
Abstract
Background:
This study aimed to determine whether combination S-1 plus cisplatin (CDDP) therapy, the most widely used therapy for Japanese patients with advanced gastric cancer, and the novel oral antiangiogenic agent TSU-68 could contribute to gastric cancer treatment.
Methods:
Ninety-three patients with chemotherapy-naïve unresectable or recurrent advanced gastric cancers were randomised into two groups: TSU-68 plus S-1/CDDP (group A) and S-1/CDDP (group B) groups. Both patient groups received identical S-1 and CDDP dosages. TSU-68 was orally administered for 35 consecutive days. Group B patients received S-1 orally twice daily for three consecutive weeks, followed by intravenous CDDP on day 8. The primary endpoint was progression-free survival (PFS).
Results:
Median PFS periods were 208 and 213 days in groups A and B, respectively (P=0.427). Median survival periods for groups A and B were 497.0 and 463.5 days, respectively (P=0.219). No statistically significant differences were noted for PFS, survival or the adverse event (AE) incidence rate. All AEs were expected according to previous reports for TSU-68, TS-1, and CDDP.
Conclusion:
Combination therapy involving TSU-68, S-1, and CDDP was safe and well tolerated in patients with chemotherapy-naïve unresectable or recurrent advanced gastric cancers. However, factors related to therapeutic efficacy should be investigated further.
Similar content being viewed by others


Main
Gastric cancer is the second most common cause of cancer death both worldwide (Jemal et al, 2011) and in Japan (Sobue et al, 2012).
Since Macdonald et al (1980) reported the use of 5-fluorouracil (5-FU), doxorubicin, and mitomycin-C combination chemotherapy (median survival time, 5.5 months) for the treatment of unresectable, advanced, or recurrent gastric cancers in 1980, multidrug chemotherapies, particularly those that include 5-FU, have been the most widely used therapies worldwide. Currently, the employed regimens differ among geographic regions. For example, epirubicin, cisplatin (CDDP), and 5-FU; epirubicin, oxaliplatin, and capecitabine (EOX); and docetaxel, CDDP, and 5-FU chemotherapies are primarily used in the control arms of clinical studies in Western countries, whereas 5-FU and CDDP chemotherapy is primarily used in non-Western countries. Thus, no global consensus has been reached on a standard therapy.
In Japan, the clinical development of chemotherapies for unresectable, advanced, or recurrent gastric cancers has progressed for many years, and many clinical studies have been conducted using TS-1 (S-1), a fluoropyrimidine anticancer drug that is produced in Japan. When compared with continuous intravenous 5-FU infusion, 5-FU/CDDP did not significantly increase life expectancy (Ohtsu et al, 2003); since then, 5-FU alone has been used as a reference arm. Nevertheless, the American Society of Clinical Oncology reported the results from two Japanese phase III clinical studies (Japan Clinical Oncology Group (JCOG) 9912 (Boku et al, 2009) and S-1 Plus cisplatin vs S-1 in RCT in the Treatment for Stomach cancer (SPIRITS) (Koizumi et al, 2008)) in 2007. Japan Clinical Oncology Group 9912 demonstrated that S-1 capsule monotherapy was not inferior to continuous intravenous 5-FU infusion in terms of overall survival (OS). In addition, the SPIRITS trial reported a significantly prolonged OS with S-1/CDDP therapy and a better (prolonged by >1 year) OS than that with S-1 alone. Therefore, a first-line standard chemotherapy was established in Japan.
The median survival period achieved in the SPIRITS trial was 13.0 months; therefore, further improvements to the therapeutic results are necessary. In recent years, the use of a fluoropyrimidine anticancer drug in combination with molecular targeted agents has been studied, and vascular endothelial growth factor (VEGF) is assumed to be closely related to tumour proliferation in gastric cancers (Laird et al, 2000). The use of bevacizumab, a monoclonal antibody that targets VEGF A, was evaluated in combination with capecitabine and cisplatin as a first-line therapy for advanced gastric cancer (Ohtsu et al, 2011). In that study, the progression-free survival (PFS) and overall response rates (ORRs) were significantly improved with bevacizumab; however, no survival benefit related to this drug was noted. On the other hand, ramucirumab, a monoclonal antibody that targets VEGF receptor 2, significantly prolonged OS when used as a second-line monotherapy for advanced gastric or gastroesophageal junction adenocarcinoma (Fuchs et al, 2013).
