
Abstract
Background:
Emerging studies have shown the potential benefit of arming oncolytic viruses with therapeutic genes. However, most of these therapeutic genes are placed under the regulation of ubiquitous viral promoters. Our goal is to generate a safer yet potent oncolytic herpes simplex virus type-1 (HSV-1) for cancer therapy.
Methods:
Using bacterial artificial chromosome (BAC) recombineering, a cell cycle-regulatable luciferase transgene cassette was replaced with the infected cell protein 6 (ICP6) coding region (encoded for UL39 or large subunit of ribonucleotide reductase) of the HSV-1 genome. These recombinant viruses, YE-PC8, were further tested for its proliferation-dependent luciferase gene expression.
Results:
The ability of YE-PC8 to confer proliferation-dependent transgene expression was demonstrated by injecting similar amount of viruses into the tumour-bearing region of the brain and the contralateral normal brain parenchyma of the same mouse. The results showed enhanced levels of luciferase activities in the tumour region but not in the normal brain parenchyma. Similar findings were observed in YE-PC8-infected short-term human brain patient-derived glioma cells compared with normal human astrocytes. intratumoural injection of YE-PC8 viruses resulted in 77% and 80% of tumour regression in human glioma and human hepatocellular carcinoma xenografts, respectively.
Conclusion:
YE-PC8 viruses confer tumour selectivity in proliferating cells and may be developed further as a feasible approach to treat human cancers.
Similar content being viewed by others
Main
Oncolytic herpes simplex virus type-1 (HSV-1) viruses are typically mutated or deleted in viral genes that encode viral replications; the absence of these viral proteins leads to an abortive replication cycle in normal cells. However, in tumour cells, the mutant virus can selectively replicate because the missing viral protein function is provided by the tumour. For example, the UL39 gene encodes the large subunit of ribonucleotide reductase (ICP6), which is required for efficient viral DNA synthesis (Goldstein and Weller, 1988). The ribonucleotide reductase has cellular homologues that could provide the missing function in trans. In addition, the proteins are generally elevated in dividing tumour cells but are expressed at low levels in normal cells. Thus, HSV-1 mutant with UL39 gene deleted (e.g., hrR3) can replicate selectively in tumour cells. Another common virus gene deletion involves the γ34.5 gene (also known as RL-1). In wild-type virus, this gene encodes ICP34.5 protein that counteracts the effect of dsRNA-dependent protein kinase (PKR) phosphorylation (Chou et al, 1990). Active PKR phosphorylates eIF2α and shutdown of viral protein synthesis. Hence, ICP34.5 enables viral protein expression that ultimately leads to cell death. ICP34.5 also binds to the C-terminal domain of Beclin1 and inhibits the cell autophagy response (Orvedahl et al, 2007). Conversely, mutant viruses deleted in γ34.5 (e.g., HSV1716) are unable to replicate and infected cells are spared. However, in tumour cells, that contains high levels of active MAP/ERK kinase (MEK), PKR activation is blocked and allowed a higher degree of viral replication than in normal cells (Smith et al, 2006).
Tumour cell killing can be mediated through attenuation of two or more viral genes such as γ34.5 and UL39 gene (e.g. G207) or similar to G207 with an additional deletion in ICP47 locus (e.g., G47Δ). Oncolytic HSV-1 can also be armed with the prodrug converting gene (Yamada et al, 2012), immunomodulatory molecules (Fukuhara et al, 2005), immune stimulant gene (Liu et al, 2003) or antiangiogenesis agents (Yoo et al, 2012) to augment the efficacy of the viruses beyond their oncolytic abilities. The first oncolytic virus that contains an active gene to enter clinical trial is the oncolytic HSV-1, OncoVexGM-CSF. It contains double deletion in γ34.5 and ICP47 genes, and the cytomegalovirus (CMV) promoter-driven granulocyte–macrophage colony-stimulating factor is inserted into the γ34.5 gene region (Liu et al, 2003). intratumoural administration into melanoma lesions have resulted in an encouraging 28% objective response rate in a Phase II clinical trial and has successfully entered into Phase III clinical trials (Kaufman and Bines, 2010).
As greater number of oncolytic variants arises through the advancement in recombineering technology, it is important to build in additional safety features. At present, most of the therapeutic genes in oncolytic viruses are expressed by ubiquitous viral promoters. For example, the antiangiogenic gene Vstat120 is placed under the regulation of HSV-1 immediate early IE4/5 promoter in oncolytic HSV-1 (Hardcastle et al, 2010). Likewise, in another oncolytic HSV-1, MGH2, the cytochrome P450 and secreted intestinal carboxylesterase genes are under the regulation of IE4/5 and CMV promoter, respectively (Tyminski et al, 2005). The non-tumour selective activities of these promoters may be the main reason that limits the potential of oncolytic viruses to arm with more potent therapeutic gene that induce direct cell killing, but are currently limited to using prodrug-activated therapeutic genes.
In the present study, we have generated a new oncolytic virus that prevents expression of transgene in post-mitotic or quiescent cells. This is achieved through transcriptional regulation of the gene of interest in a cell cycle-dependent manner, which has been shown to work well in the context of replication-deficient HSV-1 amplicon vectors (Ho et al, 2004; Wang et al, 2007; Ho et al, 2010; Sia et al, 2013). In short, in proliferating cells, the binding of transactivator fusion proteins, Gal4 NF-YA, to the Gal4-binding sites located upstream of the minimal cyclin A2 promoter will drive the transcription of the reporter or therapeutic gene (Supplementary Figure S1A). In non-dividing or quiescent cells, the binding of the Gal4 NF-YA is prevented by repressor proteins occupying the CDE/CHR site, thus suppressing transcriptional activation of the reporter or therapeutic gene (Supplementary Figure S1B). Herein, we have inserted this cell cycle-regulatable luciferase transgene cassette into the UL39-deleted oncolytic HSV-1 and demonstrated that the transcriptional activities of these newly generated viruses correlated with the cycling tumour cells in vivo based on the dual-color tracking system introduced to the virus (i.e., luciferase activities) and the tumour cells (red fluorescence). Efficient antitumour effects were subsequently demonstrated in hepatocellular carcinoma and human glioma subcutaneous xenograft mouse models. When equal amounts of the viruses were inoculated into pre-established intracranial glioma (i.e., proliferating) and the non-tumour-bearing contralateral brain (i.e., post-mitotic), luciferase activities were enhanced in the tumour region compared with the normal brain parenchyma of the same animal. Taken together, these viruses could potentially be of clinical relevance for further development against human cancer treatment.
Materials and methods
Cell culture
PLC/PRF/5 is a human hepatocellular carcinoma cell line that was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Vero cell is an African green monkey kidney cells kindly provided by Sandri-Goldin RM, University of California, Irvine, CA. ΔGli36 cells (kindly provided by Dr Esteves M, University of Massachusetts) are stable clones derived from human glioma cells and overexpressed a truncated mutant epidermal growth factor receptor (EGFR variant III). EGFR variant III accounts for ∼40%–50% of gliomas in patients (Nishikawa et al, 1994). ΔGli36-DsRed2 and PLC/PRF/5-DsRed2 were DsRed2 expressing stable clonal cell lines stably transfected with pcDNA3-DsRed2 containing DsRed2 gene regulated by CMV promoter. Normal human astrocytes (NHA) were purchased from Lonza (Basel, Switzerland) and cultured in Astrocyte Basal Medium supplemented with recombinant human EGF, insulin, ascorbic acid, gentamycin sulphate, amphotericin, L-glutamine and FBS as recommended by the supplier. For the rest of the cells, they were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS; HyClone Laboratories, Logan, UT, USA), penicillin (100 U ml−1; Life Technologies, Grand Island, NY, USA), streptomycin (100 μg ml−1; Life Technologies) and L-glutamine (2 mM; Sigma-Aldrich, St. Louis, MO, USA). PLC/PRF/5 cells were further supplemented with a non-essential amino acid (0.1 mM; Life Technologies) and sodium pyruvate (1 mM; Life Technologies). ΔGli36 cells were cultured in the presence of puromycin (1 μg ml−1; Sigma-Aldrich). All DsRed2-expressing stable clones were cultured in the presence of 500 μg ml−1 of Geneticin (Life Technologies). All cells were maintained at 37 °C in a humidified incubator with 5% CO2.
