
Abstract
Background:
Panobinostat, a pan-deacetylase inhibitor, overcomes imatinib resistance in preclinical models of gastrointestinal stromal tumours (GIST). Here we determined the maximum tolerated dose (MTD) and dose-limiting toxicities (DLT) of panobinostat in combination with imatinib (IM) for treatment of patients with refractory GIST.
Methods:
Following a 7-day run-in phase of IM (400 mg per day), escalating doses of panobinostat were added following a ‘3 plus 3’ design. Twelve heavily pretreated GIST patients were enrolled in two dose levels.
Results:
Most common adverse events were thrombocytopenia, anaemia, fatigue, creatinine elevation, nausea, emesis and diarrhoea. Twenty micrograms of panobinostat and 400 mg IM were declared the MTD. Pharmacologically active concentrations of panobinostat and IM were achieved as evidenced by histone H3 acetylation in blood mononuclear cells in vivo and inhibition of the IM-resistant KIT (D816) mutation in vitro. In FDG-PET-CT scans after IM run-in and following 3 weeks panobinostat treatment, 1 out of 11 evaluable patients showed a metabolic partial response, 7 patients were metabolically stable and 3 patients progressed. Longest treatment duration was 17 weeks (median 6).
Conclusion:
Panobinostat and IM can be administered at doses achieving target inhibition in vivo. Further clinical exploration of patients with treatment-refractory GIST is warranted. Correlative studies in this trial may help to optimise dosing schedules in GIST.
Similar content being viewed by others


Main
Activating mutations of KIT or platelet-derived growth factor receptor A (PDGFRA) represent the key oncogenic events in gastrointestinal stromal tumors (GIST) (Hirota et al, 1998; Heinrich et al, 2003). Imatinib mesylate (IM) is a small molecule inhibitor of KIT and PDGFRA that provides long-lasting responses in most patients (82–86%), with a median overall survival of 5 years (Verweij et al, 2004; Blanke et al, 2008a, 2008b). Despite long-lasting responses, most patients eventually acquire resistance and progress. Salvage treatments such as sunitinib and regorafenib may still achieve objective remissions (7% and 4.5%), but median time to progression is only in the range of 6 months (Demetri et al, 2006, 2012). Therefore, alternative treatment strategies for IM-resistant GIST are needed.
Panobinostat (LBH589) is a novel cinnamic hydroxamic acid inhibitor of histone deacetylases (HDACi), which is active on HDAC class I and II isoforms at nanomolar concentrations (Geng et al, 2006). Inhibition of HDACs has been effective in several human cancers, in particular haematologic malignancies, in clinical trials (Giles et al, 2006). Since 2006, two HDACi, vorinostat (SAHA) and romidepsin, have received FDA approval for the treatment of advanced cutaneous T-cell lymphoma (Mann et al, 2007; Xu et al, 2007; Whittaker et al, 2010). We have preclinically evaluated the activity of different classes of HDACi in GIST. The pan-HDACi panobinostat (LBH589) potently inhibited the growth of GIST cell lines in vitro (IC50 values between 80 and 100 nM) (Mühlenberg et al, 2009). Interestingly, panobinostat activity was only seen in KIT-positive GIST models indicating target specificity. This was mediated by transcriptional repression of KIT and by acetylation and subsequent inhibition of heat-shock protein 90 (HSP90), a critical chaperone protein for KIT stabilisation and non-histone client of HDAC6 (Mühlenberg et al, 2009). Additive antitumoral efficacy of panobinostat and IM was observed in IM-resistant GIST models in vitro and in vivo (Floris et al, 2009a).
Against this background, we conducted a phase I study of panobinostat in combination with IM for treatment of patients with metastatic GIST who had progressed on IM and sunitinib.
