lead to changes in Wnt signalling in the parental DLD-1 cells, but, in the DLD-1 p53 SIL/+ cells, Wnt signalling activity is decreased.
Next, we investigated the role of SMAD4 in the BMP–Wnt interaction. First, we compared the effect of BMP signalling on Wnt signalling activity in HCT116 (SMAD4 positive) versus HCT116 SMAD4−/− cells. Loss of SMAD4 leads to a major increase in Wnt signalling and reversal of the BMP/Wnt interaction with BMP activation now leading to an activation of Wnt signalling (Figure 3C). This was measured both by WRE-luc and nuclear localisation of β-catenin. The reversal of the BMP–Wnt interaction due to loss of SMAD4 is further confirmed by the treatment of HCT116 SMAD4−/− cells with the BMP inhibitor noggin, which results in reduced nuclear and increased membranous β-catenin (Supplementary Figure 5A).
The role of SMAD4 was further investigated by stable lentiviral shRNA-mediated knockdown of SMAD4 in LS174T cells and treatment with 100 ng ml−1 BMP2. BMP inhibits Wnt signalling in the control shRNA LS174T cell line, which expresses SMAD4 (Figure 3E). Knocking down SMAD4 switches BMP signalling from inhibiting to enhancing Wnt signalling. The effect of BMP signalling on Wnt signalling in SMAD4-negative cells can also be seen by the increase in mRNA expression of the Wnt signalling components AXIN2 and c-MYC (Supplementary Figure 5B). We conclude that the BMP–Wnt interaction is dependent on the SMAD4 and p53 status.
BMP activation influences Wnt target gene expression to a greater extent in SMAD4-deficient cells
To obtain insight into which Wnt pathway-associated genes are affected by the reversal of the BMP–Wnt interaction due to SMAD4 loss, we performed a Wnt signalling RT–PCR array while activating the BMP pathway in two isogenic cell lines with or without SMAD4. Figure 4 demonstrates that the activation of BMP signalling in SMAD4-positive versus SMAD4-negative cells leads to a completely different expression pattern of Wnt signalling-associated genes in the two cell types. It is also notable that BMP pathway activation leads to larger changes in Wnt gene expression in SMAD4-negative cells than in SMAD4-positive ones. One of the genes that is upregulated when activating BMP signalling in SMAD4-positive cells is CTNNBIP1. The CTNNBIP1 gene encodes for the β-catenin-interacting protein 1, which binds β-catenin to prevent interaction with TCF, thereby inhibiting Wnt signalling (Tago et al, 2000; Yemelyanova et al, 2011). The activation of BMP signalling in SMAD4-negative HCT116 cells leads to the upregulation of DVL1. DVL1 encodes for the protein Segment polarity protein dishevelled homologue (DVL1), which prevents the GSK3β/APC/Axin complex from degrading β-catenin. The full gene lists with fold changes can be found in Supplementary Tables 3 and 4.

BMP activation influences Wnt target gene expression to a greater extent in SMAD4-deficient cells. HCT116 and HCT116 SMAD4−/− cells were transfected with either BMPR2 (to activate BMP signalling) or the empty control vector pcDNA4/TO. After 24 h, RNA was isolated and a Human WNT Signalling Targets RT2 Profiler PCR Array from SABIOsciences was used. The graphs show the expression patterns of the control samples (x axis) versus the BMPR2 transfected samples (y axis). The lines indicate a 2-fold change compared to controls.