Isolation of primary glioma cells
Primary human glioma cells were isolated from the brain tumour tissues of patients undergoing brain tumour surgery at the National Neuroscience Institute, Singapore following approval from the SingHealth Centralized Institutional Review Board, and with patient informed consent. The collected tissue was rinsed with PBS and separated into small pieces in the presence of complete medium (Astrocyte Basal Medium (ABM) supplemented with 10% FBS, Penicillin/Streptomycin, normocin and L-Glucose; Cambrex Bio Science Walkersville, Inc., Walkersville, MD, USA). The tissue suspensions were first passed through a 5-ml serological pipette, followed by a 1-ml pipette and finally a flame-polished pasteur pipette until no clumps were visible. Following trypsin digestion, the homogenate was filtered through a 70-μm cell strainer (BD Biosciences, San Jose, CA, USA) and then subjected to centrifugation. The collected cells were cultured in complete ABM for immunohistochemistry staining or for cell viability assay.
Construction of recombinant viruses
Recombinant HSV-1 was generated by homologous recombination in Escherichia coli SW102 and gal selection/counterselection (Warming et al, 2005) using a BAC-cloned HSV-1 strain F genome; denoted as pYEbac102 (Ichikawa and Chiocca, 2001). Detailed methods are described in Additional Methods.
In vitro virus replication assay
Vero, PLC/PRF/5 and ΔGli36 cells (5 × 105) were seeded in six-well plates. After 24 h, cells were infected with either YE-102 or YE-PC8 at an MOI of 2. The supernatant and cells were collected at 24 h post infection and exposed to three cycles of freeze-thaw. The viral yield in each well was determined using plaque assay in Vero cells as described in Additional Methods.
In vitro cell cytotoxicity assay
Vero, PLC/PRF/5 and ΔGli36 (1 × 105 cells) were seeded in 24-well plates. After 24 h, cells were infected with either YE-102 or YE-PC8 viruses at an MOI of 0.1. For primary human glioma or NHA, 1 × 104 cells were seeded in 96-well plates and infected with YE-PC8 at MOI of 0.01, 0.1 and 1. After 72 h post infection, the viable cells were determined using Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instructions. The cell survival percentage was calculated by normalising the value of virus-infected cells to that of uninfected cells. For combination treatment, temozolomide (TMZ; Temodal; Schering Plough, Belgium) was dissolved in DMSO (Sigma-Aldrich) to produce a 100 mM stock solution for in vitro experiments. ΔGli36 cells were seeded as mentioned above. Twenty-four hours later, cells were treated with 150 μ M TMZ and infected with YE-PC8 at an MOI of 2.5. After 72 h post treatment, cell viability was measured by normalising the value of treated cells to that of untreated cells. All data were presented as mean±s.e.m. of quadruplicate experiments.
Cell cycle regulation analysis
ΔGli36 (5 × 105 cells) were incubated either in DMEM containing 10% FBS or DMEM containing 0.5% FBS with 50 μ M lovastatin to induce G0/G1 growth arrest. After 40 h of incubation, cells derived from a representative dish was quantified to determine the amount of virus used for infection. YE-PC8 viruses were subsequently allowed to adsorb at 4 °C to the remaining triplicate dishes for an hour at an MOI of 2.0. YE-PC8-infected ΔGli36 cells, which were previously incubated in standard tissue culture medium, were cultured in either DMEM containing 10% FBS or DMEM containing 10% FBS and 150 μg ml−1 phosphonoacetic acid (PAA). Likewise, YE-PC8-infected ΔGli36 cells that were previously incubated in the presence of lovastatin were incubated in the presence of DMEM containing 0.5% FBS with 50 μ M lovastatin. At different time points after infection, cells were collected for real-time quantitative PCR assay or luciferase activity measurement. To compare the cell cycle-regulated promoter activity of YE-PC8 with matching control virus (denoted as YE-CMV-Luc), ΔGli36 (5 × 105 cells) were infected with YE-PC8 or YE-CMV-Luc for an hour at an MOI of 0.05 before replenished with DMEM containing 10% FBS or DMEM containing 0.1% FBS. These cells were subjected to luciferase assay after 48 h post infection.
Animal studies
All animal experiments were approved by the SingHealth Institutional Animal Care and Use Committee (IACUC), Singapore (Protocol no. 2010/SHS/609) and were carried out in an Agri-Food and Veterinary Authority (AVA) of Singapore licensed and Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) international accredited animal research facility under the close supervision of a licensed veterinarian. Inbred severe combined immunodeficient (SCID) and nude mice (female, 4–6 weeks) were purchased from Animal Resources Centre (Canning Vale, Australia). Immunocompetent male BALB/c and C3He mice were purchased from Biomedical Sciences Institutes, Singapore. For in vivo monitoring of tumour growth and viral kinetics, 5 × 106 of ΔGli36-DsRed2 cells (ΔGli36 cells stably expressed the DsRed2 protein) were subcutaneously inoculated in the right flank of SCID mice. Fourteen days after tumour inoculation, YE-PC8 viruses (2 × 105 PFU) were injected into the tumour tissues. At the indicated time points, luciferase activities mediated by YE-PC8 and tumour DsRed2 fluorescence signals were acquired as previously described using the Xenogen IVIS Lumina (Caliper Life Sciences, Hopkinton, MA, USA) system (Sia et al, 2012). Representative animal was also killed at day 4, and tumour was collected for immunofluorescence analysis as described in Additional Methods. To evaluate the viral kinetics in tumour vs post-mitotic brain cells, orthotopic glioma model was established in nude mice as previously described (Ho et al, 2010). One week post tumour cell implantation, equal amount of viruses were inoculated to the tumour-bearing region and the contralateral normal brain parenchyma of the same animal using similar co-ordinates. Next day, bioluminescence images were acquired as previously described (Sia et al, 2010). For survival analysis in orthotopic glioma model, the mice were separated into three groups and injected with equal volume of viral supernatant or vehicle HBSS 1 week post tumour implantation. Viral supernatant consists of 5 × 105 PFU of either YE-102 or YE-PC8 in a total volume of 10 μl HBSS. To evaluate the pathogenicity of YE-102 and YE-PC8 viruses, increasing dosages of viruses from 5 × 104 to 1 × 109 PFU were either administered systemically into immunocompetent BALB/c mice or intracerebrally into immunocompetent C3He mice. For efficacy studies in subcutaneous xenograft model, ΔGli36-DsRed2 and PLC/PRF/5-DsRed2 tumour xenograft on nude mice were used as the models. To establish the PLC/PRF/5-DsRed2 tumour, 1 × 107 of PLC/PRF/5-DsRed2 cells (PLC/PRF/5 cells stably expressed the DsRed2 protein) were first injected subcutaneously on 6-week-old female SCID mice. After 1 month, when the tumour size was about 1000 mm3, the PLC/PRF/5-DsRed2 tumour was collected and minced with scissors before subcutaneously implanting in the right flank of 6-week-old female nude mice. For ΔGli36-DsRed2 tumour, 5 × 106 ΔGli36-DsRed2 cells were subcutaneously inoculated in the right flank of nude mice. When the tumour sizes reach about 300 mm3, three doses of YE-PC8 (5 × 106 PFU) viruses were intratumourally injected into the tumour at day 1, 30 and 53. HBSS was injected in control mice to serve as negative control. At selected time points, tumour volumes were measured with a caliper, using the formula V (volume)=π/6 × [L × (W)2], where L is length and W is width. At the same time point, mice were also subjected to non-invasive bioluminescence imaging as described above. All animals in the experiments were monitored every 24 h for general health status and body weight, no more that 20% weight loss over an extended period or more that 10% weight loss over a short period in accordance with IACUC policy. The animals were killed after they reached this humane end point.
