
Abstract
Background:
Polo-like kinase 1 (Plk1) has an important role in mitosis. Volasertib (BI 6727), a potent and selective cell cycle kinase inhibitor, induces mitotic arrest and apoptosis by targeting Plk; this phase I study sought to determine its maximum tolerated dose (MTD) in Asian patients with advanced solid tumours.
Methods:
Patients were enrolled simultaneously into two 3-week schedules of volasertib: a 2-h infusion on day 1 (schedule A) or days 1 and 8 (schedule B). Dose escalation followed a 3+3 design. The MTD was determined based on dose-limiting toxicities (DLT) in the first treatment course.
Results:
Among 59 treated patients, the most common first course DLTs were reversible thrombocytopenia, neutropenia and febrile neutropenia; MTDs were 300 mg for schedule A and 150 mg for schedule B. Volasertib exhibited multi-exponential pharmacokinetics (PK), a long terminal half-life of ∼135 h, a large volume of distribution (>3000 l), and a moderate clearance. Partial responses were observed in two pre-treated patients (ureteral cancer; melanoma). Volasertib was generally well tolerated, with an adverse event profile consistent with its antimitotic mode of action and a favourable PK profile.
Conclusions:
These data support further development of volasertib and a harmonised dosing for Asian and Caucasian patients.
Similar content being viewed by others


Main
The polo-like kinase (Plk) family of proteins is comprised of five highly conserved serine/threonine protein kinases that have a key role in such cellular processes as cell division and checkpoint regulation of mitosis (Medema et al, 2011). Of the five Plks identified in humans to date, polo-like kinase 1 (Plk1) is the most extensively characterised. Plk1 is involved in regulating multiple essential steps of mitosis including mitotic entry, centrosome maturation and separation, formation of the bipolar spindle, metaphase to anaphase transition, and initiation of cytokinesis (Strebhardt, 2010; Medema et al, 2011). Plk1 was reported to be overexpressed in a range of human cancers, including non-small cell lung, prostate, ovarian, breast, colorectal cancer, and acute myeloid leukaemia (Takahashi et al, 2003; Rudolph et al, 2009; Medema et al, 2011), and high levels of Plk1 expression were reported to be correlated with poor prognosis in various cancers (Strebhardt, 2010). Preclinical studies have demonstrated the functional relevance of Plk1 as a potential therapeutic target in cancer. Plk1 inhibition leads to a disruption in spindle assembly causing a distinct mitotic arrest phenotype (‘Polo-arrest’) and subsequent apoptosis (Spankuch-Schmitt et al, 2002; Liu and Erikson, 2003; Rudolph et al, 2009). As a result, Plk1 is emerging as a promising novel target for anticancer therapy.
Volasertib (Boehringer Ingelheim (BI) 6727; an investigational agent currently in clinical development) is a potent and selective Plk inhibitor that induces mitotic arrest and apoptosis (Rudolph et al, 2009). The chemical structure of volasertib has been previously published (Rudolph et al, 2009). In preclinical studies, volasertib demonstrated broad antitumour activity in multiple cancer models and a promising pharmacokinetic (PK) profile indicative of high and sustained exposure in tumour tissue (Rudolph et al, 2009). In a phase I trial of Caucasian patients with solid tumours, volasertib administered on day 1 of a 3-week treatment course demonstrated antitumour activity with a generally manageable safety profile (Schöffski et al, 2012).
As population-specific data are critically needed to optimise dosing from a safety perspective owing to potential ethnic differences, this phase I study of escalating doses of volasertib administered to Asian patients with advanced solid malignancies was conducted. The purpose of this study was to confirm in Asian cancer patients the maximum tolerated dose (MTD) previously established in Caucasians, and to establish the MTD of an alternative administration schedule. The safety, tolerability, PK, and efficacy of volasertib were assessed using two different schedules with identical cycle lengths (21 days): volasertib was administered once on day 1 (schedule A) and volasertib was administered once a week on days 1 and 8 (schedule B).