TSU-68 (orantinib) is a novel oral antiangiogenic agent that has been shown to inhibit the tyrosine phosphorylation of VEGF receptor 2, platelet-derived growth factor (PDGF) receptor 6, and fibroblast growth factor (FGF) receptor 1 in vitro (Kim et al, 2009). Previously, phase I and phase II studies in patients with breast cancer, hepatocellular carcinoma (HCC), lung cancer, and colorectal cancer were conducted in Asia (Kanai et al, 2010; Okamoto et al, 2012; Shin et al, 2012; Toi et al, 2012; Inaba et al, 2013), and a phase III study was initiated in 2010 to evaluate the survival benefit of TSU-68 in patients with intermediate-stage HCC (ClinicalTrials.gov Identifier: NCT01465464). As part of the clinical development of TSU-68, a combination of S-1/CDDP therapy, the most widely used therapy in Japan for patients with advanced gastric cancers, and TSU-68, which has antiangiogenic effects, was expected to be an effective gastric cancer treatment. Consequently, we conducted a phase II randomised study to compare the effects of a combination therapy with 3 agents—TSU-68, S-1, and CDDP—with the effects of S-1/CDDP therapy with regard to the PFS to improve the therapeutic results of first-line standard chemotherapies.
Materials and methods
Patients
The patients included in the study were ⩾20 years with (1) histologically or cytologically confirmed adenocarcinoma, (2) unresectable or recurrent gastric cancer, and (3) no prior systemic treatment. Recurrent patients were eligible if the last dose of postoperative adjuvant chemotherapy had been received at least 180 days before the start of the study. Other eligibility criteria included an Eastern Cooperative Oncology Group performance status of 0–1 and adequate functioning of the major organs, along with the following laboratory values: haemoglobin, ⩾8.0 g dl−1, neutrophil count, ⩾1500 mm−3, platelet count, ⩾100 000 mm−3, serum creatinine, ⩽the reference value at the study center, and serum bilirubin (TBIL), ⩽1.5 mg dl−1. Other laboratory criteria included a creatinine clearance of ⩾60 ml min−1, serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels of ⩽100 U l−1, and an alkaline phosphatase (ALP) level that was 2.5-fold less than the reference value at the study center. For patients with liver metastases, those with AST, ALT, and ALP values that were 5-fold less than the reference values at the study center were eligible. In addition, patients were required to have target tumours that were measurable by computed tomography, magnetic resonance imaging, or radiography in accordance with the Response Evaluation Criteria in Solid Tumours (RECIST), ver. 1.0. All patients were required to provide written consent. This study was implemented in accordance with Good Clinical Practice (GCP) guidelines and the Declaration of Helsinki.
Design
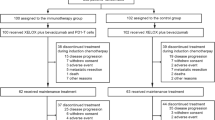
This was a phase II, multicenter, randomised, controlled study to estimate the efficacy of TSU-68 plus S-1/CDDP therapy vs S-1/CDDP therapy. Randomisation was performed according to the minimisation method, using ‘unresectable gastric cancer’, ‘recurrent gastric cancer with postoperative adjuvant chemotherapy’, and ‘recurrent gastric cancer without postoperative adjuvant chemotherapy’ as the stratification factors. Eligible patients were randomly assigned to either the TSU-68 plus S-1/CDDP (group A) or the S-1/CDDP (group B) groups at a ratio of 1 : 1 (Figure 1).

Study design. Two patients were excluded from the full analysis set by an independent data monitoring committee.