Statistical analysis
Data presented throughout this study are mean values±s.e. of the mean. Survival curves were generated by the Kaplan–Meier method using GraphPad Prism (version 3.03; GraphPad Software, La Jolla, CA, USA). Statistical significance was determined by unpaired t-test, and values of P<0.05 were considered significant.
Additional methods
Additional methodology is described in Supplementary Information—Materials and Methods.
Results
Construction and characterisation of cell cycle-specific transcriptionally regulated oncolytic herpes simplex virus type-1
We used a bacterial artificial chromosome (BAC)-cloned HSV-1 strain F genome (Tanaka et al, 2003) and homologous recombination system in E. coli and a gal-positive/negative selection method (Warming et al, 2005), which has been described in additional methods, to introduce the desired mutation into pYEbac102. Briefly, we have adopted the gal selection method to first insert the gal expression cassette into the coding region of ICP6 of pYEbac102 (Figure 1A) to generate pYEbacgalK (Figure 1B). In the second step, the gal cassette is substituted by the DNA fragment containing the cell cycle-regulatory elements with the luciferase gene (4.5-kbp). Thereafter, Gal-negative clones were selected by resistance to 2-deoxy-galactose (DOG) on minimal plates with glycerol as the carbon source. Positive colonies, denoted as pYEbacPC8 (Figure 1C), were selected based on PCR screening against the luciferase gene. As shown in Figure 2A, HindIII digestion of pYEbacPC8 plasmid DNA produced unique bands of 6.4 kbp, 2.2 kbp and 2.0 kbp, whereas digestion of pYEbac102 produced a band of 9.4 kbp. This difference in digestion pattern between pYEbacPC8 and parental pYEbac102 was due to the substitution of the ICP6 sequence with the cell cycle-regulatory elements. The pYEbacPC8 construct was further confirmed by DNA sequencing of the regions where the mutation was introduced (data not shown).

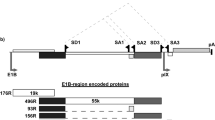
Schematic representation of the various recombinant HSV-1 bacterial artificial chromosome (BAC) and viral genomes. (A) pYEbac102 contains wild-type HSV-1 genome, and the BAC vector sequence was flanked by two loxP sites. pYEbacPC8 was subsequently derived from pYEbac102 by a two-step homologous recombination of BAC DNA. (B) In the first step, the infected cell protein 6 (ICP6) coding region in pYEbac102 was replaced by galactokinase (galK) cassette to generate pYEbacgalK. (C) In the second step, galK cassette was replaced by the cell cycle regulatory elements (consisted of cytomegalovirus promoter (CMVp), fusion protein of Gal4 DNA binding domain with nuclear factor Y, alpha (Gal4 NF-YA), synthetic poly A (P(A)), 8 × Gal4-binding site (Gal4 bs), cyclin A promoter (CycAp) and luciferase reporter gene (Luc)) to generate pYEbacPC8. (D) YE-102 recombinant viruses were derived from pYEbac102 by Cre-digestion to remove the BAC vector sequence. (E) Likewise, the BAC vector sequence in pYEbacPC8 was removed to generate YE-PC8 recombinant viruses. (F) YE-CMV-Luc was used as a control virus where the cell cycle-dependent promoter from Gal4 NF-YA to CycAp was removed using the similar strategy to construct YE-PC8.

Confirmation of recombinant HSV-1 viruses. (A) 2 μg HSV-1 BAC DNA or 1 μg viral genome DNA was digested with HindIII and separated by 0.5% agarose gel. pYEbacPC8 and YE-PC8 showed three unique bands of 6.4 kbp, 2.2 kbp and 2.0 kbp, which resulted from the substitution of ICP6 with the cell cycle regulatory elements. There was also downward shift of the 8.7-kbp band in viral genome DNA compared with BAC DNA, due to the removal of BAC vector. (B) pYEbac102 BAC DNA (lane 1), pYEbacPC8 BAC DNA (lane 2), YE-102 viral genome DNA (lane 3) and YE-PC8 viral genome DNA (lane 4) were digested with HindIII. An ICP6-specific probe hybridised to the 9.4-kbp fragment in pYEbac102 and YE-102, with no hybridisation to pYEbacPC8 or YE-PC8. On the other hand, a luciferase-specific probe recognised the 2.0-kbp fragment in pYEbacPC8 and YE-PC8 but did not hybridise to any fragment in pYEbac102 or YE-102. A BAC vector probe hybridised to the 8.7-kbp fragment in pYEbac102 and pYEbacPC8, with no hybridisation to YE-102 or YE-PC8 viral genome DNA. (C) ΔGli36 cells cultured in standard medium were infected with YE-PC8 at an MOI of 2.0. The luciferase activities of YE-PC8 were measured at different time points after infection. (D) Parental ΔGli36 cells were incubated either in DMEM containing 10% FBS (denoted as control); DMEM containing 10% FBS and 150 μg ml−1 PAA (denoted as PAA) or DMEM containing 0.5% FBS with 50 μ M lovastatin to induce G0/G1 growth arrest (denoted as Lovastatin). After 40 h of incubation, the respective dishes were infected in triplicate with YE-PC8 for an hour at an MOI of 2.0. The cells were then replaced with specific tissue culture media as previously described, i.e., control, PAA or Lovastatin. At different time points, cells were collected for quantitation of relative copy-number of viral genome by normalising the copy-number of luciferase gene to that of β-actin in each sample or for (E) luciferase reporter activities measurement. (F) In vitro comparison of cell cycle-dependent gene regulation on YE-PC8 with matched control virus (YE-CMV-Luc) under proliferation and serum starvation conditions. ΔGli36 cells were infected with YE-PC8 or YE-CMV-Luc for an hour at MOI of 0.05. The cells were then replaced with either DMEM containing 10% FBS (denoted as control) or DMEM containing 0.1% FBS (denoted as serum starvation). After 48 h post infection, cells were collected and subjected to luciferase assay. All data were presented as mean±s.e.m., n=3.
To rescue recombinant viruses and release the viral genome from the BAC backbones, the co-integrated HSV-1 BAC DNA was co-transfected with a Cre recombinase-expressing plasmid (p116) into Vero cells. Thus, the prokaryotic plasmid backbone sequences, which were flanked with the two unidirectional loxP sites, were excised from the co-integrated plasmid. The progeny virus was collected after 3 days and was plaque-purified twice in Vero cells. Viruses isolated from a single plaque were further propagated in Vero cells by infection at low MOI. The viruses rescued from pYEbac102 and pYEbacPC8 were denoted as YE-102 (Figure 1D) and YE-PC8 (Figure 1E), respectively. The viral DNA was extracted from purified virions as described in additional methods and subjected to similar restriction enzyme digestion with HindIII. The results showed that the digestion pattern of the viral genome DNA was nearly the same with its parental BAC DNA except a slight downward shift in the 8.7-kbp bands, which was due to the removal of the BAC vector plasmid sequence (Figure 2A). Genomic structures of HSV-1 BAC DNA and rescued viruses were determined by Southern blot (Figure 2B). Hybridisation probes that recognise the ICP6, luciferase and BAC vector sequences were prepared using the Roche DIG labelling system. HindIII-digested HSV-1 BAC and viral genome DNAs were subjected to hybridisation and detection using the Roche system. Results showed that the ICP6 probe specifically hybridised to the 9.4-kbp fragment of pYEbac102 and YE-102 wherever the luciferase probe specifically hybridised to the 2.0-kbp fragment of pYEbacPC8 and YE-PC8. Also, the BAC vector probe hybridised to the 8.7-kbp fragment of the HSV-1 BAC DNA, whereas it did not hybridise to any DNA fragment of the recombinant virus genome. All these results demonstrated successful substitution of ICP6 with cell cycle-regulatory elements and excision of the BAC vector plasmid sequence in the newly generated recombinant YE-PC8 viruses.