BMP signalling influences 5-FU chemosensitivity dependent on the SMAD4 and p53 status
It has been shown that patients with CRC with low levels of SMAD4 protein expression respond poorly to 5-fluorouracil (5-FU; Alhopuro et al, 2005). Also, the inactivation of SMAD4 leads to an increase in 5-FU resistance (Papageorgis et al, 2011). The inactivation of p53 has also been shown to result in a poorer response to chemotherapy in vivo (Lowe et al, 1994) and in vitro (Bunz et al, 1999), although some reports show otherwise(Hawkins et al, 1996). The use of BMPs has been proposed as a means of combatting chemoresistance in several cancer types including CRC (Piccirillo et al, 2006; Lombardo et al, 2011). We tested the influence of BMP activation on 5-FU chemosensitivity in SMAD4 and p53 inactivated cell lines. In SMAD4 and WT p53 expressing parental HCT116 cell line, an increase in chemosensitivity can be seen when BMP signalling is activated (Figure 5A). In the p53 mutated HCT116 R248 cell line, a slight reduction in chemosensitivity can be seen when BMP is activated, but not in the HCT116 p53−/− cells (Figure 5B and C). In the HCT116 SMAD4−/− cells, BMP signalling activation results in less chemosensitivity, especially at high concentrations of 5-FU (Figure 5D). These results suggest that activation of BMP signalling can increase the effects of chemotherapy as others have suggested but only in cancers that express SMAD4 and WT p53.

BMP pathway activation or inhibition differentially affects chemosensitivity dependent on the mutation status of p53 and SMAD4. (A–D) HCT116, HCT116 SMAD4−/−, HCT116 R248 (p53 mutated) and HCT116 p53−/− cells were transfected with BMPR2 (to activate BMP signalling) or the empty control vector pcDNA4/TO. The next day, cells were treated with different concentrations of 5-FU. After 24 h of 5-FU treatment, the cell viability was measured. (E) HCT116 SMAD4−/− cells were treated with different concentrations of LDN-193189 (BMP pathway inhibitor) for 4 days and afterwards cell viability was measured. (F) HCT116 SMAD4−/− cells were treated with 5 nm LDN-193189 (BMP pathway inhibitor) for 4 days. Afterwards, the cells were washed and serum starved for 8 h. Subsequently, the cells were treated with different concentrations of 5-FU. After 24 h, the cell viability was measured. (G) A scheme showing how BMP signalling influences the chemosensitivity dependent on the p53 and SMAD4 status. (All the experiments were performed in triplicate. Mean±s.e.m. is shown, *P<0.05, **P<0.01, ***P<0.001)
We subsequently investigated the effect of BMP pathway inhibition by treating cell lines with the BMPR1A (ALK3) inhibitor LDN-193189. After 4 days of treatment with 5 nM LDN-193189, a significant decrease in viability can be seen in SMAD4−/− cells (Figure 5E). At a dose of 5 nM, the kinase inhibitor LDN-193189 is a highly specific inhibitor of BMPR1A and results in specific downregulation of BMP signalling compared with TGFβ signalling as measured by reporter assays. Higher concentrations only result in less specificity (Supplementary Figure 6A–C). Pre-treating HCT116 SMAD4−/− cells for 4 days with 5 nM of LDN-193189 followed by subsequent 5-FU treatment resulted in a stronger decrease in viability (Figure 5F). In summary, in HCT116 cells in which both SMAD4 and p53 are wild type, activation of BMP signalling results in an increase in chemosensitivity, whereas BMP inhibition has no effect. In HCT116 SMAD4 wild type, p53 mutant cells, activating or inhibiting BMP signalling has no effect on chemosensitivity, whereas in HCT116 SMAD4 cells, BMP activation increases chemoresistance and BMP inhibition increases chemosensitivity (Figure 5G).
Discussion
The β-catenin paradox cannot be explained by mutations within the Wnt signalling pathway, as APC/CTNNB1 mutations are clonal within a tumour. On the basis of several observations made in previously published studies, we hypothesised that the β-catenin paradox may at least, in part, be explained by the effects of SMAD4 loss or p53 mutations on the ability of BMP signalling to suppress Wnt signalling in colorectal cancer cells at the tumour invasive front.
Further evidence to support the likely involvement of the BMP pathway in determining the levels of Wnt signalling in tumour cells at the invasive front is the finding that the combination of extensive stroma at the invasive front and loss of SMAD4 in the tumour leads to a poorer prognosis (Mesker et al, 2009). One potential explanation for this is that SMAD4-independent tumour-stroma signalling drives invasion and metastasis, both of which are driven by enhanced Wnt signalling.
