
Abstract
Background:
This open-label, multicentre, phase 2 trial evaluated the efficacy and tolerability of the mammalian target of rapamycin inhibitor ridaforolimus in women with advanced endometrial cancer.
Methods:
Women with measurable recurrent or persistent endometrial cancer and documented disease progression were treated with ridaforolimus 12.5 mg intravenously once daily for 5 consecutive days every 2 weeks in a 4-week cycle. The primary end point was clinical benefit response, defined as an objective response or prolonged stable disease of 16 weeks or more.
Results:
In all, 45 patients were treated with single-agent ridaforolimus. Clinical benefit was achieved by 13 patients (29%), including 5 (11%) with confirmed partial responses and 8 (18%) with prolonged stable disease. All patients with clinical benefit response received ridaforolimus for more than 4 months. In this heavily pretreated population, the 6-month progression-free survival was 18%. Ridaforolimus was generally well tolerated: adverse events were predictable and manageable, consistent with prior studies in other malignancies. Overall, the most common adverse events were diarrhoea (58%) and mouth sores (56%); most common grade 3 or higher adverse events were anaemia (27%) and hyperglycaemia (11%).
Conclusion:
Single-agent ridaforolimus has antitumor activity and acceptable tolerability in advanced endometrial cancer patients. Further clinical evaluation of ridaforolimus is warranted.
Similar content being viewed by others


Main
More than 46 000 new cases of endometrial cancer were diagnosed in the United States in 2011, with nearly 8100 deaths (Siegel et al, 2011). Deaths due to endometrial cancer have been on the rise, with the rate doubling over the past 20 years (Sorosky, 2008). Most deaths are due to metastatic or recurrent disease, where survival is poor even with current standard chemotherapy (Pectasides et al, 2007). Response rates with first-line chemotherapy in women with advanced or recurrent endometrial cancer are low, ranging from 20 to 30% for single agents and up to 40 to 60% for combination regimens (Fleming, 2007; Pectasides et al, 2007; Dellinger and Monk, 2009). For women with advanced disease, efficacy after first-line taxane-based regimens is extremely poor (Dellinger and Monk, 2009; Dizon, 2010). Improved treatment options are needed with an increased emphasis on improving the therapeutic index.
The mammalian target of rapamycin (mTOR) is a downstream effector of the phosphoinositide 3-kinase (PI3K)/AKT pathway and thereby serves as a key regulator of cell growth and division (Dancey, 2010; Shorning et al, 2011). Many tumours, including endometrial cancer, have dysregulated PI3K/AKT function (Dedes et al, 2011). Loss of function of the tumour suppressor phosphatase and tensin homologue (PTEN) and gain-of-function mutations of the PIK3CA gene, both of which increase activity of the PI3K/AKT pathway, have been reported in endometrial cancer patients (Salvesen et al, 2004; Hayes et al, 2006; Janku et al, 2011). Moreover, activation of the PI3K pathway in uterine epithelium via biallelic loss of PTEN or selective AKT activation appears to be sufficient for initiating endometrial cancer in preclinical models (Daikoku et al, 2008; Memarzadeh et al, 2010). On the basis of these observations, mTOR inhibition may provide a rational approach to treatment of endometrial cancer.
Several mTOR inhibitors, including rapamycin and its analogues everolimus, temsirolimus, and ridaforolimus, have been evaluated in cancer patients (Choi et al, 2010). Both everolimus and temsirolimus have shown promising results in patients with advanced or recurrent endometrial cancer (Slomovitz et al, 2010; Temkin et al, 2010; Oza et al, 2011a). Ridaforolimus has also been evaluated in multiple clinical trials, with antitumor activity seen in a variety of solid and haematologic malignancies (Mita et al, 2008; Rizzieri et al, 2008; Chawla et al, 2012; Seki et al, 2012). Preclinical studies with ridaforolimus demonstrated antiproliferative activity in endometrial tumour cell lines AN3CA and HEC-1B (Squillace et al, 2011). In a phase 1 clinical trial, treatment with ridaforolimus resulted in prolonged partial response (PR) in a patient with mixed Müllerian uterine cancer (Mita et al, 2008). Here we describe results from a phase 2 trial designed to assess the efficacy and safety of intravenously administered single-agent ridaforolimus in patients with recurrent or persistent endometrial cancer.