Materials and Methods
Trial design
This was an open-label phase I dose-escalation study of volasertib in patients with advanced cancer conducted at two sites in Taiwan (NCT00969553). The primary end point was the determination of the MTD, defined as the highest dose of volasertib studied for which the incidence of dose-limiting toxicity (DLT) was less than two of six patients in the first treatment course, for the two dosing schedules. Secondary end points included the incidence and intensity of adverse events (AEs), objective tumour responses, progression-free survival (PFS), response duration, and PK. The study was conducted in accordance with the principles laid down by the Declaration of Helsinki and approved by the independent ethics committees and/or institutional review boards of the participating centres.
Patient selection
Patients aged ⩾18 years of age with histologically or cytologically confirmed diagnosis of advanced, non-resectable, and/or metastatic solid cancer, who had failed conventional treatment, or for whom no therapy of proven efficacy existed, or who were not amenable to established forms of treatment, were eligible for inclusion. Other eligibility criteria included the Eastern Cooperative Oncology Group performance status ⩽2, recovery from toxicities from previous treatments, adequate bone marrow, liver, and renal function (absolute neutrophil count ⩾1500 mm−3, platelet count ⩾100 000 mm−3, total bilirubin ⩽1.5 mg dl−1 (⩽26 μmol l−1, SI unit equivalent), aspartate aminotransferase and/or alanine aminotransferase ⩽2.5 times the upper limit of normal (ULN, if related to liver metastases <5 × ULN), and serum creatinine ⩽1.5 × ULN), a life expectancy ⩾12 weeks, no chemo-, radio-, immuno-, molecular-targeted, or investigational therapy ⩽4 weeks before treatment with the study drug (except steroids and bisphosphonates), and no known history of relevant QT-prolongation. All patients were required to provide written informed consent consistent with International Conference on Harmonisation-Good Clinical Practice and local legislation.
Treatment and dose-escalation
Patients were sequentially assigned to one of the two volasertib dosing schedules and a 3+3-dose escalation design (Kang and Ahn, 2002) was used to evaluate the MTD in each treatment arm. Volasertib was administered by intravenous infusion for over 2 h using two schedules: day 1 every 3 weeks (schedule A) or day 1 and day 8 every 3 weeks (schedule B). The preplanned dose levels were 100, 200, 250, and 300 mg (in case of good tolerability, the dose could be further escalated up to 400 mg) for schedule A and 50, 100, 150, and 200 mg (with escalation to 250 mg in case of good tolerability) for schedule B.
Cohorts of three to six patients were enrolled sequentially into escalating dose tiers of volasertib. Dose escalation was conducted for each treatment group following a 3+3-design, where cohorts of three patients were treated per dose until the highest dose (MTD) was found at which no more than one out of six patients experienced a DLT in the first treatment course. If one of the three patients experienced a DLT in the first course, three additional patients were enrolled to the cohort. A DLT was defined as the following events: drug-related neutropenia Common Terminology Criteria for Adverse Events (CTCAE) grade 4 for ⩾7 days or any grade drug-related febrile neutropenia or neutropenic infection; drug-related grade 4 thrombocytopenia; or drug-related grade ⩾3 non-haematologic toxicity (except untreated nausea, vomiting, or diarrhoea). The following events also constituted a DLT for schedule B only, if considered to be drug-related and present on day 8: grade ⩾3 neutropenia, grade ⩾3 thrombocytopenia, or grade ⩾2 non-haematologic toxicity (except alopecia, untreated nausea, vomiting, or diarrhoea). During course 1, the use of growth factors for the treatment of haematotoxicity was not encouraged except for life-threatening circumstances.
In the dose escalation part of this study, an intra-patient dose escalation from lower dose levels of volasertib to higher dose levels (without exceeding the MTD or defined maximal dose) was allowed, if supported by safety observations, to increase the probability of clinical benefit (good tolerability resulting in a permissible dose escalation was defined as non-haematological drug-related AEs grade <2 and haematological drug-related AEs grade <3). All decisions regarding intra-patient dose escalation were made only in agreement between the investigator and sponsor. Patients experiencing DLTs were eligible for further treatment at one dose tier below. Treatment was discontinued if the DLT was not reversible. After determination of the MTD, or on reaching the highest pre-specified dose level, a total of up to 18 patients were entered at the MTD in order to gain additional safety data. Patients were treated as long as clinically benefitting or until occurrence of an intolerable AE or consent withdrawal.