Patients and methods
Patients
Patients with metastatic GIST (aged 18 years and older) refractory to IM and sunitinib were enrolled in the trial following written informed consent. Additional main inclusion criteria were Eastern Cooperative Oncology Group performance score 2 or lower, absolute neutrophil count 1500 per litre, platelets 100 000 per litre, serum transaminase activity <2.5 × ULN, total bilirubin <1.5 × ULN, serum creatinine <1.5 × ULN or a creatinine clearance of ⩾60 ml min−1, fasting serum cholesterol ⩽350 mg dl−1, triglycerides ⩽300 mg dl−1, proteinuria ⩽1 g in 24 h, no prior thromboembolic disease, no history of haematemesis or haemoptysis. Patients with a history of cardiac disease (congestive heart failure (> New York Heart Association class 2), active coronary artery disease, cardiac arrhythmias requiring antiarrhythmic therapy other than beta blockers or digoxin, uncontrolled hypertension; myocardial infarction less than 6 month before study entry) were excluded. There was no limit on the number of previous anticancer regimens. The institutional review boards of both sites approved the study. A central pathology review that included KIT and PDGFRA mutational testing was performed for all patients from archival tissue by one of the investigators (FG).
Study design and treatment
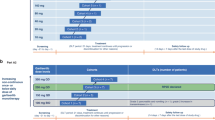
WTZ-GIST-09-01 was an open-label, single-arm, bicentric phase I trial to determine the maximum-tolerated dose (MTD) and dose-limiting toxicities (DLT) of panobinostat in combination with IM. In addition, the study assessed the safety, tolerability, activity, pharmacokinetics and pharmacodynamic activity of the combination. Given a low risk of QTc prolongation, a strict ECG screen at baseline and during treatment was incorporated in the protocol. To obtain a preliminary signal for clinical activity, a prespecified expansion of the MTD cohort to a maximum of nine patients was implemented. IM was administered orally at a dose of 400 mg once daily. As patients may respond to an imatinib rechallenge (http://meetinglibrary.asco.org/content/116140-132), the baseline FDG-PET scan was performed following a 7-day run-in phase, and escalating doses of panobinostat (three times per week for 3 out of 4 weeks) were added. One treatment cycle consisted of 29 days. The dose escalation for panobinostat followed the strict traditional escalation rule (STER). Using STER, patients are treated in cohorts of three, each receiving the same dose. If no DLT occurs, the next cohort of three will receive the next higher dose. If one DLT occurs, the cohort is expanded to three other patients. If two or more DLTs occur, escalation stops and the dose is de-escalated to the next lower level (Chin and Lee, 2008). We selected 20 mg as the starting dose following the discussion with the panobinostat development team from Novartis, based on the existing data on combination trials with panobinostat (Drappatz et al, 2012). WTZ-GIST-09-01 (EudraCT number: 2009-011417-24; German Clinical Trials Register: DRKS00000657) was approved by the institutional ethics committees of the two participating centers and the German Federal Regulatory Drug Agency (BfArM; 4035954).
Dose-limiting toxicities
DLTs were defined from the safety profile of cycle 1 for each dose level. At dose level 1, panobinostat 20 mg was given as a three-times-per-week (MWF schedule) oral dose for 3 out of 4 weeks. Panobinostat doses were increased by 10 mg if no DLT emerged. Toxicity was graded according to the National Cancer Institute Common Toxicity Criteria (NCI-CTC version 3.0). Haematological DLTs were defined as ⩾CTCAE grade 3 neutropenia lasting for >7 days, CTCAE grade 3 thrombocytopenia lasting for >7 days, CTCAE grade 4 thrombocytopenia and neutropenic fever (ANC (including bands) <1.0 × 109 l−1, fever ⩾38.5 °C). Non-haematologic DLT was defined as any grade 3 or 4 toxicity occurring during cycle 1.
Pharmacokinetics and biomarkers
Blood sampling for pharmacokinetic (PK) and pharmacodynamic (PD) studies was performed on day 1, 8, 15, 22 and 29. For PK analyses, blood was withdrawn into EDTA tubes and centrifuged immediately at 2000 × g for 10 min at 4 °C. For extraction of panobinostat and IM, 400 μl plasma was mixed with 600 μl acetonitrile. Free IM, N-desmethyl-IM (CGP074588) and panobinostat were measured by liquid chromatography coupled to a photodiode array (LC/PDA) spectrometer (Acquity Ultra Performance LC system, Waters GmbH, Eschborn, Germany). Pharmacokinetics were calculated using the TOPFIT 2.0 software (Thomae GmbH, Biberach, Germany). A concentration/time plot for IM and desmethyl-IM was generated. The pharmacokinetics of IM and desmethyl-IM have been described by a two-compartment distribution.