Materials and Methods
Study design
This was a phase 2, open-label, single-arm, multicentre study with planned enrolment of approximately 44 patients at nine centres in the United States and Europe (http://clinicaltrials.gov/ct2/show/NCT00122343; Protocol 019). Intravenous ridaforolimus was administered at a fixed dose of 12.5 mg over 30 min once daily for 5 consecutive days every 2 weeks, with each treatment cycle defined as a 4-week period (i.e., two courses of ridaforolimus). This intravenous dose was identified as the recommended phase 2 dose in a previous phase 1 trial of ridaforolimus in patients with advanced malignancies (Mita et al, 2008). Patients were scheduled to receive 2–6 cycles of treatment, but were allowed to continue treatment with ridaforolimus if they maintained at least stable disease (SD) and acceptable tolerability. Consistent with other phase 2 trials in this clinical setting, palliative and supportive care were permitted during the study, but concurrent treatment with other anticancer modalities was prohibited. Other investigational drugs or devices were also prohibited, as were herbal preparations or related over-the-counter preparations containing herbal ingredients known to affect cytochrome P450-3A isoenzymes (e.g., St John’s wort). Patients were followed on study for 24 months after their last dose of ridaforolimus.
The study protocol and patient informed consent forms were approved by each local institutional review board or independent ethics committee before patients were enroled. The study was conducted in accordance with the ethical principles originating in the Declaration of Helsinki, and in compliance with International Conference on Harmonisation, Good Clinical Practices, and regulatory guidelines.
Patients
Women aged 18 years or older with histologically confirmed endometrial cancer who had recurrent or persistent disease and documented disease progression (within 3 months before entry) were eligible if they had at least one measurable lesion based on Response Evaluation Criteria in Solid Tumours (RECIST) guidelines (Eisenhauer et al, 2009); Eastern Cooperative Oncology Group performance status 0–2; minimum expected life expectancy of 3 months; and adequate renal, hepatic, and bone marrow function. Patients were to be excluded if they had received more than two prior regimens of cytotoxic chemotherapy or targeted therapy – patients could have undergone surgery and/or received prior radiotherapy and still be included in the study. Patients who had received any therapy with rapamycin or a rapamycin analogue were also excluded. Other exclusion criteria were the presence of brain metastases, anticancer treatment within 4 weeks before the first dose of ridaforolimus (⩾6 weeks for nitrosourea or mitomycin), ongoing toxicity associated with prior anticancer therapy, another primary malignancy within 3 years of the trial (except for non-melanoma skin cancer), uncontrolled cardiovascular disease, or active infection requiring systemic therapy. Pregnant or lactating women were also excluded.
Trial end points and assessments
The primary end point was the clinical benefit response (CBR), defined as a complete response or PR or prolonged SD lasting at least 16 weeks as assessed by modified the RECIST 1.0 criteria. Secondary end points of the trial included progression-free survival (PFS), safety, and tolerability. Time to disease progression, overall survival, and duration of response were planned secondary analyses but were not performed in the trial. Target and non-target lesions were assessed at baseline and after every two cycles (8 weeks) of ridaforolimus therapy. Patients received treatment with ridaforolimus until disease progression or other discontinuation criteria were met. Safety was assessed by routine physical and laboratory evaluations. Patients were monitored for adverse events (AEs) and disease progression throughout the study and at follow-up visits up to 24 months after the last dose of ridaforolimus. All study drug-related AEs were followed until resolution or until administration of another anticancer therapy.
Statistical analyses
This study used Simon’s optimal two-stage design, which was formulated to distinguish a favourable true CBR rate of ⩾35% from a null rate of ⩽15% with 90% power, at a significance level of 0.05. In total, 19 patients were to be enroled in the first stage. Further enrolment was to be discontinued if CBRs were observed in three or fewer of the first 19 patients. However, if four or more patients achieved CBRs, 25 additional patients were to be enroled. The regimen was to be considered effective if a CBR rate of 25% or greater was observed (i.e., 11 or more CBRs among 44 patients).
All patients receiving at least one dose of ridaforolimus comprised the intent-to-treat (ITT) population, and all were assessed for safety. The severity of AEs was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE, version 3.0). The frequency of AEs categorised by severity grades and changes in laboratory tests were evaluated using descriptive statistics.