Assessments
Safety measurements included monitoring of AEs, vital signs, electrocardiograms (ECGs), laboratory evaluations, and patient performance status. The incidence and intensity of AEs during the course of the study were recorded and classified according to CTCAE version 3.0. AEs were coded centrally using the Medical Dictionary for Regulatory Activities. The durations of cytopenias reported as DLTs were calculated based on corresponding lab values. Triplicate digital 12-lead resting ECGs were performed at baseline (defined as the mean of the triplicate at the time point closest to, but before, the start of the infusion), before infusion, and after (1, 2, 3, 8, and 24 h) volasertib infusion.
Blood samples for the detection of volasertib in plasma were obtained predose and at up to seven time points (1, 2, 2.5, 3, 4, 8, and 24 h) after a 2-h infusion of volasertib. Samples were analysed by a validated high-performance liquid chromatography, tandem mass spectrometry assay. Standard non-compartmental PK evaluation was assessed using WinNonlin (v5.2, Pharsight Co., St Louis, MO, USA).
Objective tumour response was assessed by tumour measurements using computed tomography or magnetic resonance-imaging scans, performed at baseline and at the end of every other treatment course, and evaluated according to Response Evaluation Criteria in Solid Tumours (RECIST) 1.0 (Therasse et al, 2000). PFS was analysed using the Kaplan–Meier method (Kaplan and Meier, 1958).
Results
Patient demographics and disposition
A total of 59 patients (32 in schedule A, 27 in schedule B) were enrolled between 28 August 2009 and 10 March 2011. All patients received previous anticancer therapies; many patients were heavily pre-treated with 46.9% (schedule A) and 40.7% (schedule B) previously treated with at least three chemotherapy regimens (Table 1). As of 27 October 2011, 31 patients in schedule A (96.9%) and 25 patients in schedule B (92.6%) had discontinued treatment, the majority owing to progressive disease (65.6% and 77.8%, respectively). Three patients (one patient in schedule A and two in schedule B) were still receiving treatment as of 27 October 2011.
Treatment exposure
Patients received between 1 and 23 courses of treatment with volasertib, with a median (range) of 4 (1, 16) courses initiated in schedule A and 3 (1, 23) courses initiated in schedule B. The median total exposure time in both treatment schedules was 85 days, which is consistent with a median of four courses based on a course length of 21 days. The median total dose in both treatment schedules was 900 mg. Overall, 12 patients had a dose escalation, 10 (31.3%) in schedule A and two (7.4%) in schedule B. Ten patients that continued treatment following recovery from a DLT had a dose decrease per study protocol, seven (21.9%) in schedule A and three (11.1%) in schedule B.
DLTs, safety, and tolerability
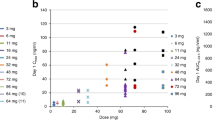
In schedule A, one DLT was reported in the 250 mg dose group (n=6) during course 1. No DLTs were reported following dose escalation to 300 mg (n=6). Therefore, the volasertib dose was escalated to 350 mg (n=3). All three patients at this dose had DLTs in the first treatment course (Table 2). The patients recovered from their DLTs and all three were subsequently treated at a reduced dose without recurrence of DLT. On the basis of DLTs reported, the MTD for volasertib was determined to be 300 mg for schedule A.
In schedule B, two out of three patients in the 200-mg dose group had DLTs in the first treatment course at this dose. Both patients recovered from DLT and received further treatment with volasertib at a reduced dose without recurrence of DLT. The MTD for volasertib was determined to be 150 mg for schedule B. The DLTs typically occurred on treatment days 8–15 and did not cause significant delays in subsequent treatment.
