
Abstract
Background:
Soft tissue sarcomas (STS) are rare tumours for which treatment options are limited in the advanced setting. Histone deacetylase inhibitors have shown activity in preclinical models of STS.
Methods:
We conducted a single-arm, open-label, multicentre phase II study to assess the efficacy and tolerability of panobinostat given orally, 40 mg thrice weekly in patients with advanced pretreated STS. The primary endpoint was the 3-month progression-free rate.
Results:
Forty-seven STS patients were enrolled between January 2010 and December 2010. Median age was 59 (range 21–79) years, 22 (47%) patients were males. Panobinostat dose was lowered to 20 mg thrice weekly after nine patients were enrolled, based on the recommendation of an independent safety committee. The most common grade 3/4 adverse events were thrombocytopenia, fatigue, lymphopenia and anaemia. Forty-five patients were evaluable for the primary endpoint. Among them, nine patients (20%, 95% CI (10–35%)) were progression-free at 3 months. No partial response was seen, but 17 patients (36%) had stable disease (SD) as their best response. Six patients were progression-free at 6 months.
Conclusion:
Panobinostat was poorly tolerated at 40 mg thrice a week. Efficacy in unselected advanced STS was limited, although some patients had prolonged SD.
Similar content being viewed by others
Main
Soft tissue sarcomas (STS) are rare malignant tumours of mesenchymal origin. Despite adequate local therapy, as many as 50% of patients will relapse, either locally or with metastasis. Treatment of patients with advanced disease is challenging because of the limited number of therapeutic options and their limited efficacy. Although some patients may be salvaged with surgery, chemotherapy using doxorubicin-based regimens is in most cases indicated for patients with advanced or recurrent STS. Median progression-free survival (PFS) with doxorubicin-based regimens is in most cases less than or equal to 6 months (Le Cesne et al, 2000). Most patient die of their disease within 12–15 months following the diagnosis of advanced disease, and the overall survival rate is close to 8% at 5 years (Blay et al, 2003). These figures have only modestly improved over the years (Italiano et al, 2010). Therefore, novel agents with different mechanisms of action are needed and may improve the outcome of patients with advanced STSs.
Inhibition of histone deacetylase (HDAC) provides a novel approach for cancer treatment. HDACs were initially discovered through their HDAC activity. Histones are part of the core proteins of nucleosomes, and acetylation and deacetylation of these proteins have a role in the regulation of gene expression. Several lines of evidence suggest that aberrant recruitment of HDAC and the resulting modification of chromatin structure may have a role in changes in gene expression seen in transformed cells. However, it soon became clear that histone only accounted for a limited number of HDAC substrates (Bolden et al, 2006). Other substrates of HDACs potentially relevant to cancer therapy include p53, the chaperone protein HSP90 and tubulins. Several HDAC inhibitors are currently in early clinical development and involve structurally different classes of agents. Romidepsin (FK228/debsipeptide), which is a cyclic tetrapeptide, was among the first agents to demonstrate clinical activity, and was approved for the treatment of cutaneous T-cell lymphoma (CTCL). Vorinstat, a hydroxamic acid class HDAC, is also approved for the treatment of CTCL. Other classes of HDAC inhibitors that have reached clinical development include short-chain fatty acid structures, such as butyrate and valproic acid, and benzamide-based drugs, such as MS-275. These classes of compounds differ with regards to their substrate selectivity and specificity, which in turn impacts their anti-tumour activity and mechanism of action. Most hydroxamic acid class compounds have activity on class I and II (and IV to a lesser extent) HDACs, whereas romidepsin, for example, is more selective of class I HDAC. Panobinostat is a pan-HDAC inhibitor (class I and II), chemically related to vorinostat, with demonstrated activity in Hodgkin’s lymphoma (Dickinson et al, 2009; Younes et al, 2012) and T-cell lymphoma (Ellis et al, 2008). Panobinostat was postulated to have activity in sarcomas by being able to arrest gene transcription through the inhibition of HDAC, to cause the misfolding of important proteins for sarcoma biology, such as fusion proteins or proteins from overexpressed genes (HDM2, cdk4, AKT etc.) via the disruption of HSP90 functioning (Bolden et al, 2006). Mechanisms similar to leukaemia-associated fusion proteins, which have been shown to recruit HDAC to repress haematopoietic differentiation (Claus and Lubbert, 2003) might be involved in translocation-associated sarcomas. HDAC inhibitors were shown to have activity on several preclinical models of translocation-related sarcomas, such as Ewing’s sarcomas (Sakimura et al, 2005; Sonnemann et al, 2007; Owen et al, 2008), clear cell sarcoma (Dickinson et al, 2009), endometrial stromal sarcoma (ESS; Hrzenjak et al, 2006, 2008), rhabdomyosarcoma and synovial sarcoma (Kutko et al, 2003; Ito et al, 2004).