We hypothesised that candidate mutations underlying the β-catenin paradox are likely to be those that occur at the same stage in tumour progression as the appearance of this phenomenon itself. High levels of Wnt signalling are seen in cells that have acquired the ability to invade and metastasise. This occurs at the transition from advanced adenoma to invasive carcinoma and the two most frequently occurring mutations at this stage are SMAD4 and p53 mutations. This choice of candidate mutations is further supported by previous studies showing that SMAD4 loss is associated with elevated levels of Wnt activity in CRC cell lines (Freeman et al, 2012) and that p53 mutations increase Wnt signalling activity in vitro (Kim et al, 2011).
The BMP pathway counteracts Wnt signalling in the normal colonic epithelium as has been shown by BMP pathway manipulation in transgenic mouse models (Haramis et al, 2004; He et al, 2004). We hypothesised that SMAD4 or p53 mutations may influence the way BMP signalling activity modulates Wnt signalling activity despite APC/β-catenin mutations and provide a molecular explanation for the β-catenin paradox.
We performed our initial analysis in archival human colorectal cancer specimens in tissue sections at the invasive front. We assessed nuclear β-catenin, SMAD4 and p53 expression using immunohistochemistry and found an association between SMAD4 loss and/or aberrant p53 expression and a high level of nuclear β-catenin (representing high Wnt activity). This provides evidence in vivo to support previous evidence in vitro that SMAD4 and p53 can alter Wnt signalling activity, revealing for the first time a connection between SMAD4, p53 and Wnt signalling in the invasive front of CRC tissue.
To investigate this further, we activated BMP signalling using transient transfection of BMPR2 in a set of isogenic cell lines in which p53 and SMAD4 have been genetically manipulated. Although transient transfection of BMPR2 is a rather artificial method to activate the BMP pathway, we have previously shown that it is a reliable way of achieving robust activation of BMP signalling in all CRC cell lines by circumventing the loss of BMPR2 seen in a proportion of CRCs and variations in expression levels of BMP ligands and inhibitors (Kodach et al, 2008). To avoid the possibility of bias arising from the use of a single method of BMP activation, we also performed similar experiments using BMP2 ligands in cells that we have previously shown to have normal BMP receptor levels.
We found an interesting correlation between the p53 and SMAD4 status and the ability of the BMP signalling pathway to modulate Wnt signalling activity. Wild-type p53 is necessary for BMP to inhibit Wnt signalling and loss of SMAD4 completely reverses the BMP–Wnt interaction switching inhibition into activation. It has been previously described that the effect of cancer signalling pathways can change due to the mutation or loss of oncogenes outside the main signalling pathway. This is illustrated by studies showing that the Transforming Growth Factor-β (TGF-β) signalling pathway can either inhibit or promote migration/invasion dependent on the presence of wild-type or mutant p53 (Adorno et al, 2009).
We also found that the BMP signalling pathway can modulate the chemosensitivity of 5-FU based on the SMAD4 or p53 status. The similarity between the effects of BMP signalling on both Wnt signalling activity and chemosensitivity can be explained by the previously observed correlation between β-catenin levels and chemoresistance (Sinnberg et al, 2011).
Our study has several limitations. The study in patient tissue is limited by the fact that it is difficult to assess BMP signalling activity in CRC tissue especially when this is SMAD-independent. We observed an abundance of BMP ligand expression at the invasive front both in the stroma and the tumour cells, which would suggest that active BMP signalling is more or less ubiquitous at the invasive front of CRC. However, we have not analysed the expression of a large number of other BMP ligands and inhibitors as it is currently impossible to deduce the integrated effect on pathway activity even with a much more comprehensive analysis. Many of the effects of BMP signalling on Wnt signalling seem to be SMAD4-independent, as we describe in our study. Although canonical SMAD-dependent BMP activity can be assessed by nuclear pSMAD1,5,8 localisation using immunohistochemistry as we have performed previously, there is no equivalent for the assessment of SMAD-independent BMP activity.