According to the protocol, enrolment was continued at a dose of 300 mg in schedule A and 150 mg in schedule B to gain more safety information at the MTD levels. In total, 17 patients received the 300 mg dose and 16 received the 150 mg dose. Four patients experienced DLTs (grade 4 neutropenia for ⩾7 days; grade 4 febrile neutropenia; and grade 4 thrombocytopenia) during course 1 in the expansion cohort for schedule A. None of the patients in the expansion cohort of schedule B experienced DLTs (Table 2).
The most common all grade and grade ⩾3 AEs, irrespective of relatedness, in schedules A and B were neutropenia, leukopenia, anaemia, and thrombocytopenia (Tables 3 and 4). The most common drug-related AEs (in schedule A and B, respectively) were neutropenia (56.3% and 33.3%), leukopenia (56.3% and 33.3%), thrombocytopenia (62.5% and 14.8%), and anaemia (40.6% and 18.5%). The incidence of drug-related haematological side effects appeared to be dose-related in both dose schedules.
Mild and transient increases from baseline in QTcF (QT interval, corrected for heart rate according to Fridericia’s formula) intervals were observed. At the MTD levels, the maximal mean change (±s.d.) from baseline was 21.0 (±10.6) and 14.9 (±11.7) ms observed 2 h post-administration during the first two courses for schedule A (300 mg) and schedule B (150 mg), respectively. The maximal mean change of QTcF was observed at the end of infusion (that is, 2 h post start of administration), and the magnitude of the effect was comparable across all courses. By the 8-h post-dose assessment in the first course, the values returned to baseline levels. No related clinically relevant cardiac AEs related to a prolonged QT interval were reported. There were three deaths during the course of the study. All three were due to the underlying cancer and none were considered related to volasertib.
At the MTD of schedule A (300 mg), 11 of 17 patients had a change of neutrophil counts from grade 0 or 1 at baseline to a worst case of grade 3 or 4 with treatment. Four patients had a DLT related to neutropenia; two of these patients developed transient febrile neutropenia (3 and 4 days) as a DLT, which did not lead to study discontinuation. Nine patients had a change of platelet grading from grade 0 or 1 at baseline to a worst case of grade 3 or 4 with treatment. Two patients had thrombocytopenia (grade 4) as DLT in the first course; both events were transient (3 and 8 days) and did not lead to treatment discontinuation. At the MTD of schedule B (150 mg), three of 16 patients had a change of neutrophil grading from grade 0 or 1 at baseline to a worst case of grade 3 or 4 with treatment. None of these changes met the criteria for DLT. None had a change of platelet counts from 0 to 1 at baseline to a worst case of grade 3 or 4 with treatment. There were no DLTs of thrombocytopenia (grade 4) observed in schedule B. On the basis of these results, volasertib was considered tolerable in Asian patients with solid cancer at the determined doses. In addition, among patients who had intra-individual dose escalation, three patients in schedule A and one patient in schedule B, respectively, had been treated at the MTD and were escalated from the MTD to a higher dose (up to 350 mg and 200 mg, respectively). No DLTs were observed in this cohort.
PK
Volasertib exhibited multi-exponential plasma concentration profiles (Figure 1). The volume of distribution at steady state (Vss >3000 l) was large with a moderate clearance of >770 ml min−1. An apparent terminal half-life (t1/2) of ∼135 h was observed (Table 5). No accumulation was observed when volasertib was administered on day 1 and day 8 in schedule B.

Plasma concentration–time profile of volasertib following a 2-h intravenous infusion of 300 mg (day 1, schedule A) and 150 mg (days 1 and 8, schedule B) of volasertib.