Acetylation of histone H3 in peripheral blood mononuclear cells (PBMNC) was assessed as a PD marker of panobinostat activity in vivo. Protein lysates were prepared from PBMNC and subjected to electrophoresis and immunoblotting as previously described (Rubin et al, 2001; Duensing et al, 2004). Changes in protein expression were visualised by chemiluminescence and captured and quantified using a FUJI LAS3000 system with Science Lab 2001 ImageGauge 4.0 software (Fujifilm Medial Systems, Stamford, CT, USA). To identify the lowest plasma level of panobinostat that would induce acetylation, PBMNC from two healthy volunteers were incubated ex vivo with increasing concentrations of panobinostat. In addition, IM-sensitive GIST-T1 (exon 11-mutant) cells and the highly IM-resistant subline GIST-T1-juke (exon 11 and exon 17 D816E double mutant) were incubated in vitro with panobinostat at concentrations that were achieved in study patients as confirmed by our PK analyses.
Evaluation of clinical activity
Full-dose CT scans were performed at least 14 days before randomisation and at least every 56 days. Metabolic imaging studies were conducted at baseline after the run-in period with IM (day 7) before administration of the first dose of panobinostat to exclude metabolic effects by IM rechallenge. All patients were instructed to fast for 6 h before intravenous administration of 18 F-FDG (median: 273 MBq, range: 250–450 MBq). Patients with a blood glucose level exceeding 150 mg dl−1 were not included in the study. Whole-body PET/CT scans were obtained using a PET/CT system (mCT; Siemens Molecular Imaging, Eschborn, Germany) after a median of 65 min (range: 50–80 min) post injection of 18F-FDG.
PET scans were repeated on day 29 of the first cycle. In case of clinical progression, a full-dose PET-CT was performed instead. Metabolic response was defined according to the EORTC-PET Study Group criteria; radiologic response was evaluated per RECIST. Patients with clinical, radiological and/or metabolic response or disease stabilisation were continued on trial medication until progression. In the follow-up phase of the study, CT scans were repeated every 56 days.
Results
Patient characteristics
In all, 12 patients (median 56 years, range 34–74) with metastatic GIST were enrolled at two sarcoma centers. All patients were eligible and evaluable for toxicity. Patients had been heavily pretreated with a median of five treatment lines (range 3–11) before study entry. Clinical and epidemiological data and treatment outcome of the study patients are summarised in Table 1.
Safety
All patients experienced at least one adverse event (AE), the majority of AEs were graded mild. The most common AEs were thrombocytopenia (92%), fatigue (67%), creatinine elevation (58%), nausea (50%), anaemia (42%), diarrhoea (42%) and vomiting (33%; Table 2). The majority of events (thrombocytopenia, nausea, diarrhoea) occurred in the third week of panobinostat treatment, but quickly resolved during week 4, in which no panobinostat was given. Although AE grades 3 or 4 were uncommon, the MWF schedule for 3 weeks was associated with severe thrombocytopenia, which was the dose-limiting toxicity at dose level 2 (30 mg panobinostat). Grade 3 and 4 toxicities that were clearly attributable to study treatments were thrombocytopenia, neutropenia and nausea. After panobinostat rechallenge (3 patients at panobinostat 30 mg, 1 patient at 20 mg) at reduced dose, thrombocytopenia was <grade III except for one patient (grade 3) in cycle 2. This resolved after further dose reduction. There were no treatment-related deaths, but one patient died of progressive disease shortly after discontinuation of treatment.