In groups A and B, S-1 was administered at a dose of <40 mg m−2. The S-1 dose was calculated according to the patient’s body surface area as follows: <1.25 m2, 40 mg; 1.25–1.5 m2, 50 mg; and >1.5 m2, 60 mg. S-1 was orally administered twice daily for three consecutive weeks. CDDP was administered at a dose of 60 mg m−2 by intravenous infusion on day 8. The duration of each cycle was 5 weeks (35 days). In group A, 400 mg of TSU-68 was orally administered twice daily (total daily dosage, 800 mg) for five consecutive weeks. The treatments were continued until 1 of the following occurred: progressive disease (PD), unacceptable toxicity, withdrawal of patient consent (regardless of toxicity), or termination of treatment at the discretion of the attending physician.
The protocol was approved by the Institutional Review Board of each study center. The Independent Data Review Committee evaluated safety throughout the study period. This study was conducted in compliance with the Declaration of Helsinki and the Japanese GCP Guidelines.
Endpoints and evaluation methods
The primary endpoint was PFS, which was defined as the period from the day of enrolment to the day on which (1) radiological or clinical progression was evident, (2) subsequent treatment was indicated, or (3) the patient died. The earliest day among these defined days was considered. If patients were lost to follow-up because of second-line treatment or a transfer to another hospital, their data were censored. Tumours were measured every 5 weeks until the onset of PD. All measured images were assessed by a Central Imaging Review Committee in accordance with the RECIST (New Guideline 2000; Therasse et al, 2000).
Secondary endpoints were the antitumour effect (ORR), OS, safety, pharmacokinetics (PK), and the relationship between angiogenesis-related factors and efficacy. To determine safety, blood tests, biochemical analyses, and urinalyses were performed and subjective as well as objective findings were followed-up throughout the study period. Adverse events (AEs) were graded in accordance with the National Cancer Institute Common Toxicity Criteria ver. 3.0.
In the patients who were included in the PK evaluation on day 8, the PK of TSU-68 after repeated administration of TSU-68 (400 mg per dose) on day 8, the PK of tegafur (FT), 5-FU, 5-chloro-2,4-dihydroxypyridine (CDHP), and potassium oxonate (Oxo) after the repeated administration of S-1 (50–60 mg per dose), and the PK of the total and free platinum levels after the administration of CDDP (60 mg m−2) were investigated.
Angiogenesis-related factors were tested at baseline levels and before the start of the next cycle. The following angiogenesis-related factors were measured: PDGF-AA, PDGF-BB, soluble vascular cell adhesion molecule-1, soluble endothelial-leukocyte adhesion molecule-1 in the serum and plasma, and interleukin-8 with enzyme-linked immunosorbent assays (ELISAs; BioSource Europe, Nivelles, Belgium); plasma tissue plasminogen activator (t-PA) with a soluble t-PA ELISA kit (Oncogene Science, Cambridge, MA, USA); and plasma plasminogen activator inhibitor-1, acidic FGF, VEGF, VEGF soluble receptor type 2, hepatocyte growth factor, VEGF-C, VEGF soluble receptor type 3, and the lactate dehydrogenase isozyme.
Statistical analyses
The SPIRITS trial that was conducted in Japan showed that the median PFS achieved with S-1/CDDP was 6 months. According to this result, the PFS with TSU-68+S-1/CDDP was estimated to be 9 months. This would have a significant clinical impact on systemic therapy for advanced gastric cancer. We assumed that a total of 86 patients (two groups) would be necessary to demonstrate the superiority of TSU-68+S-1/CDDP at a power of 80% and a one-sided significance level of 20% with unstratified log-rank tests at the end of the follow-up period (Rubenstein et al, 2005). After considering possibilities such as ineligible patients, we determined that 92 patients were required for the study.
We used a full analysis set (FAS), defined as patients who met the eligibility criteria, for the primary analyses of efficacy and safety.
To compare the PK parameters of S-1 and CDDP between groups A and B, the Wilcoxon test was performed for the maximum drug concentration time (tmax), and the Student’s t-test or Aspin–Welch test was performed for parameters other than the tmax after logarithmic transformation.