Transgene expression mediated by YE-PC8 viruses is mediated by cell cycle-regulatory elements
Next, the viral kinetics of YE-PC8 in cycling vs quiescent cells was determined. Luciferase activities mediated by YE-PC8 (MOI=2) were measured during the first 20 h post infection. The recombinant viruses, YE-PC8, could confer luciferase activities as early as 4 h post infection in actively proliferating tumour cells. This activity remained stable after reaching a plateau at 12 h post infection (Figure 2C). Thus, time points of 6 h and 12 h were chosen for subsequent characterisation studies of YE-PC8 viruses.
Next, human glioma ΔGli36 cells were cultured in control media (for normal cell growth), lovastatin (to arrest cells in the G0/G1 phase but not interfering with HSV-1 IE, early gene activities) or PAA (to inhibit of HSV-1 DNA polymerase and block viral DNA synthesis). Inhibition of host cell division by lovastatin was confirmed by the reduced levels of cell proliferation marker, Ki67 proteins and S-phase regulator cyclins, cyclin A proteins (Supplementary Figure S2). Inhibition of viral DNA replication by PAA was shown by the low viral copy-number as determined by real-time quantitative PCR (Figure 2D). After 40 h of incubation, the respective dishes were infected in triplicate with YE-PC8 for an hour at an MOI equivalent of 2.0. The cells were then replenished with fresh media corresponding to control media, lovastatin or PAA. At 6 or 12 h post infection, the cells were collected, and luciferase activities were measured. In YE-PC8-infected host cells treated with lovastatin, YE-PC8 failed to replicate as shown by low viral copy-number (Figure 2D; solid circle) and low luciferase activity (Figure 2E). When replication of YE-PC8 was inhibited by PAA, the viral copy-number remained low (Figure 2D; solid triangle). However, the luciferase activities mediated by YE-PC8 in the presence of PAA were insignificantly different from YE-PC8 transduced but untreated control cells (Figure 2E), indicating that the luciferase activity was independent of viral DNA replication, rather, it was regulated by the cell cycle-regulatory elements.
To further confirm that the luciferase activities mediated by YE-PC8 is transcriptionally regulated, we generated an additional control virus (denoted as YE-CMV-Luc; Figure 1F) whereby the cell cycle-regulatory elements were removed, leaving the luciferase reporter under the regulation of the CMV promoter. The cells were first infected with YE-PC8 and control viruses at an MOI of 0.05 for 1 h. The cells were then replenished with either complete control medium or serum starved to induce G1 arrest, respectively, and the cell cycle profiles were confirmed by flow cytometry analysis (data not shown). The evaluation of the gene expressions was then carried out at longer time point after 48 h post infection using luciferase assays. The results showed that the luciferase activities mediated by YE-PC8 were significantly reduced by about 50% when the infected cells were G1 arrested (Figure 2F). In contrast, cells infected by YE-CMV-Luc control virus showed no significant difference in gene expression level between proliferating and serum-starved cells. Taken together, these results confirmed that transgene expression mediated by YE-PC8 was regulated by the cell cycle-regulatory elements.
In vitro growth characteristic and in vivo cytotoxicity analysis of the recombinant viruses
To investigate the growth properties of recombinant viruses, we determined the yield of progeny virus after infection of different cell types with control and YE-PC8 viruses at an MOI of 2. At 24 h after infection, the viral yields in infected cells were determined using plaque assay in Vero cells. The levels of YE-PC8 viral replication vary in different cell types. Wild-type YE-102 viruses were more efficient in producing progeny virus (∼2-fold in Vero, 2.2-fold in PLC/PRF/5 and 2.7-fold in ΔGli36 cells) compared with mutant YE-PC8 (Figure 3A).

Characterization of YE-102 and YE-PC8 viruses in vitro. (A) Viral replication assay. Cells were infected with either YE-102 or YE-PC8 at MOI of 2, and viral yields were determined at 24 h post viral infection. (B) Cell cytotoxicity assay. Cells were infected with either YE-102 or YE-PC8 viruses at an MOI of 0.1. Cell viabilities were determined after 72 h post viral infection, and cell survivals were expressed as a percentage normalised to uninfected cells. All data were presented as mean±s.e.m., n=3.
Next, the cytotoxic effect of the viruses (YE-102 or YE-PC8) was determined in these cells at an MOI of 0.1. Uninfected cells were used as negative control. Three days later, cytotoxicity was measured by using a Cell Counting Kit-8. In Vero cells, there was no significant difference in the number of viable cells between YE-102- and YE-PC8-infected cells (Figure 3B; P>0.05). By contrast, the reduction in cell viability in HCC (i.e., PLC/PRF/5) and human glioma (i.e., ΔGli36) was significant (P<0.05).
To further evaluate the pathogenicity of YE-102 and YE-PC8 viruses, increasing dosage of viruses was administered systemically into immunocompetent BALB/c mice (n=5 per group per virus type) or intracerebrally into immunocompetent C3He mice (n=3 per group per virus type). YE-PC8 was found to be safe up to doses as high as 108 PFU (systemically) and 106 PFU (intracerebrally) (Table 1).
Dual imaging of both host cellular proliferation and transcriptional activities of YE-PC8 viruses
In an attempt to correlate the transcriptional activities of the viruses to the host cell proliferation, the viral kinetics and tumour growth were independently monitored using luciferase activities and DsRed2 fluorescence as outputs in a subcutaneous xenograft mouse model. Although it is ideal to show the viral kinetics in human glioma cells in an intracranial mouse model, we were unable to monitor the tumour growth based on DsRed2 signals because of the high level of absorption of light by the brain parenchyma tissues and the low depth of penetration through the cranium of the mice (as shown in Supplementary Figure S3). Small amount of viruses (2 × 105 PFU) was injected intratumourally, as otherwise the shrinkage in tumour volume would mask our attempt to correlate the transcriptional activities of the viruses to the proliferating tumour cells. During this period of time, the tumour cells actively proliferated as shown by the increasing DsRed2 fluorescence intensity (Figure 4A and B). Increasing tumour cell proliferation corresponded to an increase in luciferase activities up to day 4 (Figure 4A and C), indicating a proliferation-dependent increase in viral replication activities. This was confirmed by the high levels of HSV-1 viral envelope glycoproteins, gC, detected in tumours of representative animal at day 4 (Figure 4D). However, at day 6 and 8, although the DsRed2 signals continued to increase, the luciferase activities declined. The attenuated luciferase signals may be attributed to poorer tissue penetration and greater absorbance of luciferase luminescence light by haemoglobin (Colin et al, 2000) as the tumours became larger and vascularised, a trade-mark of HCC. Thus, a more sophisticated bioimaging system will be needed to provide a visual correlation between viral activities and host cell proliferation over longer time point.

Dual imaging of host cellular proliferation and transcriptional activities of YE-PC8 viruses. (A) ΔGli36-DsRed2 tumours were established in the right flank of SCID mice that were intratumourally injected with an equal amount of YE-PC8 viruses (2 × 105 PFU) directly into the tumours. The DsRed2 fluorescence images were acquired as an indication of tumour growth (represented by top panel). Luciferase activities mediated by YE-PC8 in tumour tissues were imaged at indicated time points after vial injection (represented by the bottom panel). Corresponding quantification of (B) DsRed2 fluorescence signals and (C) luciferase signals. Data were presented as mean±s.e.m., n=4. (D) Tumour sections removed at day 4 post viral administrations and incubated with an anti-gC HSV-1 polyclonal antibody and isotypic control. Scale bar=50 μm.