Pharmacokinetics
Plasma concentrations of IM, desmethyl-IM (IM-metabolite) and panobinostat were determined in 14 treatment courses from 12 patients. Results are summarised in Table 3. The mean peak concentrations (Cmax) of panobinostat were 12.6, 16.6 and 15.6 ng ml−1 (Days 1, 8 and 15) at dose level 1 (n=8), which was declared the MTD (Table 3, Figure 1A). Because of low plasma concentrations of panobinostat, only the Cmax was calculated. The lower limit of detection for panobinostat in plasma was 2 ng ml−1, and the lower limit of quantification was 4 ng ml−1. The mean Cmax for IM was 2068 ng ml−1 during the 7-day run-in period, which was not significantly different from the Cmax of 2564 ng ml−1 during combination therapy (P=0185, t-test). Mean IM trough levels during the run-in period and during combination therapy amounted 1085 ng ml−1 and 1290 ng ml−1 (P=0.125). The terminal half-life for IM in the presence of panobinostat was 20.5 h, whereas the terminal half-life of IM alone is approximately 18 h (Peng et al, 2005). Actual plasma levels for desmethyl-IM, the main metabolite of IM, were as predicted, thus making a pharmacodynamic interaction of panobinostat on IM metabolism highly unlikely (Figure 1B).

Pharmakokinetic studies and biomarker analyses. (A) Cmax diagram for panobinostat at day 1 (first dose panobinostat), day 8 and day 15. The black lines within the boxes indicate the median values, the red lines the mean values. (B) Calculated fit curves for IM and d-desmethyl IM. (C) Immunoblot analyses for acetylated histone H3 from lysates of PBMNCs taken at various time points from selected patients with corresponding panobinostat plasma levels. LLQ=lower level of quantification.
Pharmacodynamic biomarkers for target inhibition in vivo
We assessed the acetylation of histone H3 (H3) in PBMNCs obtained from five patients treated at dose level 1 (MTD) as a pharmacodynamic biomarker for the activity of panobinostat (Figure 1C). Peak H3 acetylation was observed between 4 and 8 h following the first oral dose of panobinostat in four of five patients. H3 acetylation was maintained for 24 h in three patients and decreased at later time points in most patients (not shown). H3 acetylation correlated with detectable plasma levels of panobinostat, although not quantitatively (Figure 1C).
To corroborate these findings, we incubated PBMNCs from healthy donors with panobinostat ex vivo for 8 h. At concentrations of 5 ng ml−1 and above, H3 acetylation became detectable (Figure 2A). Time course experiments with GIST-T1 cells (IM-sensitive cell line) confirmed that H3 acetylation persisted for 8 h after removal of panobinostat and completely disappeared within 24 h (Figure 2B). Incubating GIST-T1 cells and IM-resistant GIST-T1 (KIT D816E mutant) with panobinostat and IM at those concentrations clinically achieved in study patients treated at the MTD (Cmax and ½ Cmax) demonstrated complete inhibition of KIT phosphorylation (Figure 2C).

Preclinical revalidation of clinical biomarkers and tumour-specific target inhibition. (A) Immunoblot analyses for acetylated histone H3 from lysates of PBMNCS from healthy volunteers incubated ex vivo with increasing concentrations of panobinostat. (B) Immunoblot analysis of GIST-T1 cells treated with panobinostat (Pan, 50 nm) for 8 h following a time course after withdrawal of treatment (WD). Continuous treatment with panobinostat at 16 h and 32 h are shown in the right columns. (C) Immunoblot analyses of imatinib-sensitive GIST-T1 and imatinib-resistant GIST-T1R treated with IM, panobinostat or with IM and panobinostat combined.
Clinical activity
No objective responses per RECIST were observed (8 SD, 3 PD). Most patients had extensive metastatic disease, and a decrease in size or change of density was rarely seen in single lesions. Changes in metabolic activity of tumour lesions were assessed by comparing the individual patients’ 18FDG-PET scans obtained at the end of the IM run-in with those obtained on day 29 of cycle 1. Eleven patients were evaluable for PET response, and one patient treated at dose level 1 achieved a metabolic partial response (mPR) as defined by EORTC-PET Study Group criteria (Young et al, 1999). Another seven patients exhibited metabolic disease stabilisation, and three patients had metabolically progressive disease (Figure 3A). The median treatment duration was 6 weeks. The patient achieving a mPR was treated for 17 weeks (Table 1; Figure 3B). He had been diagnosed with metastatic disease 6 years before study enrolment and already received imatinib, sunitinib, nilotinib, imatinib rechallenge, radiation therapy to the large tumour bulk and sorafenib. We attribute his disease stabilisation to the panobinostat addition as he was treated with imatinib at 800 mg qd−1 before the study and enrolled into the trial due to progressive disease at that dose.