Results
Patient background
Between December 2008 and February 2012, a total of 93 patients (group A, n=46; group B, n=47) from a total of 14 centres in Japan were enroled and randomised in this study (Figure 2). One patient from group A was found to be ineligible, and 1 patient from group B did not receive treatment. Therefore, a total of 91 patients (group A, n=45 and group B, n=46) were included in the FAS that was used for efficacy and safety analyses. There were no significant imbalances in the patient background characteristics at enrolment between the two groups (Table 1). The percentages of patients with 1, 2, or ⩾3 organs with infiltration and/or metastasis were 46.7%, 40%, and 13.3%, respectively, in group A and 41.3%, 50%, and 8.7%, respectively, in group B. None of the patients had locally advanced disease alone. Peritoneal metastases were noted in 15 (33.3%) group A patients and 15 (32.6%) group B patients. Histologically, diffuse-type and intestinal-type adenocarcinomas were noted in 23 (48.9%) and 22 (51.1%) group A patients, respectively, and in 20 (54.3%) and 25 (43.5%) group B patients, respectively. Gastrectomies had been performed in 6 (19.6%) group A patients and in 9 (13.3%) group B patients before enrolment. Postoperative adjuvant chemotherapy was administered to 4 (10.9%) group A patients and 5 (8.9%) group B patients.

CONSORT diagram. A total of 93 patients (group A, n=46; group B, n=47) were randomised. One patient from group A did not receive treatment, and 1 patient from group B was ineligible. Therefore, a total of 91 patients (group A, n=45; group B, n=46) were included in the FAS used for the efficacy and safety analyses.
Efficacy
Progression-free survival
The median PFS were not significantly different between the two groups (group A, 208.0 days; group B, 213 days; P=0.424; Figure 3).

Kaplan–Meier analysis of PFS and OS. Of the 93 total patients, 46 were placed into group A and 47 were placed into group B. The median PFS times of the patients in group A and group B were 208.0 days (95% CI: 141.0–274.0 days) and 213.0 days (95% CI: 178.0–309.0 days), respectively. The HR for radiological progression or death in group A was 1.23 (95% CI: 0.74–2.05). The median OS times of the patients in group A and group B were 497.0 days (95% CI: 371.0–635.0 days) and 463.5 days (95% CI: 359.0–554.0 days), respectively. The HR for death in group A was 0.74 (95% CI: 0.46–1.19).
The hazard ratio (HR) was 1.23 (95% confidence interval (CI): 0.74–2.05).
Survival
All follow-up investigations were completed at the time of data cutoff in April 2012, which was 1 year and 8 months after the last patient enrolment. Outcomes were confirmed in all patients (100%). Of the 91 patients in the FAS, 33 of the 45 (73.3%) group A patients and 38 of the 46 (82.6%) group B patients died. The median OS periods were 497.0 days in group A and 463.5 days in group B. The 1-year survival rates were 66.7% in group A and 63.0% in group B. The 2-year survival rate was 30.4% in group A and 22.4% in group B. The survival rates in group A were not significantly different from those in group B (P=0.213) (Figure 3).
The HR was 0.74 (95% CI: 0.46–1.19).
Best overall response
Twenty-eight of the 45 group A patients achieved a partial response (PR), and thus the response rate was 62.2% (95% CI: 46.5–76.2%). Twenty-six of the 46 group B patients achieved a PR, and thus the response rate was 56.5% (95% CI: 41.1–71.1%). The response rate in group A was not significantly different from that in group B (P=0.671).