Results
Patient baseline characteristics and disposition
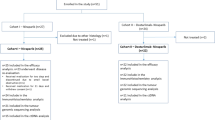
A total of 45 patients, with a median age of 66.7 (range: 28–89) years, were enroled in the trial and received at least one dose of ridaforolimus (Table 1). The majority of patients had endometrioid adenocarcinoma (64%); 22% had serous tumours, and 11% had carcinosarcomas. Most patients had metastatic disease in lymph nodes (56%) or in the lung (51%). The study population was heavily pretreated, with 26 patients (56%) having received two prior systemic regimens. In addition, a protocol violation occurred (as patients with two or more prior cytotoxic chemotherapy or targeted therapy regimens were excluded from the study): three patients (7%) received three or more prior systemic regimens. Ridaforolimus was administered for a median duration of 61 days (range: 1–425 days) and at a median cumulative dose of 250 mg (range: 12.5–1750 mg). All patients had discontinued ridaforolimus by the time of the analysis. In all, 33 patients (73%) were taken off this protocol owing to documented progressive disease, four patients (8%) were taken off this protocol owing to AEs (of which one was considered by the investigator to be related study treatment), four patients (8%) made a voluntary decision to stop treatment (i.e., withdrew consent), and the remaining four patients (8%) discontinued at the discretion of the investigator.
Efficacy
Of the 45 patients in the ITT population, 13 (29%) achieved CBR, including 5 (11%) with confirmed PR and 8 (18%) with SD lasting 16 weeks or longer (Table 2). Clinical benefit response was achieved in 10 of 29 patients (34%) with endometrioid tumours, in 2 of 10 patients (20%) with papillary serous carcinomas, and in the lone patient with mixed epithelial histology. None of the five patients with carcinosarcomas had a CBR. Overall, the 13 patients who achieved a CBR received ridaforolimus for more than 4 months (Figure 1). The efficacy of ridaforolimus is illustrated by the case of a 59-year-old woman diagnosed with papillary serous endometrial cancer with multiple metastases, who achieved a confirmed PR after four cycles of therapy (Figure 2). A waterfall plot of the best overall response from baseline for each patient is shown in Figure 3. The 6-month PFS was determined to be 18% following treatment with ridaforolimus.

Time on trial with ridaforolimus.

Patient aged 59 years with papillary serous endometrial cancer with multiple metastases treated with ridaforolimus. Baseline scan revealed a 25 × 23 mm mass in the right lung, which decreased to 14 × 13 mm after cycle 2 of therapy, and by cycle 4 was confirmed as a PR by RECIST guidelines.

Waterfall plot showing distribution of the best percentage change in target lesion size from baseline for an individual patient (n=35; not evaluable, n=10). The lines (–30 and +20%) indicate the region with change from baseline that typically represent SD based on RECIST guidelines.
Safety
All patients had at least one AE regardless of relationship to study treatment. The most common AEs were mouth sores (stomatitis and mucosal inflammation), anaemia, fatigue, diarrhoea, nausea, and vomiting (Table 3). Treatment-related AEs occurred in 42 patients (93%); the most common were mouth sores (56%), anaemia (42%), fatigue (40%), diarrhoea (31%), nausea (29%), vomiting (27%), asthenia (24%), and anorexia (22%). In total, 23 patients (51%) experienced serious AEs (SAEs), with seven patients (16%) having SAEs attributable, by the investigator, to treatment. These included three patients with anaemia (two with grade 3 and one with grade 2) and individual patients with deep vein thrombosis (grade 3), vomiting (grade 2), dehydration (grade 3), and stomatitis (grade 2). Each of these events was self-limiting except for one case of anaemia that led to clinical sequelae. A total of 15 patients (33%) experienced at least one treatment-related AE that led to a dose modification. One patient (2%) discontinued treatment with ridaforolimus owing to a treatment-related AE (grade 3 worsening of interstitial lung disease) and three additional patients (7%) discontinued treatment owing to AEs not attributed to study drug (grade 2 infection; grade 3 sepsis; and grade 3 mood alteration, respectively).
None of the patients died during treatment with ridaforolimus. In all, 24 patients (53%) died during the follow-up period after ridaforolimus was discontinued, including four patients (17%) who died within 30 days of receiving the last dose of ridaforolimus. All four of these deaths were due to progressive disease.