Clinical activity of panobinostat and imatinib in GIST. (A) Water fall plot of the relative change in SUV of baseline scans compared with the scans at the end of cycle 1. (B) FDG-PET-CT images of a patient with advanced metastatic GIST at baseline after imatinib run-in (left) and after 3 weeks panobinostat (right) resulting in a partial metabolic remission.
Discussion
In spite of the vast improvement in the treatment of metastatic GIST since the introduction of IM, most patients eventually develop resistance and progress. The mortality of IM-resistant GIST patients amounts 10% per year, which highlights the need for effective salvage treatments (Blanke et al, 2008a). Secondary mutations of the KIT kinase domain are the major mechanism of resistance to IM and sunitinib (Heinrich et al, 2006), thus providing an opportunity for the development of better KIT inhibitors. However, the genomic heterogeneity of these resistance mutations poses a pharmacological challenge to develop selective KIT inhibitors that inhibit all possible mutations (Wardelmann et al, 2005). In contrast to ATP-competitive drugs, such as IM, sunitinib or regorafenib, allosteric KIT inhibitors exhibited an improved capability to inhibit various secondary KIT mutations including those affecting exon 17 (for example, point mutations of D816). However, the clinical utility of these compounds still remains to be proven (Heinrich et al, 2010).
Modulating the KIT chaperone heat-shock protein 90 (HSP90) is an alternative strategy to overcome IM resistance in GIST. Pharmacologic HSP90 inhibitors effectively inhibit KIT in vitro at concentrations that can be achieved in patients regardless of secondary KIT mutations (Bauer et al, 2006; Wagner et al, 2008). Unfortunately, encouraging results from early clinical trials were followed by unexpected toxicities, which were observed in the subsequent phase III trial (Demetri et al, 2010). Hence, the therapeutic potential of HSP90 inhibitors in the treatment of GIST remains unclear and is subject of ongoing clinical trials. Recently, HDACi such as SAHA or panobinostat were shown to have potent antitumoral activity in GIST models in vitro and in vivo (Mühlenberg et al, 2009; Floris et al, 2009b). Mechanistically, HDACi in part act by indirect inhibition of KIT via acetylation and functional inhibition of HSP90 and by transcriptional repression of KIT itself (Mühlenberg et al, 2009; Floris et al, 2009b). Combining HDACi with IM further enhanced the activity, which was even observed in IM-resistant GIST models (Mühlenberg et al, 2009; Floris et al, 2009b). These findings provided the rationale for the present phase I study. Here we aimed to identify a feasible dose and schedule for combination therapy of GIST with panobinostat and IM. We further explored pharmacodynamic effects of plasma levels of both drugs, which could be achieved in the clinic.
Dose levels of the trial were chosen based on clinical and pharmacological considerations. Notably, the MTD had to be defined after only 2 dose levels following a classical ‘3 plus 3’ design. The dose-limiting toxicity at dose level 2 (30 mg panobinostat and 400 mg IM) was thrombocytopenia, which correlated well with DLTs seen in other phase I trials with panobinostat (Fukutomi et al, 2012; Morita et al, 2012). Although most of the latter trials targeted malignancies affecting the bone marrow, haematotoxicity also is important in GIST, a cancer not known for common bone marrow involvement. Notably, haematologic toxicity, especially in the third week of panobinostat, requiring dose interruptions was still observed at the MTD in subsequent cycles. In this combination trial, we did not observe additional toxicities as compared with panobinostat or IM monotherapy trials in patients with other types of cancer (Fukutomi et al, 2012). Common toxicities included fatigue, nausea, changes in appetite or dysgeusia. These toxicities peaked during the third week of combination therapy. Notably, severe hepatotoxicity, as seen with ansamycin-based HSP90 inhibitors in GIST, did not occur. On the basis of the literature, panobinostat and IM could have a two-way pharmacokinetic interaction. CYP3A4 appears to be the main enzyme responsible for the oxidative metabolism of panobinostat in human liver microsomes. Panobinostat itself has been found to be a weak inhibitor of CYP2D6 and a weak inhibitor of CYP3A4/5. In contrast, IM is a potent inhibitor of CYP3A4 and may increase substrate levels 2- to 3.5-fold (Hamberg et al, 2011; Filppula et al, 2012). In the present trial, we observed an increase of the mean Cmax of IM by 23%. This is suggestive of an interaction. However, because of the limited number of patients, these findings did not reach statistical significance. Fukutomi et al (2012) recently reported Cmax plasma levels for panobinostat monotherapy of 10 ng /ml−1 when given at a dose of 20 mg thrice weekly . Panobinostat Cmax levels in our combination study ranged from 13 to 16 ng ml−1, which may hint at an increase in exposure of panobinostat resulting from the coadministration of IM. This could not be formally proven as the study design did not provide a period of panobinostat monotherapy for comparison. Nonetheless, we did not find evidence for a disproportionately strong interaction.