Safety
The AEs that occurred in this study are shown in Table 2. The main AEs that occurred at least 10% more frequently in group A than in group B were as follows: changes in the ALT, AST, and ALP levels, vomiting, diarrhoea, pigmentation abdominal pain, oedema, and urine colour change. The main AEs that occurred at least 10% less frequently in group A than in group B were as follows: neutropenia, changes in the leukocyte, TBIL, and creatinine levels, and stomatitis. The incidence rates of Grade 3 or higher AEs were the same in both groups; however, anorexia and changes in the haemoglobin and platelet levels occurred more frequently in group A than in group B. Specific changes observed in group A and group B patients were as follows: haemoglobin, 48.9% and 26.1%, respectively; platelet, 24.4% and 6.5%, respectively; anorexia, 17.8% and 8.5%, respectively.
In addition, no treatment-related deaths were noted in either group. Only 1 of the 46 patients (2.2%) in group B died within 90 days after enrolment, while only 2 (4.4%) died of aspiration and hypoxia during the study period.
Treatment continuity
The mean actual dose intensity of each drug in groups and B was as follows: S-1, 80.3% and 83.0%, respectively; CDDP, 89.6% and 92.0%, respectively; and TSU-68, 72.9% in group A. The median relative dose intensity (RDI) for S-1 was 85.6% in group A and 88.7% in group B. The median RDI for CDDP was 92.5% in group A and 92.9% in group B. Reasons for treatment discontinuation in groups A and B were as follows: PD, 69.6% and 78.7%, respectively; AEs (mainly bone marrow depression), 15.2% and 6.4%, respectively; withdrawal of consent, 4.3% and 4.3%, respectively; and indications for surgery, 10.9% and 4.3%, respectively. A total of 97.8% and 91.2% of patients in groups A and B, respectively, received second-line chemotherapy. At the end of the study, CPT-11-containing regimens were given to 37.8% and 42.9% of the patients in groups A and B, respectively, and taxane-containing regimens were given to 26.7% and 28.9% of the patients in groups A and B, respectively.
Subgroup analyses
Subgroup analyses of the patient backgrounds revealed no prolongation of PFS in any of the subgroups (Figure 4). In addition, neither the baseline nor the post-treatment measurements of the angiogenesis-related factors correlated with efficacy (data not shown).

Forest plot for PFS. No prolongation of PFS was observed in any of the subgroups.
Pharmacokinetics
The pharmacokinetic parameters of TSU-68, S-1, and CDDP are shown in Table 3.
The mean maximum drug concentration (Cmax) and the area under the curve of the plasma concentration vs time from 0 to the final time point (AUC0–last) for TSU-68 were 4.46 μg ml−1 and 23.23 μg h−1 ml−1, respectively. These values were not significantly different from the previously reported results for TSU-68 monotherapies and combination therapies (Kanai et al, 2010; Murakami et al, 2011; Ueda et al, 2011; Okamoto et al, 2012; Toi et al, 2012).
For S-1, the Cmax and AUC0–last of the FT were significantly lower in group A than in group B, and the half-life (t1/2) was significantly shorter in group A than in group B. However, no significant difference was noted between the two groups with regard to the Cmax or the AUC0–last of 5-FU. The AUC0–last of CDHP and Oxo were significantly lower in group A than in group B.
For CDDP, the Cmax and the AUC0–last of free platinum were significantly lower in group A than in group B.
Discussion
The median PFS was 208.0 days (95% CI: 141.0–274.0 days) in group A and 213.0 days (95% CI: 178.0–309.0 days) in group B.
According to the Central Imaging Review Committee, none of the patients in either group achieved a complete response. A total of 28 patients in group A and 26 patients in group B achieved a PR. The response rate was 62.2% (95% CI: 46.5–76.2%) in group A and 56.5% (95% CI: 41.1–71.1%) in group B. No additional TSU-68 effect was demonstrated.
The median survival period was 497.0 days (95% CI: 371.0–635.0 days) in group A and 463.5 days (95% CI: 359.0–554.0 days) in group B. Beyond the median point, differences in the survival curve indicated that a small number of patients in group A tended to have prolonged survival; however, per the stratified analyses, no correlation with efficacy was observed.
No statistically significant differences were noted for any of the endpoints, which included PFS, response rate, and survival.