Discussion
Patients with endometrial cancers in the second-line setting or beyond have few effective treatment options. This phase 2 clinical trial demonstrates that single-agent ridaforolimus has antitumor activity in women with advanced endometrial cancer, most of whom had received two prior chemotherapy regimens. The study met its primary end point, as 29% of patients achieved a CBR (11% confirmed PR, 18% SD lasting 16 weeks or longer), thereby exceeding the Simon 2-stage criteria (i.e., CBR rate ⩾25%) established for efficacy. Ridaforolimus also showed an acceptable toxicity profile with the most common AEs (e.g., mouth sores, anaemia, and fatigue) being predictable, manageable, and consistent with those observed in previous trials with ridaforolimus and in studies with other mTOR inhibitors (Dancey and Monzon, 2011; Pilotte et al, 2011). One-third of the patients had treatment-related AEs that led to dose delay or reduction. Four patients were taken off protocol owing to AEs, but in only one case the AE was considered by the investigator to be treatment related.
Several other mTOR inhibitors have been investigated in women with advanced endometrial cancer. In a recently published phase 2 study among patients with recurrent endometrial carcinoma treated with a 10-mg oral daily dose of everolimus, 6 of 28 evaluable patients (21%) achieved a CBR; fatigue, anaemia, pain, lymphopenia, and nausea were reported as the most common AEs (Slomovitz et al, 2010). In another phase 2 trial, treatment with 25 mg intravenous temsirolimus administered weekly as monotherapy to patients with recurrent or metastatic endometrial cancer produced confirmed PRs in 4 of 29 chemotherapy-naïve evaluable patients (14%) and 1 of 25 evaluable patients (4%) who had been previously treated with chemotherapy (Oza et al, 2011a). The chemotherapy-naïve subset also had higher SD rates with longer median response duration than the chemotherapy-treated subset. Median PFS was 7.3 months in the chemotherapy-naïve group compared with 3.3 months in the chemotherapy-treated group. The most common AEs reported with temsirolimus were pneumonitis, mucositis, fatigue, gastrointestinal disorders, and pain.
The results of this study with ridaforolimus combined with the findings of the earlier phase 2 studies with everolimus and temsirolimus suggest that mTOR inhibitors have single-agent clinical benefit in advanced and recurrent endometrial cancer. Unfortunately, predictive factors have not yet been identified to select patients most likely to benefit from mTOR inhibitor therapy. For example, in a phase 2 trial with temsirolimus, loss of PTEN and other molecular markers of the PI3K/AKT/mTOR pathway did not appear to correlate with clinical outcomes (Oza et al, 2011a). Similarly, in a preclinical setting (i.e., fresh endometrial cancer tumour explants), PTEN and AKT status did not predict the antiproliferative effects of rapamycin (Bae-Jump et al, 2010). Predictive markers were not evaluated in this study owing to the limited availability of archival and biopsy tumour specimens in the study cohort.
The profile of mTOR inhibitors as monotherapy suggests that they may be amenable for use in combination with other active agents. In support of this approach, rapamycin showed synergistic effects in vitro in endometrial cancer cell lines when tested in combination with paclitaxel or cisplatin (Bae-Jump et al, 2009; Shafer et al, 2010). The clinical significance of these findings remains to be determined, along with careful evaluation of the tolerability of mTOR inhibitor–chemotherapy combinations. Temsirolimus was evaluated in combination with topotecan in women with advanced gynaecologic tumours, including endometrial cancer, in a phase 1 study; the combination was successful in treating a subset of patients without prior pelvic radiotherapy, but it was not tolerated in patients with prior pelvic radiotherapy (Temkin et al, 2010).
The results of this study indicate that ridaforolimus has antitumor activity in women with advanced or recurrent endometrial cancer. Studies with both intravenous (ridaforolimus and temsirolimus) and oral (everolimus) formulations of mTOR inhibitors have demonstrated clinical benefit in this patient population. No differences in treatment outcomes have been formally investigated between intravenous and oral formulations. Therefore, with similar clinical benefits and safety profiles observed across the mTOR studies, administration of an oral formulation would most likely be preferred by patients and their treating physicians. Preliminary results of a phase 2 trial testing the oral formulation of ridaforolimus, administered at 40 mg per day once daily for 5 days followed by a 2-day rest period, suggest that orally administered ridaforolimus is also effective in women with advanced endometrial cancer (Oza et al, 2011b). On the basis of these findings, mTOR inhibition with ridaforolimus is a potential therapeutic option in endometrial cancer and warrants further study as a single agent or in combination with other agents. Future studies evaluating predictive biomarkers will also provide critical data to help identify patients who may benefit from treatment with mTOR inhibitors.