Antitumour activity
Two partial responses were observed in this study. A patient with ureteral urothelial carcinoma (wild-type FGFR3) was treated with volasertib (350 mg in schedule A) started in July 2010 (two courses), and achieved a partial response. This patient had initially been treated with surgery without adjuvant therapy in July 2008 and subsequently with chemotherapy after presentation with bone metastases in July 2009 (gemcitabine and cisplatin from August 2009) resulting in a partial response. The patient was diagnosed with bone and liver metastases in June 2010 before enrolment in this study. The patient discontinued volasertib after a second occurrence of DLT (grade 4 thrombocytopenia) at 300 mg. A second patient with melanoma and lymph node involvement (wild-type BRAF, NRAS, and cKIT) was treated with volasertib (150 mg in schedule B) and achieved a partial response. The patient had previously been treated with surgery in December 2008 and then presented with lung and lymph node metastases in September 2010 before enrolment in this study. The patient received nine courses of volasertib before discontinuing owing to disease progression.
Overall, 26 (44.1%) patients showed stable disease as their best overall response (14 (43.8%) patients in schedule A and 12 (44.4%) patients in schedule B). Eleven patients have been treated for 10 courses or more, and one patient with bladder urothelial cancer with lymph node metastasis was treated for 23 courses. This patient was initially treated at 50 mg in schedule B and her disease remained stable for >8 months, before the dose was escalated to 150 mg after disease progression. The median PFS over all dose groups was 49 days (range, 30–274 days) in schedule A and 56 days (range, 8–499 days) in schedule B.
Discussion
This study was the first to assess the MTD, safety, and efficacy of volasertib in a day 1 and day 8 dosing schedule, and also the first to investigate the MTD of volasertib in Asian patients with cancer. The MTD of volasertib as a 2-h infusion was determined to be 300 mg when administered on day 1 of a 3-week treatment course (schedule A) and 150 mg when administered on days 1 and 8 of a 3-week treatment course (schedule B). The MTD determined in schedule A of this study is the same as the recommended phase II dose determined in a previous phase I study in Caucasian patients with solid tumours (Schöffski et al, 2012), indicating that for future development Asian and Caucasian patients can be enrolled in global clinical trials without the need for dose adaptations.
Considering the potent antimitotic mode of action and compared with conventional mitosis inhibitors, volasertib was generally well tolerated up to the MTDs determined in this study and its safety profile was comparable to that observed in the phase I/II trials in Western populations with solid tumours (Schöffski et al, 2012; Stadler et al, 2014). The drug-related AEs reported as dose limiting were typically haematological including grade 4 thrombocytopenia, grade 4 neutropenia for ⩾7 days, and/or neutropenia complicated by infections (febrile neutropenia), and were transient and clinically manageable with an established standard of care. As most neutropenias were transient, prophylactic treatment with a granulocyte colony-stimulating factor was not indicated. There were fewer patients with a DLT or grade ⩾3 haematological AEs at the MTD in schedule B than in schedule A, although no formal comparisons were made between the two volasertib dose schedules. These findings may have implications for dosing in future trials of volasertib in patients with solid tumours. A dose-finding study of weekly volasertib with growth factor support may achieve a higher MTD with potentially higher efficacy.
Volasertib had antitumour activity in this cancer patient population comparable to that observed in other phase I/II trials in Western populations (Schöffski et al, 2012; Stadler et al, 2014). Volasertib demonstrated multi-compartmental PK behaviour with an apparent half-life of 135 h, moderate clearance and a large volume of distribution. It is likely that the high volume of distribution (>3000 l) and long t1/2 (135 h) of volasertib resulted in an increased and more sustainable tumour exposure than in previous studies with BI 2536, a dihydropteridinone derivative similar to volasertib. This is in contrast to studies of BI 2536, which demonstrates an inferior PK profile (lower volume of distribution (1000 l) and shorter t1/2 (15 h)) compared with volasertib and did not demonstrate objective responses in patients with solid tumours (Mross et al, 2008; Hofheinz et al, 2010; Frost et al, 2012; Schöffski et al, 2012). Volasertib is primarily eliminated unchanged with metabolism representing a relatively minor elimination route as indicated by low exposures of the main metabolite (data not shown). Therefore, PK drug–drug interactions are not expected with concomitant medications.