Efficient suppression of tumour growth mediated by YE-PC8 viruses
To investigate the antitumour effect mediated by YE-PC8, the higher viral dose was administered into human glioma-derived xenograft (ΔGli36; Figure 5A) and human hepatocellular carcinoma-derived xenograft (PLC/PRF/5; Figure 5B). The time of injection was determined according to the decrease in luciferase activities (data not shown), which indirectly corresponded to the extent of viral replication. In total, three injections were administered (5 × 106 PFU per injection) at day 1, day 30 and day 53. Significant suppression of tumour growth was observed in both subcutaneous mouse xenograft models as shown by corresponding images in representative animals. The reduction in tumour volume corresponded with a decrease in the fluorescence signal intensity (data not shown). At the end point of the experiment, the mean tumour volume in human glioma and HCC xenograft animals injected with YE-PC8 viruses was 5-fold and 10-fold smaller than the corresponding untreated group (P<0.0005 and P<0.005, respectively). These results demonstrated that YE-PC8 could mediate an effective antitumour effect regardless of the tumour types.

Efficacy of YE-PC8 viruses in human glioma and hepatocellular carcinoma subcutaneous xenograft models. (A) In vivo killing efficacy of YE-PC8 after three intratumoural injection doses of viruses (5 × 106 PFU per dose; red arrows indicate the time of viral injection) into the ΔGli36-DsRed2 human glioma tumour xenograft. Representative treated animals imaged under standard conditions as described in materials and methods were shown on the right panel. (B) Similar experiment was performed using PLC/PRF/5-DsRed2 human hepatocellular carcinoma tumour xenograft. Data were presented as mean±s.e.m., n=7. Hank’s balanced salt solution (HBSS) was used as negative control. The experiments were stopped, and all mice were killed at humane end point when the tumour burden was >10% body weight or tumour size reached about 2000 mm3 in average.
Treatment with YE-PC8 viruses prolong survival of intracranial glioma-bearing mice
In addition to subcutaneous tumour models, we evaluated the efficacy and the ability of YE-PC8 to confer proliferation-dependent transgene expression in nude mice-bearing intracranial human glioma xenografts. One week post tumour cell implantation, the mice were inoculated with similar doses of YE-PC8 in the tumour-bearing region of the brain and the contralateral normal brain parenchyma of the same mouse. Tumour formation was confirmed using haematoxylin and eosin staining in representative animals (Figure 6A). The results showed that tumours injected with YE-PC8 produced enhanced luciferase signal (right hemisphere; Figure 6B) compared with the control normal region (left hemisphere; Figure 6B). The trend is similar in three tested viral dosages of 5 × 104 PFU (6.3-fold higher luciferase activity in tumour versus normal), 5 × 105 PFU (2.6-fold) and 5 × 106 PFU (2.5-fold) (Figure 6B and C), indicative of higher levels of viral replication in tumour region compared with normal region of the brain. Next, YE-PC8 or control virus (YE-102) was administered in a single dose (5 × 105 PFU) to nude mice 7 days after intracranial tumour cell implantation. Survival analysis showed a significant increase in the survival of mice treated with YE-PC8 compared with YE-102 or HBSS-treated tumours (log-rank test, P<0.0001) (Figure 6D). Collectively, these results demonstrated that functional YE-PC8 viruses in orthotopic glioma mice confer survival advantage.

Transgene expression mediated by YE-PC8 is restricted to intracranial glioma cells and not post-mitotic brain cells. (A) H&E staining for normal brain tissue and human ΔGli36 glioma tumour sections. N indicates the normal brain region in the left brain (left panel). T indicates tumour region implanted in the right brain (middle panel). Interface of normal and tumour region was shown in right panel. (B) Non-invasive imaging of luciferase activities after 24 h post intracranial injection of 5 × 106, 5 × 105 or 5 × 104 PFU of YE-PC8 at both normal brain region (left brain) and tumour region (right brain). Data were presented as mean±s.e.m., n=2. (C) Corresponding quantification of photon emitted from the injection sites in (B). (D) Survival of mice harbouring the orthotopic ΔGli36 glioma after treatment with YE-PC8. Wild-type YE-102 HSV-1 virus and HBSS were also intracranially injected as control. Data were presented as mean±s.e.m., n=7.
YE-PC8 infects and kills human glioma obtained from surgical specimens
Short-term human glioma cell cultures were derived from surgical biopsy specimen obtained from local patients with consent. These cells expressed glial fibrillary acidic protein (GFAP) (Figure 7Ai) and high levels of proliferative markers, Ki67 (Figure 7Aii). Three days post infection of these glioma cells with YE-PC8 at various MOIs, we observed a dose-dependent decrease in cell viability (Figure 7B). Although a similar trend was observed in normal human astrocytes, the extent of cell killing was not as drastic as those observed with the primary tumour cultures. As most oncolytic viruses kill tumour cells through a unique mechanism different from those of small molecule chemotherapeutic agent, we asked whether YE-PC8 could mediate greater tumour cell killing in the presence of TMZ, an alkylating agent commonly used for the treatment of human gliomas. Human glioma ΔGli36 cells were incubated with TMZ and infected with YE-PC8 at an MOI of 2.5. Cell viability was assessed using a CCK-8 assay 72 h post treatment. The results showed that the combination treatment of TMZ and YE-PC8 resulted in greater cell killing than viruses or TMZ alone, suggesting the existence of a combinational effect (Figure 7C). In addition, western blot analysis showed that the incubation with TMZ does not affect the expression of the viral protein, ICP4 (Figure 7D). Taken together, these results indicated that YE-PC8 was effective in killing primary human glioma cells.

Efficacy of YE-PC8 in short-term GBM patient-derived glioma cells. (A) Immunocytochemistry staining on short-term GBM culture for (i) GFAP and (ii) Ki67 expression. (iii) Similar concentrations of mouse IgG antibodies were used as negative control. (B) Cell cytotoxicity of YE-PC8 at MOI of 0.01, 0.1 and 1 in primary human glioma vs NHA. (C) Combination treatment of YE-PC8 at MOI of 2.5 and TMZ at 150 μ M. All cell viabilities were determined after 72 h post treatment, and cell survivals were expressed as a percentage normalised to untreated cells. All data were presented as mean±s.e.m., n=4. (D) Western blot analysis on the presence of HSV-1 virion via expression of HSV-1 ICP4 (175 kDa) for combination treatment as in panel (C). Tubulin (55 kDa) was used as loading control.
Discussion
Oncolytic viruses are attractive agents for the treatment of human cancers because they can be genetically engineered at various stages including during virion adhesion and entry into cells, viral transcription or replication for specific targeting and killing of tumour cells. For example, HSV-1 can be retargeted to human epidermal growth factor receptor 2 (HER2), a receptor overexpressed in breast cancers (Menotti et al, 2008), or IL-13Ralpha2 commonly overexpressed in brain tumours (Zhou and Roizman, 2006). Tumour cell killing efficacy can also be enhanced by insertion of therapeutic genes into the genome of these oncolytic viruses. However, most of these therapeutic genes are placed under the regulation of ubiquitous viral promoters, which are activated immediately post viral infection, and may pose a hazard to normal cells. As a proof-of-principle, the reporter luciferase gene was placed under a cell cycle-dependent promoter. We reasoned that when the luciferase gene is eventually replaced by the appropriate therapeutic gene, therapeutic efficacy and safety should be enhanced. Herein, the results showed that luciferase activities mediated by the newly generated viral vector, YE-PC8, is dependent on the proliferation status of the host cells based on the inhibition studies of HSV-1 replication by PAA. Further, intratumoural inoculation of the viruses in the tumour region yielded elevated luciferase activities compared with similar amount of viruses inoculated into the post-mitotic normal brain parenchyma of the same animal, indicating that these viruses confer transgene expression in a cell cycle-dependent manner.