Change history
19 March 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bae-Jump VL, Zhou C, Boggess JF, Gehrig PA (2009) Synergistic effect of rapamycin and cisplatin in endometrial cancer cells. Cancer 115: 3887–3896
Bae-Jump VL, Zhou C, Boggess JF, Whang YE, Barroilhet L, Gehrig PA (2010) Rapamycin inhibits cell proliferation in type I and type II endometrial carcinomas: a search for biomarkers of sensitivity to treatment. Gynecol Oncol 119: 579–585
Chawla SP, Staddon AP, Baker LH, Schuetze SM, Tolcher AW, D’Amato GZ, Blay JY, Mita MM, Sankhala KK, Berk L, Rivera VM, Clackson T, Loewy JW, Haluska FG, Demetri GD (2012) Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J Clin Oncol 30: 78–84
Choi CH, Lee JS, Kim SR, Kim TJ, Lee JW, Kim BG, Bae DS (2010) Clinical significance of pmTOR expression in endometrioid endometrial carcinoma. Eur J Obstet Gynecol Reprod Biol 153: 207–210
Daikoku T, Hirota Y, Tranguch S, Joshi AR, DeMayo FJ, Lydon JP, Ellenson LH, Dey SK (2008) Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res 68: 5619–5627
Dancey J (2010) mTOR signaling and drug development in cancer. Nat Rev Clin Oncol 7: 209–219
Dancey JE, Monzon J (2011) Ridaforolimus: a promising drug in the treatment of soft-tissue sarcoma and other malignancies. Fut Oncol 7: 827–839
Dedes KJ, Wetterskog D, Ashworth A, Kaye SB, Reis-Filho JS (2011) Emerging therapeutic targets in endometrial cancer. Nat Rev Clin Oncol 8: 261–271
Dellinger TH, Monk BJ (2009) Systemic therapy for recurrent endometrial cancer: a review of North American trials. Expert Rev Anticancer Ther 9: 905–916
Dizon DS (2010) Treatment options for advanced endometrial carcinoma. Gynecol Oncol 117: 373–381
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New Response Evaluation Criteria in Solid Tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45: 228–247
Fleming GF (2007) Systemic chemotherapy for uterine carcinoma: metastatic and adjuvant. J Clin Oncol 25: 2983–2990
Hayes MP, Wang H, Espinal-Witter R, Douglas W, Solomon GJ, Baker SJ, Ellenson LH (2006) PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin Cancer Res 12: 5932–5935
Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, Naing A, Falchook GS, Moroney JW, Piha-Paul SA, Wheler JJ, Moulder SL, Fu S, Kurzrock R (2011) PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther 10: 558–565
Memarzadeh S, Zong Y, Janzen DM, Goldstein AS, Cheng D, Kurita T, Schafenacker AM, Huang J, Witte ON (2010) Cell-autonomous activation of the PI3-kinase pathway initiates endometrial cancer from adult uterine epithelium. Proc Natl Acad Sci USA 107: 17298–17303
Mita MM, Mita AC, Chu QS, Rowinsky EK, Fetterly GJ, Goldston M, Patnaik A, Mathews L, Ricart AD, Mays T, Knowles H, Rivera VM, Kreisberg J, Bedrosian CL, Tolcher AW (2008) Phase I trial of the novel mammalian target of rapamycin inhibitor deforolimus (AP23573; MK-8669) administered intravenously daily for 5 days every 2 weeks to patients with advanced malignancies. J Clin Oncol 26: 361–367
Oza AM, Elit L, Tsao MS, Kamel-Reid S, Biagi J, Provencher DM, Gotlieb WH, Hoskins PJ, Ghatage P, Tonkin KS, Mackay HJ, Mazurka J, Sederias J, Ivy P, Dancey JE, Eisenhauer EA (2011a) Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: a trial of the NCIC Clinical Trials Group. J Clin Oncol 29: 3278–3285
Oza AM, Poveda A, Del Campo JM, Richard A, Dorer DJ, Haluska F (2011b) A randomized phase II trial of ridaforolimus compared with progestin or chemotheraphy in female adult patients with advanced endometrial carcinoma. Presented at the 47th Annual Meeting of the American Society of Clinical Oncology. Chicago, IL, USA, 3–7 June