On the basis of these data, we hypothesised that panobinostat could have clinical activity in patients with advanced, pretreated STS. In the absence preclinical data indicating a lack of activity in non-translocation-related sarcomas at the time the protocol was written, all histotypes were eligible, with the aim of generating data for subsequent studies on selected subtypes.
Patients and methods
Patients
This study enrolled adult patients (aged 18 years and older) who had a histological confirmation of advanced STS, excluding gastrointestinal stromal tumours. Patients with bone sarcomas, such as osteosarcomas and Ewing’s sarcomas (both bone and extraskeletal), were excluded. Patients were required to be pretreated with an anthracycline-containing regimen for STS, to have received at least one line of therapy for advanced disease and to have documented disease progression according to RECIST within 6 months before study entry. Other inclusion criteria included ECOG performance status 0, 1 or 2, adequate organ function (bone marrow, liver and renal function), normal serum levels of potassium, magnesium and calcium, and no significant heart disease. Patients pretreated with an HDAC inhibitor, patients receiving a non-substitutable treatment known to prolong the QT interval, and pregnant or breast-feeding women were excluded. Patients were to have stopped any previous systemic anticancer treatment at least five half lives before study entry. After amendment 4, the number of previous lines of therapy was limited to 3 prior cytotoxic regimens.
Study treatment
Panobinostat was administered orally on a thrice-a-week schedule (Monday, Wednesday and Friday (MWF)). The initial dose was 40 mg thrice a week. This dose was selected based on emerging data from phase I studies of panobinostat in patients with solid tumours or haematological malignancies that were near completion at the time of protocol writing (Prince et al, 2007). Dose reductions to 30 and 20 mg thrice a week were allowed. However, because of excessive toxicity in the first nine patients who received study treatment and based on the recommendation from an independent safety review committee, the study protocol was amended (amendment 4) to reduce the dose to 20 mg thrice a week. PK-PD data from phase I studies indicated sustained histone acetylation at doses of 20 mg MWF and higher (Prince et al, 2007). Following amendment 4, dose reductions to 20 mg twice a week and 20 mg thrice a week every other week were allowed. Treatment was continued until disease progression, unacceptable toxicity despite dose adjustments, and decision of the treating physician or patient withdrawal of consent.
Study procedures
The study was approved by the Lyon-Est Ethics committee and the French regulatory authorities (Agence Nationale de Sécurité du Médicament). The ESTIM LBH study was registered at clinicaltrials.gov under trial number NCT01136499. All patients provided written informed consent before any study-related procedure. Patients had full history, physical examination, 12-lead ECG (triplicate), an assessment of cardiac function (left ventricular ejection fraction either by echocardiogram of MUGA-scan), haematology and serum biochemistry at baseline. Physical examinations were repeated at day one of every treatment cycle. Haematological analysis and 12-lead ECGs were repeated at days 1, 5, 8, 15 and 22 of cycle 1 and at days 1 and 15 of cycle 2. Haematological analysis was done at days 1 and 15 of each subsequent treatment cycles, whereas ECGs were done at day 1 of subsequent cycles. Serum biochemistry tests were repeated at days 1, 8, 15 and 22 of cycle 1 and at day 1 of subsequent cycles. Tumour assessments (using preferably contrast computed tomography (CT) of the thorax, abdomen and pelvis) were done at baseline, cycle 2 day 15 (6 weeks of treatment start), cycle 4 day 1 (12 weeks of treatment start) and every 8 weeks thereafter. All patients were required to have measurable disease per RECIST 1.0. All imaging assessments were centrally reviewed by two independent radiologists who were blinded to all clinical data.
Study design and statistical methods
ESTIM-LBH was an open-label, multicentre, single-arm phase II study. The primary endpoint was the progression-free rate at 3 months based on the review by the central review committee.
The sample size was estimated using the following parameters, where π is the true probability of success (3-month progression-free rate):
-
π0=0.20 (the largest progression-free rate below which LBH589 would be considered as ineffective) and
-
π1=0.40 (the smallest progression-free rate above which the PANOBINOSTAT would be considered as effective) based on the analysis of second-line agents by Van Glabbeke et al (2002).
On the basis of a Fleming-A’Hern single-stage design for phase II trials, and assuming a type-I error α of 0.05 with 90% power, 47 patients were needed to reject the null hypothesis H0: π⩽π0 vs the alternative hypothesis H1: π⩾π1 in a one-sided situation. After adjusting the sample size by 5% to account for non-evaluable patients (patients stopping the treatment early for toxicity), enrolment of 50 patients was required.