There are several studies that use regulatable system for controlling the replication-conditional virus for cancer therapy. The general approaches taken include placing the essential genes for viral replication under a tissue-specific promoter. Alternatively, the viral replication is controlled using tetracycline operator/repressor or equivalent systems. In the first approach, nonspecific toxicity to normal tissues due to leaky promoters has remained a problem (Yang et al, 2003). In a recent report, reversion of the ICP4 gene to its wild-type configuration in the context of an oncolytic HSV-1 vector was reported, indicating that such recombinant viruses are genetically unstable (Longo et al, 2011). By contrast, the efficiency of tetracycline-regulated gene expression in recombinant HSV-1 vectors varies, in part, due to the extent of transactivating immediate early gene products of HSV-1, which can interfere with the integrity of tetracycline-regulated transgene expression (Herrlinger et al, 2000). While efforts continue to improve upon the efficiency of these regulatory systems, we have focused our attention to developing a transcriptionally regulated oncolytic HSV-1 system that mediates transgene expression in a cell cycle-dependent manner. We have previously demonstrated that cell cycle-dependent regulation is functional in the context of a replication incompetent HSV-1 amplicon viral vector (Ho et al, 2004; Lam et al, 2007; Sia et al, 2013).
Using BAC recombineering, the ICP6 gene was successfully replaced with similar cell cycle-regulatory elements as in previous studies (Ho et al, 2004). The resultant recombinant viruses, YE-PC8, conferred comparable luciferase activity profiles as another reported oncolytic mutant HSV-1 (denoted as rHsvQ1-CMV-Luc). In the latter construct, the luciferase gene placed under the regulation of CMV promoter was also inserted into the ICP6 locus of a HSV-1 (F strain) (Terada et al, 2006). Using inhibitors against viral replication (PAA) and cellular division (lovastatin), the differentiated luciferase activities mediated by YE-PC8 viruses in proliferating and G1-arrested cells were clearly attributed to the control of cell cycle-regulatory elements instead of differential viral replication (Figure 2E). These cell cycle regulatory elements have facilitated the ability of YE-PC8 viruses to mediate transgene expression in accordance with the proliferation status of the host cells, but this was not observed in matched control virus where the luciferase reporter gene was placed under the regulation of the ubiquitous CMV promoter (Figure 2F). Given that matched control and YE-PC8 viruses differ in promoter strength, we have chosen to administer an equal amount of YE-PC8 into pre-established intracranial glioma vs contralateral post-mitotic brain parenchyma. Similar results were obtained in that luciferase activities were higher in actively proliferating glioma tumours than the corresponding post-mitotic normal brain parenchyma (Figure 6B). Taken together, these data provided support that the cell cycle regulatory elements are truly functional in the context of an oncolytic HSV-1.
We further investigated the replication potential of YE-PC8 viruses and found that they replicated 51%±1.9, 45%±2.2 and 37%±2.7 less efficiently in Vero, PLC/PRF/5 and ΔGli36 cells, respectively, when compared with the corresponding wild-type equivalence YE-102 viruses (Figure 3A). The reduction in progeny virus indicated that the deletion of ICP6 gene in the genome of YE-PC8 has affected to some extent the ability of these viruses to replicate. Interestingly, ICP6-defective oncolytic herpes virus has previously been reported to replicate in quiescent tumour cells carrying p16 deletions independent of cell cycle status (Aghi et al, 2008). In our study, YE-PC8 viruses could not replicate in ΔGli36 cells when the latter was induced into growth arrest state by lovastatin (Figure 2D). However, the same cells have been reported not to express p16INK4a mRNA or protein (Inoue et al, 2004), suggesting that the substitution of the ICP6 sequence with the cell cycle regulatory elements may be functioning differently from the ICP6 deleted mutant HSV-1 mutant virus. Further studies are warranted to investigate further into the role of p16 or other cellular factors that may influence the replication and cytotoxicity induced by YE-PC8 viruses.
At present, multi-targeted approach in the presence of oncolytic viruses is gaining acceptance because the mechanisms by which oncolytic viruses mediate the destruction of tumour cells are different from those of standard treatment by chemotherapy or irradiation. Thus, enhanced therapeutic efficacies have been reported when oncolytic HSV-1 is used in combination with (i) 5-fluorouracil and gemcitabine for the treatment of pancreatic cancers (Eisenberg et al, 2005); (ii) 5-fluorouracil, SN38 or oxaliplatin for the treatment of colon carcinoma (Gutermann et al, 2006); (iii) histone deacetylase inhibitors for the treatment of gliomas (Otsuki et al, 2008); (iv) taxanes for the treatment of prostate cancers (Passer et al, 2009); and (v) RNA-dependent protein kinase (PKR) inhibitor 2-aminopurine for the treatment of rectal carcinoma (Kolodkin-Gal et al, 2009). The effect of combined viral and radiation treatment is less clear in that the same mutant oncolytic HSV-1, G207, has been reported to potentiate antitumour efficacy in human colorectal cancer (Stanziale et al, 2002) but not in human prostate cancer xenografts (Jorgensen et al, 2001). The variability may be due to the different cell types, dose and time of radiation exposure. For example, Advani and colleagues have demonstrated that oncolytic efficacy can be enhanced when irradiation was delivered at a specific window of time within the HSV-1 viral replicative cycle (Advani et al, 2011). Nevertheless, significant progress has been made in recent years through viral engineering and gaining insights into how these newly engineered oncolytic viruses could bring about a greater therapeutic effect in the presence of different adjuvant therapies. In preclinical mouse model, the combined treatment of angiostatin-expressing oncolytic HSV-1 with bevacizumab could further reduce the tumour-invasive-like phenotypes and increase antiangiogenesis and survival (Zhang et al, 2012). In the ‘armed’ therapeutic viruses, the insertion of apoptosis-inducing ligand (TRAIL) could also improve cytotoxicity in human liver carcinoma (Cao et al, 2011) or human gliomas resistant to either TRAIL-mediated apoptosis or viral-mediated oncolysis (Tamura et al, 2013). TRAIL has been shown to induce cell death specifically in tumour cells, while sparing normal cells in nonhuman primates (Bellail et al, 2009). Alternatively, oncolytic viruses have been engineered to express suicide enzymes (e.g., yeast cytosine deaminase) that allow the conversion of the non-toxic prodrug (5-fluorocytosine) into a toxic metabolite (5-fluorouracil) at the site of viral replication. The solubility nature of these toxic metabolites could elicit a potent bystander effect on neighboring tumour cells that have not been actively infected, thereby increasing the overall therapeutic efficacy of oncolytic HSV-1 viruses (Yamada et al, 2012).
As a proof-of-concept, we have generated an oncolytic HSV-1 virus that replicates based on the proliferation status of the infected host cells. These viruses were shown to suppress the growth of human tumour xenografts of different origins, human glioma and human hepatocellular carcinoma (Figure 5A and B, respectively), indicating that their replicating potential is independent of tumour type. Control YE-102 viruses could not be included in these studies as the viruses were very lethal to the animals (data not shown), possibly due to the efficient penetration of these viruses into the gastroinstestinal system via the tumour vasculature as previously reported by other group (Takase et al, 1994). Using an intracranial glioma-bearing mouse model, we have demonstrated that luciferase activities were higher in actively proliferating glioma tumours than the corresponding post-mitotic normal brain parenchyma when both tumour and normal parts of the brain were injected with an equal amount of viruses (Figure 6B). Representative animal was imaged at about 6 h post viral injection, and the levels of luciferase activities acquired at low range scale were similar in both semi-hemispheres of the brain (data not shown). These viruses could also induce selective cell killing in actively proliferating patient-derived short-term glioma culture when compared with normal human astrocytes (Figure 7B). Although we have yet to incorporate an appropriate suicide gene into YE-PC8, preliminary results have demonstrated an enhanced therapeutic efficacy when human glioma cells infected with YE-PC8 were cultured in the presence of TMZ (Figure 7C). HSV-1 ICP4 is a major regulatory protein involved in the initiation of viral DNA replication and is essential for the virus life cycle (Clements et al, 1977). Our results showed that the treatment of TMZ did not affect the level of HSV-1 ICP4 proteins, suggesting that the virus should be functional in the presence of TMZ (Figure 7D).