Pectasides D, Pectasides E, Economopoulos T (2007) Systemic therapy in metastatic or recurrent endometrial cancer. Cancer Treat Rev 33: 177–190
Pilotte AP, Hohos MB, Polson KM, Huftalen TM, Treister N (2011) Managing stomatitis in patients treated with mammalian target of rapamycin inhibitors. Clin J Oncol Nurs 15: E83–E89
Rizzieri DA, Feldman E, Dipersio JF, Gabrail N, Stock W, Strair R, Rivera VM, Albitar M, Bedrosian CL, Giles FJ (2008) A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 14: 2756–2762
Salvesen HB, Stefansson I, Kretzschmar EI, Gruber P, MacDonald ND, Ryan A, Jacobs IJ, Akslen LA, Das S (2004) Significance of PTEN alterations in endometrial carcinoma: a population-based study of mutations, promoter methylation and PTEN protein expression. Int J Oncol 25: 1615–1623
Seki Y, Yamamoto N, Tamura Y, Goto Y, Shibata T, Tanioka M, Asahina H, Nokihara H, Yamada Y, Shimamoto T, Noguchi K, Tamura T (2012) Phase I study for ridaforolimus, an oral mTOR inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 69: 1099–1105
Shafer A, Zhou C, Gehrig PA, Boggess JF, Bae-Jump VL (2010) Rapamycin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and induction of apoptosis. Int J Cancer 126: 1144–1154
Shorning BY, Griffiths D, Clarke AR (2011) Lkb1 and Pten synergise to suppress mTOR-mediated tumorigenesis and epithelial-mesenchymal transition in the mouse bladder. PLoS One 6: e16209
Siegel R, Ward E, Brawley O, Jemal A (2011) Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 61: 212–236
Slomovitz BM, Lu KH, Johnston T, Coleman RL, Munsell M, Broaddus RR, Walker C, Ramondetta LM, Burke TW, Gershenson DM, Wolf J (2010) A phase 2 study of the oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer 116: 5415–5419
Sorosky JI (2008) Endometrial cancer. Obstet Gynecol 111: 436–447
Squillace RM, Miller D, Cookson M, Wardwell SD, Moran L, Clapham D, Wang F, Clackson T, Rivera VM (2011) Antitumor activity of ridaforolimus and potential cell-cycle determinants of sensitivity in sarcoma and endometrial cancer models. Mol Cancer Ther 10: 1959–1968
Temkin SM, Yamada SD, Fleming GF (2010) A phase I study of weekly temsirolimus and topotecan in the treatment of advanced and/or recurrent gynaecologic malignancies. Gynecol Oncol 117: 473–476
Acknowledgements
Medical writing and editorial assistance was provided by Joseph J Abrajano, PhD, and Kakuri M Omari, PhD, of Integrus Scientific, a division of Medicus International New York (New York, NY, USA). This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. (Whitehouse Station, NJ, USA). The authors were fully responsible for all content and editorial decisions and received no financial support or other compensation related to the development of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Colombo, N., McMeekin, D., Schwartz, P. et al. Ridaforolimus as a single agent in advanced endometrial cancer: results of a single-arm, phase 2 trial. Br J Cancer 108, 1021–1026 (2013). https://doi.org/10.1038/bjc.2013.59
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.59
Keywords
This article is cited by
-
Managing Adverse Effects of Novel Therapeutic Agents in Gynecologic Malignancies
SN Comprehensive Clinical Medicine (2023)
-
Metabolic reprogramming in macrophage responses
Biomarker Research (2021)
-
Targeted therapies in gynecological cancers: a comprehensive review of clinical evidence
Signal Transduction and Targeted Therapy (2020)
-
Targeting mTOR for cancer therapy
Journal of Hematology & Oncology (2019)
-
Pharmacogenomic analysis of patient-derived tumor cells in gynecologic cancers
Genome Biology (2019)