Our study is necessarily highly reductionist in nature. As we have outlined, there are many other molecular pathways and many other mutations that could be explored. In fact, one of the cell lines we used (RKO) is SMAD4 positive and p53 WT, but showed an increase in Wnt signalling upon BMP activation suggesting the involvement of other pathways. However, we feel that the study of the two most important signalling pathways in conjunction with two of the commonest mutations is a good starting point.
Cancer therapy is increasingly focused on the targeted pharmacological modulation of the specific molecular pathways underlying carcinogenesis. Understanding how the major pathways interact and how mutations influence, this is thus of critical importance. The clinical implications of this study are that the use of BMPs in cancer therapy, as has been proposed by previous studies, could have deleterious effects and should be targeted to individual cancers. However, interventions to modulate BMP signalling have the potential to enhance the effect of conventional chemotherapy dependent on the mutational status of p53 and SMAD4. This may be especially important in SMAD4-negative CRCs, which are aggressively invasive and metastatic cancers with a poor prognosis that respond poorly to current therapy.
Change history
06 January 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, Parenti AR, Rosato A, Bicciato S, Balmain A, Piccolo S (2009) A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 137 (1): 87–98.
Alazzouzi H, Alhopuro P, Salovaara R, Sammalkorpi H, Jarvinen H, Mecklin JP, Hemminki A, Schwartz S Jr, Aaltonen LA, Arango D (2005) SMAD4 as a prognostic marker in colorectal cancer. Clin Cancer Res 11 (7): 2606–2611.
Alhopuro P, Alazzouzi H, Sammalkorpi H, Davalos V, Salovaara R, Hemminki A, Jarvinen H, Mecklin JP, Schwartz S Jr, Aaltonen LA, Arango D (2005) SMAD4 levels and response to 5-fluorouracil in colorectal cancer. Clin Cancer Res 11 (17): 6311–6316.
Auclair BA, Benoit YD, Rivard N, Mishina Y, Perreault N (2007) Bone morphogenetic protein signaling is essential for terminal differentiation of the intestinal secretory cell lineage. Gastroenterology 133 (3): 887–896.
Beck SE, Jung BH, Fiorino A, Gomez J, Rosario ED, Cabrera BL, Huang SC, Chow JY, Carethers JM (2006) Bone morphogenetic protein signaling and growth suppression in colon cancer. Am J Physiol Gastrointest Liver Physiol 291 (1): G135–G145.
Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T (2001) Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA 98 (18): 10356–10361.
Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, Williams J, Lengauer C, Kinzler KW, Vogelstein B (1999) Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest 104 (3): 263–269.
Carlotti F, Bazuine M, Kekarainen T, Seppen J, Pognonec P, Maassen JA, Hoeben RC (2004) Lentiviral vectors efficiently transduce quiescent mature 3T3-L1 adipocytes. Mol Ther 9 (2): 209–217.
Cho KR, Vogelstein B (1992) Suppressor gene alterations in the colorectal adenoma-carcinoma sequence. J Cell Biochem Suppl 16G: 137–141.
Curtin K, Slattery ML, Holubkov R, Edwards S, Holden JA, Samowitz WS (2004) p53 alterations in colon tumors: a comparison of SSCP/sequencing and immunohistochemistry. Appl Immunohistochem Mol Morphol 12 (4): 380–386.
Dennler S, Itoh S, Vivien D, Ten DP, Huet S, Gauthier JM (1998) Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 17 (11): 3091–3100.
Fodde R, Brabletz T (2007) Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol 19 (2): 150–158.
Freeman TJ, Smith JJ, Chen X, Washington MK, Roland JT, Means AL, Eschrich SA, Yeatman TJ, Deane NG, Beauchamp RD (2012) Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of beta-catenin. Gastroenterology 142 (3): 562–571.
Haramis AP, Begthel H, van den Born M, van EJ, Jonkheer S, Offerhaus GJ, Clevers H (2004) De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science 303 (5664): 1684–1686.