The potential limitation of most of the oncolytic viruses including the oncolytic HSV-1 generated in this study is probably the incapability of these viruses to efficiently spread to surrounding tumour cells after administration. The oncolytic HSV-1 may encounter overlapping barriers, including (1) small capillaries in tumour that prevent penetration; (2) dense network of extracellular matrix (ECM) and cellular compaction that prevent interstitial diffusion between solid tumours with little interstitial space; and (3) hindered by the presence of normal and healthy cells within the tumour (Smith et al, 2011). Several groups have tried to improve the virus spread in the tumour by modifying the tumour microenvironment to decrease the extracellular matrix (ECM) component with decorin (Choi et al, 2010), hyaluronidase (Ganesh et al, 2008), relaxin (Kim et al, 2006) and proteases such as metalloproteinase-9 (Hong et al, 2010), collagenase/dispase and trypsin (Kuriyama et al, 2000; McKee et al, 2006). Despite the challenges faced, we believe that it is feasible to further improve the functionalities of oncolytic HSV-1 as evidenced by the success of OncoVexGM-CSF (Kaufman and Bines, 2010).
In conclusion, we have constructed an oncolytic HSV-1 that conferred transgene expression in accordance with the extent of host cell proliferation. These recombinant viruses are cytotoxic but less virulent when compared with wild-type HSV-1 virions. At a high viral dosage, actively proliferating tumour cells were effectively suppressed in two independent subcutaneous xenograft mouse models consisting of human glioma and hepatocellular carcinoma cells. In addition, prolonged survival was observed in animals treated with YE-PC8 in intracranial glioma-bearing animals. Thus, these viruses may be further developed with appropriate therapeutic genes for cancer treatment.
Change history
07 January 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Advani SJ, Markert JM, Sood RF, Samuel S, Gillespie GY, Shao MY, Roizman B, Weichselbaum RR (2011) Increased oncolytic efficacy for high-grade gliomas by optimal integration of ionizing radiation into the replicative cycle of HSV-1. Gene Ther 18 (11): 1098–1102.
Aghi M, Visted T, Depinho RA, Chiocca EA (2008) Oncolytic herpes virus with defective ICP6 specifically replicates in quiescent cells with homozygous genetic mutations in p16. Oncogene 27 (30): 4249–4254.
Bellail AC, Qi L, Mulligan P, Chhabra V, Hao C (2009) TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev Recent Clin Trials 4 (1): 34–41.
Cao X, Yang M, Wei RC, Zeng Y, Gu JF, Huang WD, Yang DQ, Li HL, Ding M, Wei N, Zhang KJ, Xu B, Liu XR, Qian QJ, Liu XY (2011) Cancer targeting Gene-Viro-Therapy of liver carcinoma by dual-regulated oncolytic adenovirus armed with TRAIL gene. Gene Ther 18 (8): 765–777.
Choi IK, Lee YS, Yoo JY, Yoon AR, Kim H, Kim DS, Seidler DG, Kim JH, Yun CO (2010) Effect of decorin on overcoming the extracellular matrix barrier for oncolytic virotherapy. Gene Ther 17 (2): 190–201.
Chou J, Kern ER, Whitley RJ, Roizman B (1990) Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250 (4985): 1262–1266.
Clements JB, Watson RJ, Wilkie NM (1977) Temporal regulation of herpes simplex virus type 1 transcription: location of transcripts on the viral genome. Cell 12 (1): 275–285.
Colin M, Moritz S, Schneider H, Capeau J, Coutelle C, Brahimi-Horn MC (2000) Haemoglobin interferes with the ex vivo luciferase luminescence assay: consequence for detection of luciferase reporter gene expression in vivo. Gene Ther 7 (15): 1333–1336.
Eisenberg DP, Adusumilli PS, Hendershott KJ, Yu Z, Mullerad M, Chan MK, Chou TC, Fong Y (2005) 5-fluorouracil and gemcitabine potentiate the efficacy of oncolytic herpes viral gene therapy in the treatment of pancreatic cancer. J Gastrointest Surg 9 (8): 1068–1077.
Fukuhara H, Ino Y, Kuroda T, Martuza RL, Todo T (2005) Triple gene-deleted oncolytic herpes simplex virus vector double-armed with interleukin 18 and soluble B7-1 constructed by bacterial artificial chromosome-mediated system. Cancer Res 65 (23): 10663–10668.
Ganesh S, Gonzalez-Edick M, Gibbons D, Van Roey M, Jooss K (2008) intratumoural coadministration of hyaluronidase enzyme and oncolytic adenoviruses enhances virus potency in metastatic tumor models. Clin Cancer Res 14 (12): 3933–3941.
Goldstein DJ, Weller SK (1988) Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology 166 (1): 41–51.
Gutermann A, Mayer E, von Dehn-Rothfelser K, Breidenstein C, Weber M, Muench M, Gungor D, Suehnel J, Moebius U, Lechmann M (2006) Efficacy of oncolytic herpesvirus NV1020 can be enhanced by combination with chemotherapeutics in colon carcinoma cells. Hum Gene Ther 17 (12): 1241–1253.
Hardcastle J, Kurozumi K, Dmitrieva N, Sayers MP, Ahmad S, Waterman P, Weissleder R, Chiocca EA, Kaur B (2010) Enhanced antitumor efficacy of vasculostatin (Vstat120) expressing oncolytic HSV-1. Mol Ther 18 (2): 285–294.
Herrlinger U, Woiciechowski C, Sena-Esteves M, Aboody KS, Jacobs AH, Rainov NG, Snyder EY, Breakefield XO (2000) Neural precursor cells for delivery of replication-conditional HSV-1 vectors to intracerebral gliomas. Mol Ther 1 (4): 347–357.
Ho IA, Hui KM, Lam PY (2004) Glioma-specific and cell cycle-regulated herpes simplex virus type 1 amplicon viral vector. Hum Gene Ther 15 (5): 495–508.
Ho IA, Ng WH, Lam PY (2010) FasL and FADD delivery by a glioma-specific and cell cycle-dependent HSV-1 amplicon virus enhanced apoptosis in primary human brain tumors. Mol Cancer 9: 270.
Hong CS, Fellows W, Niranjan A, Alber S, Watkins S, Cohen JB, Glorioso JC, Grandi P (2010) Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Ther 17 (10): 1200–1205.
Ichikawa T, Chiocca EA (2001) Comparative analyses of transgene delivery and expression in tumors inoculated with a replication-conditional or -defective viral vector. Cancer Res 61 (14): 5336–5339.
Inoue R, Moghaddam KA, Ranasinghe M, Saeki Y, Chiocca EA, Wade-Martins R (2004) Infectious delivery of the 132 kb CDKN2A/CDKN2B genomic DNA region results in correctly spliced gene expression and growth suppression in glioma cells. Gene Ther 11 (15): 1195–1204.
Jorgensen TJ, Katz S, Wittmack EK, Varghese S, Todo T, Rabkin SD, Martuza RL (2001) Ionizing radiation does not alter the antitumor activity of herpes simplex virus vector G207 in subcutaneous tumor models of human and murine prostate cancer. Neoplasia 3 (5): 451–456.
Kaufman HL, Bines SD (2010) OPTIM trial: a Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol 6 (6): 941–949.
Kim JH, Lee YS, Kim H, Huang JH, Yoon AR, Yun CO (2006) Relaxin expression from tumor-targeting adenoviruses and its intratumoural spread, apoptosis induction, and efficacy. J Natl Cancer Inst 98 (20): 1482–1493.
Kolodkin-Gal D, Edden Y, Hartshtark Z, Ilan L, Khalaileh A, Pikarsky AJ, Pikarsky E, Rabkin SD, Panet A, Zamir G (2009) Herpes simplex virus delivery to orthotopic rectal carcinoma results in an efficient and selective antitumor effect. Gene Ther 16 (7): 905–915.