Other Plk inhibitors currently in phase I/II clinical development include GSK461364 (Olmos et al, 2011), HMN-214 (Garland et al, 2006), MK-1496 (Doi et al, 2011), NMS-1286937 (NMS-P937) (Valsasina et al, 2012), and TAK-960 (Hikichi et al, 2012). Volasertib has shown signs of antitumour activity in a variety of disease settings (Schöffski et al, 2012), such as malignant melanoma and urothelial carcinoma (Stadler et al, 2014). Two of 17 patients with advanced solid tumours achieved partial responses in a phase I study of MK-1496, which also has a long half-life (Doi et al, 2011). In contrast, no objective responses were reported to date in phase I trials of either GSK461364 (Olmos et al, 2011) or HMN-214 (Garland et al, 2006) in patients with advanced solid malignancies. Phase I trials of NMS-1286937 and TAK-960 in patients with advanced solid tumours are currently ongoing.
In conclusion, this phase I study determined that intravenous volasertib was generally well tolerated up to the identified MTDs of 300 mg (schedule A) and 150 mg (schedule B). Volasertib had a safety profile consistent with its antimitotic mode of action, and a favourable PK profile. These data support Plk as a therapeutic target, warrant further development of volasertib, and suggest a harmonised dosing for Asian and Caucasian patients.
Change history
13 May 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Doi T, Murakami H, Wan K, Miki M, Kotani H, Sakamoto N, Yamamoto N, Okano Y (2011) A first-in-human phase I dose-escalation study of MK-1496, first-in-class orally available novel PLK1 inhibitor, in patients with advanced solid tumors. J Clin Oncol 29 (15_suppl): abstract 3012.
Frost A, Mross K, Steinbild S, Hedbom S, Unger C, Kaiser R, Trommeshauser D, Munzert G (2012) Phase I study of the Plk1 inhibitor BI 2536 administered intravenously on three consecutive days in advanced solid tumours. Curr Oncol 19: e28–e35.
Garland LL, Taylor C, Pilkington DL, Cohen JL, Von Hoff DD (2006) A phase I pharmacokinetic study of HMN-214, a novel oral stilbene derivative with polo-like kinase-1-interacting properties, in patients with advanced solid tumors. Clin Cancer Res 12: 5182–5189.
Hikichi Y, Honda K, Hikami K, Miyashita H, Kaieda I, Murai S, Uchiyama N, Hasegawa M, Kawamoto T, Sato T, Ichikawa T, Cao S, Nie Z, Zhang L, Yang J, Kuida K, Kupperman E (2012) TAK-960, a novel, orally available, selective inhibitor of polo-like kinase 1, shows broad-spectrum preclinical antitumor activity in multiple dosing regimens. Mol Cancer Ther 11: 700–709.
Hofheinz RD, Al-Batran SE, Hochhaus A, Jäger E, Reichardt VL, Fritsch H, Trommeshauser D, Munzert G (2010) An open-label, phase I study of the polo-like kinase-1 inhibitor, BI 2536, in patients with advanced solid tumors. Clin Cancer Res 16: 4666–4674.
Kang SH, Ahn CW (2002) An investigation of the traditional algorithm-based designs for Phase 1 cancer trials. Drug Inf J 36: 865–873.
Kaplan EL, Meier P (1958) Nonparametric estimation from incomplete observations. J Amer Statist Assoc 53: 457–481.
Liu X, Erikson RL (2003) Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci USA 100: 5789–5794.
Medema RH, Lin CC, Yang JC (2011) Polo-like kinase 1 inhibitors and their potential role in anticancer therapy, with a focus on NSCLC. Clin Cancer Res 17: 6459–6466.
Mross K, Frost A, Steinbild S, Hedbom S, Rentschler J, Kaiser R, Rouyrre N, Trommeshauser D, Hoesl CE, Munzert G (2008) Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J Clin Oncol 26: 5511–5517.
Olmos D, Barker D, Sharma R, Brunetto AT, Yap TA, Taegtmeyer AB, Barriuso J, Medani H, Degenhardt YY, Allred AJ, Smith DA, Murray SC, Lampkin TA, Dar MM, Wilson R, de Bono JS, Blagden SP (2011) Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin Cancer Res 17: 3420–3430.
Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, Haslinger C, Garin-Chesa P, Adolf GR (2009) BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res 15: 3094–3102.
Schöffski P, Awada A, Dumez H, Gil T, Bartholomeus S, Wolter P, Taton M, Fritsch H, Glomb P, Munzert G (2012) A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur J Cancer 48: 179–186.
Spankuch-Schmitt B, Bereiter-Hahn J, Kaufmann M, Strebhardt K (2002) Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J Natl Cancer Inst 94: 1863–1877.
Stadler WM, Vaughn DJ, Sonpavde G, Vogelzang NJ, Tagawa ST, Petrylak DP, Rosen P, Lin C-C, Mahoney J, Modi S, Lee P, Ernstoff MS, Su W-C, Spira A, Pilz K, Vinisko R, Schloss C, Fritsch H, Zhao C, Carducci MA (2014) An open-label, single-arm phase II trial of the polo-like kinase inhibitor volasertib (BI 6727) in patients with locally advanced or metastatic urothelial cancer. Cancer 120: 976–982.
Strebhardt K (2010) Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 9: 643–660.
Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, Kimura M, Okano Y, Saji S (2003) Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci 94: 148–152.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 92: 205–216.
Valsasina B, Beria I, Alli C, Alzani R, Avanzi N, Ballinari D, Cappella P, Caruso M, Casolaro A, Ciavolella A, Cucchi U, De Ponti A, Felder E, Fiorentini F, Galvani A, Gianellini LM, Giorgini ML, Isacchi A, Lansen J, Pesenti E, Rizzi S, Rocchetti M, Sola F, Moll J (2012) NMS-P937, an orally available, specific small-molecule polo-like kinase 1 inhibitor with antitumor activity in solid and hematologic malignancies. Mol Cancer Ther 11: 1006–1016.
Acknowledgements
Data from this study have previously been presented at the following congresses: American Society of Clinical Oncology Annual Meeting, 2011; Annual Meeting of the Japanese Society of Medical Oncology, 2011; International Conference of Asian Clinical Oncology Society, 2012. We would like to thank the patients of the National Taiwan University Hospital and the National Cheng Kung University Hospital and their families. We also thank Dr Chiun Hsu, Department of Oncology, National Taiwan University Hospital, for his helpful discussion and comments; Su-Fen Yeh, National Clinical Trial and Research Center, National Taiwan University Hospital, for her dedicated patient care; and Mr Ricardo Schreck for data management. Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Victoria A Robb of GeoMed during the preparation of this article.
Author contributions
The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and have approved the final version.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
A-LC: Honoraria from speakers bureau, Bayer Taiwan Co., Ltd, and consultant/advisory board Eisai Inc. and Excelixis Inc. DC-LH, HF, FV, and TT: employment, Boehringer Ingelheim. JC-HY: provided uncompensated expert testimony to afatinib produced by Boehringer Ingelheim in Taiwan FDA. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Lin, CC., Su, WC., Yen, CJ. et al. A phase I study of two dosing schedules of volasertib (BI 6727), an intravenous polo-like kinase inhibitor, in patients with advanced solid malignancies. Br J Cancer 110, 2434–2440 (2014). https://doi.org/10.1038/bjc.2014.195
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.195
Keywords
This article is cited by
-
Kinome-wide CRISPR-Cas9 knockout screens revealed PLK1 as a therapeutic target for osteosarcoma
Cell Death Discovery (2023)
-
Phase 1 dose escalation trial of volasertib in combination with decitabine in patients with acute myeloid leukemia
International Journal of Hematology (2021)
-
Novel Targeted Therapeutics in Acute Myeloid Leukemia: an Embarrassment of Riches
Current Hematologic Malignancy Reports (2021)
-
Antitumor activity of the polo-like kinase inhibitor, TAK-960, against preclinical models of colorectal cancer
BMC Cancer (2018)
-
Polo-like Kinase 1 as a potential therapeutic target in Diffuse Intrinsic Pontine Glioma
BMC Cancer (2016)