With regard to the safety profile, no significant difference was observed in the AE incident rates between the groups, except for changes in ALP levels, oedema, and urine colour change (Table 2). Although the incidence of changes in the ALP levels tended to be higher in group A than in group B (44.4% and 21.7%, respectively), most of these patients with ALP level alterations had Grade 1 or Grade 2 AEs. OEdema and urine colour change are typical AEs of TSU-68, and almost of them were not severe and controlled enough. All AEs were expected according to previous reports on AEs for TSU-68, TS-1, and CDDP. The addition of TSU-68 to TS-1 plus CDDP, a standard therapy, is unlikely to induce serious or fatal events.
On the other hand, although the evaluation of the quality of life (QOL) was recently determined to be important in the evaluation of tolerability, we did not collect data on the QOL in the present study.
From the results of the TSU-68 PK profile in group A, S-1 and CDDP are unlikely to influence the PK of TSU-68. The induction of FT metabolism by TSU-68 could be a reason for the decreased AUC of FT in group A, as CYP1A2 has been reported to have a minor role in the metabolism of FT to 5-FU (Komatsu et al, 2000), and TSU-68 has the potential to induce CYP1A2 (Kitamura et al, 2008). The effects of TSU-68 on plasma exposure to CDHP and Oxo cannot be denied; however, TSU-68 had no effect on plasma exposure to 5-FU, the active ingredient of S-1. Therefore, combination therapy with TSU-68 was unlikely to affect the efficacy or safety of S-1. In the CDDP PK analysis, the plasma exposure to free platinum significantly decreased when TSU-68 was administered in combination with S-1/CDDP, but the degree of this decrease was not remarkable (∼20%). The effect of this slight decrease in platinum exposure on the efficacy and safety of CDDP is unknown. Therefore, further studies are required to investigate the interaction between TSU-68 and CDDP.
Molecular target therapies are increasingly being developed for the treatment of gastric cancer. Trastuzumab was found to induce a substantial increase in OS in HER-2-positive patients with metastatic gastric cancer when combined with chemotherapy (Bang et al, 2010). The antiangiogenic agent bevacizumab, in combination with capecitabine and cisplatin as a first-line therapy, significantly improved the PFS rate and ORR; however, no survival benefit related to this drug was noted (Ohtsu et al, 2011). Ramucirumab significantly prolonged OS when used as a second-line monotherapy (Fuchs et al, 2013). An understanding of past studies of molecular target agents is necessary for appropriate patient selection.
Taken together, our results show that a combination therapy that comprised TSU-68, TS-1, and CDDP was safe and well tolerated in patients with unresectable or recurrent gastric cancers. However, TSU-68 did not demonstrate the expected enhanced efficacy. Further studies to explore all aspects that affect efficacy are necessary.
References
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, Aprile G, Kulikov E, Hill J, Lehle M, Ruschoff J, Kang YK (2010) Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376: 687–697.
Boku N, Yamamoto S, Fukuda H, Shirao K, Doi T, Sawaki A, Koizumi W, Saito H, Yamaguchi K, Takiuchi H, Nasu J, Ohtsu A Gastrointestinal Oncology Study Group of the Japan Clinical Oncology Group (2009) Fluorouracil versus combination of irinotecan plus cisplatin versus S-1 in metastatic gastric cancer: a randomised phase 3 study. Lancet Oncol 10: 1063–1069.
Fuchs CS, Tomasek J, Cho JY, Dumitru F, Passalacqua R, Goswami C, Safran H, Dos Santos LV, Aprile G, Ferry DR, Melichar B, Tehfe M, Topuzov E, Tabernero J, Zalcberg JR, Chau I, Koshiji M, Hsu Y, Schwartz JD, Ajani JA (2013) REGARD: A phase III, randomized, double-blinded trial of ramucirumab and best supportive care (BSC) versus placebo and BSC in the treatment of metastatic gastric or gastroesophageal junction (GEJ) adenocarcinoma following disease progression on first-line platinum- and/or fluoropyrimidine-containing combination therapy. J Clin Oncol 31 (4_suppl): LBA5.