Kuriyama N, Kuriyama H, Julin CM, Lamborn K, Israel MA (2000) Pretreatment with protease is a useful experimental strategy for enhancing adenovirus-mediated cancer gene therapy. Hum Gene Ther 11 (16): 2219–2230.
Lam PY, Sia KC, Khong JH, De Geest B, Lim KS, Ho IA, Wang GY, Miao LV, Huynh H, Hui KM (2007) An efficient and safe herpes simplex virus type 1 amplicon vector for transcriptionally targeted therapy of human hepatocellular carcinomas. Mol Ther 15 (6): 1129–1136.
Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, McGrath Y, Thomas SK, Thornton M, Bullock P, Love CA, Coffin RS (2003) ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther 10 (4): 292–303.
Longo SL, Griffith C, Glass A, Shillitoe EJ, Post DE (2011) Development of an oncolytic herpes simplex virus using a tumor-specific HIF-responsive promoter. Cancer Gene Ther 18 (2): 123–134.
McKee TD, Grandi P, Mok W, Alexandrakis G, Insin N, Zimmer JP, Bawendi MG, Boucher Y, Breakefield XO, Jain RK (2006) Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res 66 (5): 2509–2513.
Menotti L, Cerretani A, Hengel H, Campadelli-Fiume G (2008) Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J Virol 82 (20): 10153–10161.
Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, Huang HJ (1994) A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA 91 (16): 7727–7731.
Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B (2007) HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1 (1): 23–35.
Otsuki A, Patel A, Kasai K, Suzuki M, Kurozumi K, Chiocca EA, Saeki Y (2008) Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol Ther 16 (9): 1546–1555.
Passer BJ, Castelo-Branco P, Buhrman JS, Varghese S, Rabkin SD, Martuza RL (2009) Oncolytic herpes simplex virus vectors and taxanes synergize to promote killing of prostate cancer cells. Cancer Gene Ther 16 (7): 551–560.
Sia KC, Chong WK, Ho IA, Yulyana Y, Endaya B, Huynh H, Lam PY (2010) Hybrid herpes simplex virus/Epstein-Barr virus amplicon viral vectors confer enhanced transgene expression in primary human tumors and human bone marrow-derived mesenchymal stem cells. J Gene Med 12 (10): 848–858.
Sia KC, Huynh H, Chinnasamy N, Hui KM, Lam PY (2012) Suicidal gene therapy in the effective control of primary human hepatocellular carcinoma as monitored by noninvasive bioimaging. Gene Ther 19 (5): 532–542.
Sia KC, Huynh H, Chung AY, Ooi LL, Lim KH, Hui KM, Lam PY (2013) Preclinical evaluation of transcriptional targeting strategy for human hepatocellular carcinoma in an orthotopic xenograft mouse model. Mol Cancer Ther 12: 1651–1664.
Smith E, Breznik J, Lichty BD (2011) Strategies to enhance viral penetration of solid tumors. Hum Gene Ther 22 (9): 1053–1060.
Smith KD, Mezhir JJ, Bickenbach K, Veerapong J, Charron J, Posner MC, Roizman B, Weichselbaum RR (2006) Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by Deltagamma(1)34.5 mutants of herpes simplex virus 1. J Virol 80 (3): 1110–1120.
Stanziale SF, Petrowsky H, Joe JK, Roberts GD, Zager JS, Gusani NJ, Ben-Porat L, Gonen M, Fong Y (2002) Ionizing radiation potentiates the antitumor efficacy of oncolytic herpes simplex virus G207 by upregulating ribonucleotide reductase. Surgery 132 (2): 353–359.
Takase H, Yamamura E, Murakami Y, Ikeuchi T, Osada Y (1994) Gastrointestinal invasion by herpes simplex virus type 1 inoculated cutaneously into the immunosuppressed mice. Arch Virol 134 (1-2): 97–107.
Tamura K, Wakimoto H, Agarwal AS, Rabkin SD, Bhere D, Martuza RL, Kuroda T, Kasmieh R, Shah K (2013) Multimechanistic tumor targeted oncolytic virus overcomes resistance in brain tumors. Mol Ther 21 (1): 68–77.
Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y (2003) Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77 (2): 1382–1391.
Terada K, Wakimoto H, Tyminski E, Chiocca EA, Saeki Y (2006) Development of a rapid method to generate multiple oncolytic HSV vectors and their in vivo evaluation using syngeneic mouse tumor models. Gene Ther 13 (8): 705–714.
Tyminski E, Leroy S, Terada K, Finkelstein DM, Hyatt JL, Danks MK, Potter PM, Saeki Y, Chiocca EA (2005) Brain tumor oncolysis with replication-conditional herpes simplex virus type 1 expressing the prodrug-activating genes, CYP2B1 and secreted human intestinal carboxylesterase, in combination with cyclophosphamide and irinotecan. Cancer Res 65 (15): 6850–6857.
Wang GY, Ho IA, Sia KC, Miao L, Hui KM, Lam PY (2007) Engineering an improved cell cycle-regulatable herpes simplex virus type 1 amplicon vector with enhanced transgene expression in proliferating cells yet attenuated activities in resting cells. Hum Gene Ther 18 (3): 222–231.
Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG (2005) Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33 (4): e36.
Yamada S, Kuroda T, Fuchs BC, He X, Supko JG, Schmitt A, McGinn CM, Lanuti M, Tanabe KK (2012) Oncolytic herpes simplex virus expressing yeast cytosine deaminase: relationship between viral replication, transgene expression, prodrug bioactivation. Cancer Gene Ther 19 (3): 160–170.
Yang CT, Song J, Bu X, Cong YS, Bacchetti S, Rennie P, Jia WW (2003) Herpes simplex virus type-1 infection upregulates cellular promoters and telomerase activity in both tumor and nontumor human cells. Gene Ther 10 (17): 1494–1502.
Yoo JY, Haseley A, Bratasz A, Chiocca EA, Zhang J, Powell K, Kaur B (2012) Antitumor efficacy of 34.5ENVE: a transcriptionally retargeted and ‘Vstat120’-expressing oncolytic virus. Mol Ther 20 (2): 287–297.
Zhang W, Fulci G, Buhrman JS, Stemmer-Rachamimov AO, Chen JW, Wojtkiewicz GR, Weissleder R, Rabkin SD, Martuza RL (2012) Bevacizumab with angiostatin-armed oHSV increases antiangiogenesis and decreases bevacizumab-induced invasion in U87 glioma. Mol Ther 20 (1): 37–45.
Zhou G, Roizman B (2006) Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc Natl Acad Sci USA 103 (14): 5508–5513.
Acknowledgements
We thank Dr. Kawaguchi Y. (Tokyo Medical and Dental University, Tokyo, Japan) for providing YEBac102; Dr. Warming S. (National Cancer Institute, Frederick, USA) for providing reagents for BAC homologous recombination and Bernd Vogt (University of Zurich, Zurich, Switzerland) for the construction of YE-CMV-Luc. We express appreciation for current and past colleagues at the National Cancer Centre Dr. Ivy Ho, Dr. Grace Wang, Ms Yulyana, Ms Seet Yi Ting and Mr. Berwini Endaya for their support and advices. This work is supported by NMRC-Hong Leong Foundation Medical Research Scientist Award and National Medical Research Council, Singapore.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Miao, L., Fraefel, C., Sia, K. et al. The potential application of a transcriptionally regulated oncolytic herpes simplex virus for human cancer therapy. Br J Cancer 110, 94–106 (2014). https://doi.org/10.1038/bjc.2013.692
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.692
Keywords
This article is cited by
-
RETRACTED ARTICLE: Transcriptional retargeting of herpes simplex virus for cell-specific replication to control cancer
Journal of Cancer Research and Clinical Oncology (2018)
-
Bovine herpesviruses induce different cell death forms in neuronal and glial-derived tumor cell cultures
Journal of NeuroVirology (2016)
-
Oncolytic viruses—immunotherapeutics on the rise
Journal of Molecular Medicine (2016)

{kind=link}
{kind=link}
{kind=link}