With regard to clinical activity, we did not observe objective radiographic responses but did observe evidence of metabolic inhibition and prolonged disease stabilisation in some patients, which is noteworthy in the light of a median of five previous treatment lines. Importantly, in vivo inhibition of HDAC in PBMNCs from study patients was demonstrated using histone H3 acetylation as a pharmacodynamic biomarker. Moreover, our correlative ex vivo and in vitro studies confirmed that target inhibition of HDACs and KIT can be achieved at plasma levels of panobinostat and IM (Cmax and ½ Cmax), which were observed in the present trial. Notably, at these concentrations, panobinostat and IM inhibited the notorious KIT D816E mutation, which is known to confer cross-resistance to IM, sunitinib, sorafenib and regorafenib in GIST (Heinrich et al, 2012; and data not shown).
In the present trial, panobinostat plasma levels were below the lowest level of quantification 24 h after the first dose. This situation was recapitulated in our preclinical models by removing panobinostat from the media from responding cells, resulting in complete reactivation of KIT signalling within 24 h. As KIT activation drives the disease, these findings would beg for a continuous treatment schedule, as used with direct KIT inhibitors such as IM. Notably, continuous panobinostat treatment for 32 h resulted in reactivation of KIT phosphorylation (Figure 2B).
We believe that these data are particularly relevant, as it shows that HDAC inhibitors may require disease-specific dosing schedules. In GIST, HDACi exert the antitumoral effect via HSP90 acetylation rather than on a complex epigenetic ‘reprogramming’ as in lymphomas. Dosing intervals should therefore be short enough to prevent the GIST cell to fully reactivate its KIT-driven cell cycle machinery.
With regard to the optimal duration of panobinostat treatment, the experience from the PANORAMA-1 trial in multiple myeloma has shown that panobinostat for three consecutive weeks is associated with unacceptable toxicity, as we have seen in our trial. Panobinostat 20 mg (thrice weekly) in a 2 week on ora 1 week off schedule has substantially reduced the rate of grade 3 thrombocytopenia in the combination trial in myeloma (personal communication Novartis) and improved the tolerability, which would be the blueprint for future studies in GIST.
In conclusion, the present schedule of panobinostat in combination with IM is tolerated in heavily pretreated patients with metastatic GIST but future studies should use a 2 week on or 1 week off schedule to improve the tolerability. Haematological toxicity represents the dose-limiting toxicity. Target inhibition was achieved at the MTD, and limited evidence for metabolic tumour inhibition was observed. Acetylation of histone H3 in PBMNCs correlated with measurable panobinostat levels and seems to be a valuable biomarker. On the basis of our correlative studies, additive effects can be expected with IM and panobinostat, and KIT inhibition may even be achieved in exon 17-mutant GIST. These findings warrant further clinical exploration of HDAC inhibitors in combination with KIT inhibitors.
Change history
04 March 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bauer S, Yu LK, Demetri GD, Fletcher JA (2006) Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res 66 (18): 9153–9161.
Blanke CD, Demetri GD, Von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, Corless CL, Fletcher CD, Roberts PJ, Heinz D, Wehre E, Nikolova Z, Joensuu H (2008a) Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 26 (4): 620–625.
Blanke CD, Rankin C, Demetri GD, Ryan CW, Von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki RG, Tanaka M, Hecht JR, Heinrich MC, Fletcher CD, Crowley JJ, Borden EC (2008b) Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 26 (4): 626–632.