Inaba F, Kanai F, Aramaki T, Yamamoto T, Tanaka T, Yamakado K, Kaneko S, Kudo M, Imanaka K, Kora S, Nishida N, Kawai N, Seki H, Matsui O, Arioka H, Arai Y (2013) A randomised phase II study of TSU-68 in patients with hepatocellular carcinoma treated by transarterial chemoembolization. Eur J Cancer 49: 2832–2840.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61: 69–90.
Kanai F, Yoshida H, Tateishi R, Sato S, Kawabe T, Obi S, Kondo Y, Taniguchi M, Tagawa K, Ikeda M, Morizane C, Okusaka T, Arioka H, Shiina S, Omata M (2010) A phase I/II trial of the oral antiangiogenic agent TSU-68 in patients with advanced hepatocellular carcinoma. Cancer Chemother Pharmacol 67: 315–324.
Kim SE, Shim KN, Jung SA, Yoo K, Lee JH (2009) The clinicopathological significance of tissue levels of hypoxia-inducible factor-1alpha and vascular endothelial growth factor in gastric cancer. Gut Liver 3: 88–94.
Kitamura R, Asanoma H, Nagayama S, Otagiri M (2008) Identification of human liver cytochrome P450 isoforms involved in autoinduced metabolism of the antiangiogenic agent (Z)-5-[(1,2-dihydro-2-oxo-3H-indol-3-ylidene)methyl]-2,4-dimethyl-1H-pyrrole-3-propanoic acid (TSU-68). Drug Metab Dispos 36: 1003–1009.
Koizumi W, Narahara H, Hara T, Takagane A, Akiya T, Takagi M, Miyashita K, Nishizaki T, Kobayashi O, Takiyama W, Toh Y, Nagaie T, Takagi S, Yamamura Y, Yanaoka K, Orita H, Takeuchi M (2008) S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol 9: 215–221.
Komatsu T, Yamazaki H, Shimada N, Nakajima M, Yokoi T (2000) Roles of cytochromes P450 1A2, 2A6, and 2C8 in 5-fluorouracil formation from tegafur, an anticancer prodrug, in human liver microsomes. Drug Metab Dispos 28: 1457–1463.
Laird AD, Vajkoczy P, Shawver LK, Thurnher A, Liang C, Mohammadi M, Schlessinger J, Ullrich A, Hubbard SR, Blake RA, Fong TA, Strawn LM, Sun L, Tang C, Hawtin R, Tang F, Shenoy N, Hirth KP, McMahon G, Cherrington (2000) SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res 60: 4152–4160.
Macdonald JS, Schein PS, Woolley PV, Smythe T, Ueno W, Hoth D, Smith F, Boiron M, Gisselbrecht C, Brunet R, Lagarde C (1980) 5-Fluorouracil, doxorubicin, and mitomycin (FAM) combination chemotherapy for advanced gastric cancer. Ann Intern Med 93: 533–536.
Murakami H, Ueda Y, Shimoyama T, Yamamoto N, Yamada Y, Arioka H, Tamura T (2011) Phase I, pharmacokinetic, and biological studies of TSU-68, a novel multiple receptor tyrosine kinase inhibitor, administered after meals with solid tumors. Cancer Chemother Pharmacol 67: 1119–1128.
Ohtsu A, Shah MA, Van Cutsem E, Rha SY, Sawaki A, Park SR, Lim HY, Yamada Y, Wu J, Langer B, Starnawski M, Kang YK (2011) Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol 29: 3968–3976.
Ohtsu A, Shimada Y, Shirao K, Boku N, Hyodo I, Saito H, Yamamichi N, Miyata Y, Ikeda N, Yamamoto S, Fukuda H, Yoshida S Japan Clinical Oncology Group Study (JCOG9205) (2003) Randomized phase III trial of fluorouracil alone versus fluorouracil plus cisplatin versus uracil and tegafur plus mitomycin in patients with unresectable, advanced gastric cancer: The Japan Clinical Oncology Group Study (JCOG9205). J Clin Oncol 21: 54–59.