Hardwick JC, van den Brink GR, Bleuming SA, Ballester I, Van Den Brande JM, Keller JJ, Offerhaus GJ, van Deventer SJ, Peppelenbosch MP (2004) Bone morphogenetic protein 2 is expressed by, and acts upon, mature epithelial cells in the colon. Gastroenterology 126 (1): 111–121.
Hawkins DS, Demers GW, Galloway DA (1996) Inactivation of p53 enhances sensitivity to multiple chemotherapeutic agents. Cancer Res 56 (4): 892–898.
He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, Tian Q, Zeng X, He X, Wiedemann LM, Mishina Y, Li L (2004) BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat Genet 36 (10): 1117–1121.
Karagiannis GS, Berk A, Dimitromanolakis A, Diamandis EP (2013) Enrichment map profiling of the cancer invasion front suggests regulation of colorectal cancer progression by the bone morphogenetic protein antagonist, gremlin-1. Mol Oncol 7 (4): 826–839.
Kim NH, Kim HS, Kim NG, Lee I, Choi HS, Li XY, Kang SE, Cha SY, Ryu JK, Na JM, Park C, Kim K, Lee S, Gumbiner BM, Yook JI, Weiss SJ (2011) p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci Signal 4 (197): ra71.
Kodach LL, Wiercinska E, de Miranda NF, Bleuming SA, Musler AR, Peppelenbosch MP, Dekker E, van den Brink GR, van Noesel CJ, Morreau H, Hommes DW, Ten DP, Offerhaus GJ, Hardwick JC (2008) The bone morphogenetic protein pathway is inactivated in the majority of sporadic colorectal cancers. Gastroenterology 134 (5): 1332–1341.
Korchynskyi O, Ten DP (2002) Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem 277 (7): 4883–4891.
Kosinski C, Li VS, Chan AS, Zhang J, Ho C, Tsui WY, Chan TL, Mifflin RC, Powell DW, Yuen ST, Leung SY, Chen X (2007) Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc Natl Acad Sci USA 104 (39): 15418–15423.
Lombardo Y, Scopelliti A, Cammareri P, Todaro M, Iovino F, Ricci-Vitiani L, Gulotta G, Dieli F, de Maria R, Stassi G (2011) Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology 140 (1): 297–309.
Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T (1994) p53 status and the efficacy of cancer therapy in vivo. Science 266 (5186): 807–810.
McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, Cabrera L, Keller E, McCauley L, Cho KR, Buckanovich RJ (2011) Human ovarian carcinoma-associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J Clin Invest 121 (8): 3206–3219.
Mesker WE, Liefers GJ, Junggeburt JM, van Pelt GW, Alberici P, Kuppen PJ, Miranda NF, van Leeuwen KA, Morreau H, Szuhai K, Tollenaar RA, Tanke HJ (2009) Presence of a high amount of stroma and downregulation of SMAD4 predict for worse survival for stage I-II colon cancer patients. Cell Oncol 31 (3): 169–178.
Papageorgis P, Cheng K, Ozturk S, Gong Y, Lambert AW, Abdolmaleky HM, Zhou JR, Thiagalingam S (2011) Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res 71 (3): 998–1008.
Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL (2006) Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444 (7120): 761–765.
Radtke F, Clevers H (2005) Self-renewal and cancer of the gut: two sides of a coin. Science 307 (5717): 1904–1909.
Rosenzweig BL, Imamura T, Okadome T, Cox GN, Yamashita H, Ten DP, Heldin CH, Miyazono K (1995) Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proc Natl Acad Sci USA 92 (17): 7632–7636.
Sinnberg T, Menzel M, Ewerth D, Sauer B, Schwarz M, Schaller M, Garbe C, Schittek B (2011) beta-Catenin signaling increases during melanoma progression and promotes tumor cell survival and chemoresistance. PLoS One 6 (8): e23429.
Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314 (5797): 268–274.