Chin R, Lee BY (2008) Principles and Practice of Clinical Trial Medicine. Academic Press, : London, UK.
Demetri GD, Le Cesne A, Von Mehren M, Chmielowski B, Bauer S, Chow WA, Rodenas E, McKee K, Grayzel DS, Kang Y (2010) Final results from a phase III study of IPI-504 (retaspimycin hydrochloride) versus placebo in patients (pts) with gastrointestinal stromal tumors (GIST) following failure of kinase inhibitor therapies. In: ASCO Gastrointestinal Cancers Symposium, Vol. 9; Orlando, FL; 22-24 January 2010 (abstract 64).
Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, Hohenberger P, Leahy M, von MM, Joensuu H, Badalamenti G, Blackstein M, Le CA, Schoffski P, Maki RG, Bauer S, Nguyen BB, Xu J, Nishida T, Chung J, Kappeler C, Kuss I, Laurent D, Casali PG (2012) Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381 (9863): 295–302.
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG (2006) Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368 (9544): 1329–1338.
Drappatz J, Lee EQ, Hammond S, Grimm SA, Norden AD, Beroukhim R, Gerard M, Schiff D, Chi AS, Batchelor TT, Doherty LM, Ciampa AS, Lafrankie DC, Ruland S, Snodgrass SM, Raizer JJ, Wen PY (2012) Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J Neurooncol 107 (1): 133–138.
Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD, Fletcher JA (2004) Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene 23 (22): 3999–4006.
Filppula AM, Laitila J, Neuvonen PJ, Backman JT (2012) Potent mechanism-based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol 165 (8): 2787–2798.
Floris G, Debiec-Rychter M, Sciot R, Stefan C, Fieuws S, Machiels K, Atadja P, Wozniak A, Faa G, Schoffski P (2009a) High efficacy of panobinostat towards human gastrointestinal stromal tumors in a xenograft mouse model. Clin Cancer Res 15 (12): 4066–4076.
Floris G, Debiec-Rychter M, Sciot R, Stefan C, Fieuws S, Machiels K, Atadja P, Wozniak A, Faa G, Schoffski P (2009b) High efficacy of panobinostat towards human gastrointestinal stromal tumors in a xenograft mouse model. Clin Cancer Res 15 (12): 4066–4076.
Fukutomi A, Hatake K, Matsui K, Sakajiri S, Hirashima T, Tanii H, Kobayashi K, Yamamoto N (2012) A phase I study of oral panobinostat (LBH589) in Japanese patients with advanced solid tumors. Invest New Drugs 30 (3): 1096–1106.
Geng L, Cuneo KC, Fu A, Tu T, Atadja PW, Hallahan DE (2006) Histone deacetylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res 66 (23): 11298–11304.
Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, Masson E, Rae P, Laird G, Sharma S, Kantarjian H, Dugan M, Albitar M, Bhalla K (2006) A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res 12 (15): 4628–4635.
Hamberg P, Woo MM, Chen LC, Verweij J, Porro MG, Zhao L, Li W, van der Biessen D, Sharma S, Hengelage T, de JM (2011) Effect of ketoconazole-mediated CYP3A4 inhibition on clinical pharmacokinetics of panobinostat (LBH589), an orally active histone deacetylase inhibitor. Cancer Chemother Pharmacol 68 (3): 805–813.
Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, Von Mehren M, Fletcher CD, Sandau K, McDougall K, Ou WB, Chen CJ, Fletcher JA (2006) Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 24 (29): 4764–4774.
Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CD, Fletcher JA (2003) PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299 (5607): 708–710.
Heinrich MC, Marino-Enriquez A, Presnell A, Donsky RS, Griffith DJ, McKinley A, Patterson J, Taguchi T, Liang CW, Fletcher JA (2012) Sorafenib inhibits many kinase mutations associated with drug-resistant gastrointestinal stromal tumors. Mol Cancer Ther 11 (8): 1770–1780.
Heinrich MC, Wise S, Hood M, Smith B, Kaufman M, Lu W, Wang Y, Griffith D, Flynn D, Fletcher JA (2010) in vitro activity of novel KIT/PDGFRA switch pocket kinase inhibitors against mutations associated with drug-resistant GI stromal tumors. Proc Am Soc Clin Oncol 28: a10007.