Okamoto I, Yoshioka H, Takeda K, Satouchi M, Yamamoto N, Seto T, Kasahara K, Miyazaki M, Kitamura R, Ohyama A, Hokoda N, Nakayama H, Yoshihara E, Nakagawa K (2012) Phase I clinical study of the angiogenesis inhibitor TSU-68 combined with carboplatin and paclitaxel in chemotherapy-naive patients with advanced non-small cell lung cancer. J Thorac Oncol 7: 427–433.
Rubinstein LV, Korn EL, Freidlin B, Hunsberger S, Ivy SP, Smith MA (2005) Design issues of randomized phase II trials and a proposal for phase II screening trials. J Clin Oncol 23: 7199–7206.
Shin SJ, Jung M, Jeung HC, Kim HR, Rha SY, Roh JK, Chung HC, Ahn JB (2012) A phase I pharmacokinetic study of TSU-68 (a multiple tyrosine kinase inhibitor of VEGFR-2, FGF and PDFG) in combination with S-1 and oxaliplatin in metastatic colorectal cancer patients previously treated with chemotherapy. Invest New Drugs 30: 1501–1510.
Sobue T, Katanoda K, Ajiki W, Tsukuma H, Ioka A (2012) Gan Tokei Hakusho 2012. pp 1–14. Shinoharashinsha Publishers: Tokyo, Japan.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92: 205–216.
Toi M, Saeki T, Iwata H, Inoue K, Tokuda Y, Sato Y, Ito Y, Aogi K, Takatsuka Y, Arioka H (2012) A multicenter phase II study of TSU-68, an oral multiple tyrosine kinase inhibitor, in combination with docetaxel in metastatic breast cancer patients with anthracycline resistance. Breast Cancer ; e-pub ahead of print 2 March 2012; doi:10.1007/s12282-012-0344-3.
Ueda Y, Shimoyama T, Murakami H, Yamamoto N, Yamada Y, Arioka H, Tamura T (2011) Phase I and pharmacokinetic study of TSU-68, a novel multiple receptor tyrosine kinase inhibitor, by twice daily oral administration between meals in patients with advanced solid tumors. Cancer Chemother Pharmacol 67: 1101–1109.
Acknowledgements
We are grateful to Dr Junji Tanaka and Dr Atsushi Sato, who served as the Central Imaging Review Committee. We are also grateful to Yutaka Ariyoshi, Tomohide Tamura, and Yuh Sakata who served as an Independent Data Review Committee. This trial was supported by Taiho Pharmaceutical Co., Ltd.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This study is registered with JAPIC Clinical Trial Information (JapicCTI- 101327).
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Koizumi, W., Yamaguchi, K., Hosaka, H. et al. Randomised phase II study of S-1/cisplatin plus TSU-68 vs S-1/cisplatin in patients with advanced gastric cancer. Br J Cancer 109, 2079–2086 (2013). https://doi.org/10.1038/bjc.2013.555
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.555
Keywords
This article is cited by
-
Comparative efficacy and tolerability of targeted and immunotherapy combined with chemotherapy as first-line treatment for advanced gastric cancer: a Bayesian network meta-analysis
Scientific Reports (2022)
-
Efficacy and safety for Apatinib treatment in advanced gastric cancer: a real world study
Scientific Reports (2017)
-
Efficacy and safety of angiogenesis inhibitors in advanced gastric cancer: a systematic review and meta-analysis
Journal of Hematology & Oncology (2016)
-
Efficacy and safety of target combined chemotherapy in advanced gastric cancer: a meta-analysis and system review
BMC Cancer (2016)
-
Optimal first-line chemotherapeutic treatment in patients with locally advanced or metastatic esophagogastric carcinoma: triplet versus doublet chemotherapy: a systematic literature review and meta-analysis
Cancer and Metastasis Reviews (2015)