Sneddon JB, Zhen HH, Montgomery K, van de Rijn M, Tward AD, West R, Gladstone H, Chang HY, Morganroth GS, Oro AE, Brown PO (2006) Bone morphogenetic protein antagonist gremlin 1 is widely expressed by cancer-associated stromal cells and can promote tumor cell proliferation. Proc Natl Acad Sci USA 103 (40): 14842–14847.
Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N (2009) A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci USA 106 (10): 3964–3969.
Tago K, Nakamura T, Nishita M, Hyodo J, Nagai S, Murata Y, Adachi S, Ohwada S, Morishita Y, Shibuya H, Akiyama T (2000) Inhibition of Wnt signaling by ICAT, a novel beta-catenin-interacting protein. Genes Dev 14 (14): 1741–1749.
van VW, Le NH, Helvensteijn W, Blonden L, Theeuwes M, Bakker ER, Franken PF, van GL, Meijlink F, van der Valk MA, Kuipers EJ, Fodde R, Smits R (2011) beta-catenin tyrosine 654 phosphorylation increases Wnt signalling and intestinal tumorigenesis. Gut 60 (9): 1204–1212.
Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, Sprick MR, Kemper K, Richel DJ, Stassi G, Medema JP (2010) Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 12 (5): 468–476.
Voorneveld PW, Kodach LL, Jacobs RJ, Liv N, Zonnevylle AC, Hoogenboom JP, Biemond I, Verspaget HW, Hommes DW, de Rooij K, van Noesel CJ, Morreau H, van Wezel T, Offerhaus GJ, van den Brink GR, Peppelenbosch MP, Ten DP, Hardwick JC (2014) Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology 1471: 196–208.e13.
Voorneveld PW, Stache V, Jacobs RJ, Smolders E, Sitters AI, Liesker A, Korkmaz S, Lam SM, de Miranda NF, Morreau H, Kodach LL, Hardwick JC (2013) Reduced expression of bone morphogenetic protein receptor IA in pancreatic cancer is associated with a poor prognosis. Br J Cancer 109 (7): 1805–1812.
Wakefield LM, Hill CS (2013) Beyond TGFbeta: roles of other TGFbeta superfamily members in cancer. Nat Rev Cancer 13 (5): 328–341.
Yemelyanova A, Vang R, Kshirsagar M, Lu D, Marks MA, Shih I, Kurman RJ (2011) Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol 24 (9): 1248–1253.
Zhou S, Buckhaults P, Zawel L, Bunz F, Riggins G, Dai JL, Kern SE, Kinzler KW, Vogelstein B (1998) Targeted deletion of Smad4 shows it is required for transforming growth factor beta and activin signaling in colorectal cancer cells. Proc Natl Acad Sci USA 95 (5): 2412–2416.
Acknowledgements
PWV and JCHH are funded by the Netherlands Digestive Diseases Foundation. LLK is funded by the Dutch Cancer Society. LJACH and MP are funded by the Alpe D’Huzes/Bas Mulder award 2011.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Voorneveld, P., Kodach, L., Jacobs, R. et al. The BMP pathway either enhances or inhibits the Wnt pathway depending on the SMAD4 and p53 status in CRC. Br J Cancer 112, 122–130 (2015). https://doi.org/10.1038/bjc.2014.560
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.560
Keywords
This article is cited by
-
The molecular mechanisms underlying the regeneration process in the earthworm, Perionyx excavatus exhibit indications of apoptosis-induced compensatory proliferation (AICP)
In Vitro Cellular & Developmental Biology - Animal (2024)
-
The Smad4-MYO18A-PP1A complex regulates β-catenin phosphorylation and pemigatinib resistance by inhibiting PAK1 in cholangiocarcinoma
Cell Death & Differentiation (2022)
-
Prostate zones and cancer: lost in transition?
Nature Reviews Urology (2022)
-
HOXA13 in etiology and oncogenic potential of Barrett’s esophagus
Nature Communications (2021)
-
Nanos3, a cancer-germline gene, promotes cell proliferation, migration, chemoresistance, and invasion of human glioblastoma
Cancer Cell International (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}