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad TG, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y (1998) Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279 (5350): 577–580.
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12 (10): 1247–1252.
Morita S, Oizumi S, Minami H, Kitagawa K, Komatsu Y, Fujiwara Y, Inada M, Yuki S, Kiyota N, Mitsuma A, Sawaki M, Tanii H, Kimura J, Ando Y (2012) Phase I dose-escalating study of panobinostat (LBH589) administered intravenously to Japanese patients with advanced solid tumors. Invest New Drugs 30 (5): 1950–1957.
Mühlenberg T, Zhang Y, Wagner AJ, Grabellus F, Bradner J, Taeger G, Lang H, Taguchi T, Schuler M, Fletcher JA, Bauer S (2009) Inhibitors of deacetylases suppress oncogenic KIT signaling, acetylate HSP90, and induce apoptosis in gastrointestinal stromal tumors. Cancer Res 69 (17): 6941–6950.
Peng B, Lloyd P, Schran H (2005) Clinical pharmacokinetics of imatinib. Clin Pharmacokinet 44 (9): 879–894.
Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, Demetri GD, Fletcher CD, Fletcher JA (2001) KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 61 (22): 8118–8121.
Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PC, Van Glabbeke M, Bertulli R, Judson I (2004) Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 364 (9440): 1127–1134.
Wagner A, Morgan JA, Rosen LS, George S, Gordeon MS, Devine CM, Van den Abbeele A, Grayzel D, Demetri GD (2008) Inhibition of heat shock protein 90 (Hsp90) with the novel agent IPI-504 in metastatic GIST following failure of tyrosine kinase inhibitors (TKIs) or other sarcomas: Clinical results from phase I trial. J Clin Oncol 26 (6): a10503.
Wardelmann E, Thomas N, Merkelbach-Bruse S, Pauls K, Speidel N, Buttner R, Bihl H, Leutner CC, Heinicke T, Hohenberger P (2005) Acquired resistance to imatinib in gastrointestinal stromal tumours caused by multiple KIT mutations. Lancet Oncol 6 (4): 249–251.
Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, Samtsov A, McCulloch W, Kim YH (2010) Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol 28 (29): 4485–4491.
Xu WS, Parmigiani RB, Marks PA (2007) Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26 (37): 5541–5552.
Young H, Baum R, Cremerius U, Herholz K, Hoekstra O, Lammertsma AA, Pruim J, Price P (1999) Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer 35 (13): 1773–1782.
Acknowledgements
We thank the teams of the Sarcoma Center at the West German Cancer Center, Essen and the Sarcoma Center Berlin/Brandenburg for their dedicated clinical care. We also wish to acknowledge the contributions of Julia Ketzer, Claudia Ose, Sarah Rauhut, Edel Ó Halloran and Simone Micheel. This study was supported by a research grant from Novartis; SB holds a Max Eder Fellowship of the Deutsche Krebshilfe (No. 109757) and received funds from the Life Raft Group Research Initiative; the West German Cancer Center is supported by a National Oncology Center of Excellence grant of the Deutsche Krebshilfe.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work has in part been presented at the 2012 Annual Meeting of the American Society of Clinical Oncology, Chicago, May 31-June 4.
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Bauer, S., Hilger, R., Mühlenberg, T. et al. Phase I study of panobinostat and imatinib in patients with treatment-refractory metastatic gastrointestinal stromal tumors. Br J Cancer 110, 1155–1162 (2014). https://doi.org/10.1038/bjc.2013.826
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.826
Keywords
This article is cited by
-
Histone deacetylase inhibitor ITF2357 leads to apoptosis and enhances doxorubicin cytotoxicity in preclinical models of human sarcoma
Oncogenesis (2018)
-
Gastrointestinale Stromatumoren
Der Gastroenterologe (2018)
-
Gastrointestinale Stromatumoren
Wiener klinisches Magazin (2018)
-
A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC
Scientific Reports (2017)
-
Fingerprinting of neurotoxic compounds using a mouse embryonic stem cell dual luminescence reporter assay
Archives of Toxicology (2017)