On the basis of preclinical studies, the type of sarcoma (translocation vs other) was recorded in order to conduct hypothesis-generating subgroup analyses, but no statistical hypothesis were made on potential differences between groups.
Results
Patients
Forty-eight patients were enrolled between January and December 2010 (12 months). One patient came off study before starting treatment because of rapid disease progression and clinical deterioration, and was excluded from further analysis. The characteristics of the 47 remaining patients are summarised in Table 1. Briefly, the median age was 59 (range 21–79) years, 25 patients (53%) were women and 43 patients (92%) had metastasis at the time of enrolment. The most common histologies were liposarcoma (LPS) (excluding myxoid LPS; n=11, 23%), leiomyosarcoma (LMS; n=10, 21%), followed by synovial sarcoma (n=6, 13%) and malignant peripheral nerve sheath tumour (n=6, 13%). All patients had measurable disease and confirmed RECIST-defined disease progression within 6 months before study entry. All patients were assessed with contrast-enhanced CT scan throughout the study. The median number of previous line of systemic therapy was 2 (range 1–7).
Efficacy
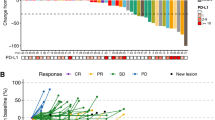
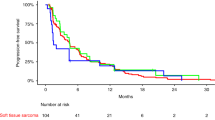
Median follow-up for surviving patients was 10 months (95% CI (8.1–12.9)). Two of the 47 treated patients were not assessable for the primary endpoint, because radiological assessment at 3 months was not available for central radiology review, leaving 45 patients included in the efficacy analysis set for the primary endpoint. Of these 45 STS patients, 9 were progression-free at 3 months (20%, 95% CI (9.6–34.6)), which corresponds to the inefficacy boundary (π0). The median PFS for the whole cohort was 1.7 months (95% CI (1.4–2.7)) (Figure 1). The 6 months PFS rate was 13.2% (95% CI (5.3–24.5)). Three-month progression-free rate among the different groups (SCT and translocation-related sarcoma) is summarised in Table 2 and PFS curves are shown in Figure 2. There was no complete or partial response. The best response was stable disease (SD) in 17 patients (36.2%) and progressive disease in 30 patients (63.8%). Changes in RECIST target lesions size at 3 months are indicated in Figure 3. Six patients were progression-free at 6 months: 1 of 11 patients with LPS (this patient had well-differentiated LPS, PFS: 9.6 months), 2 of 10 patients with LMS (PFS: 6.2 and 18.5 months), 2 of 3 patients with ESS (PFS: 9.3 and 18.3 months) and the patient with solitary fibrous tumour (SFT; PFS: 18.6 months). Median overall survival for the whole STS cohort was 10.3 months (95% CI (6.6–14.0); Supplementary Figure 1A) and was 9.5 months (95% CI (4.9–14.0)) and 11 months (95% CI (1.2—not reached)) for patients with other sarcomas and TRS, respectively (Supplementary Figure 1B).

Progression-free survival for all patients. Median: 1.7 months (95% CI (1.4–2.7)).

Progression-free survival by subgroup. Translocation-related sarcomas vs other sarcomas.

Waterfall plot. Waterfall plot of changes in target lesion size at 3 months.
Safety
Forty-seven STS patients, who received at least one dose of study drug, were included in the safety analysis. Overall, panobinostat was poorly tolerated at 40 mg thrice a week: among the first 9 patients who received study treatment, all but 1 required a dose interruption and/or dose reduction. Six of these patients had grade 3–4 thrombocytopenia. One patient came off study because of unresolved toxicity (grade 3 QTc prolongation) after more than 7 days of treatment interruption. Study enrolment was interrupted and the study was monitored by an independent safety monitoring committee (ISMC). The recommendation from the ISMC was to reduce the starting dose to 20 mg thrice a week (MWF) and limit the number of previous lines of therapy for advanced disease (excluding hormonal agents) to 3.
Forty-four of the 47 patients evaluable for toxicity (94%) reported at least one adverse event (AE), 29 (61.7%) reported at least one grade 3–4 AE, while 19 (40%) reported at least one drug-related grade 3–4 AE. The most common grade 3–4 AEs were thrombocytopenia, fatigue and lymphocytopenia, which were reported for 30, 17 and 15% of patients, respectively. The AEs occurring in more than 5% of patients are detailed in Table 3. There were three occurrences of grade-4 thrombocytopenia requiring platelet transfusion in three patients; no bleeding event was recorded. Most occurrences of grade-3 thrombocytopenia resolved within a week. Four patients had grade-3 prolongation of QTc, which lead to treatment discontinuations and dose reductions; in all but one patient, this event was less than 48 h in duration. In one patient treated at 40 mg thrice a week, the QTc recovery to grade 1 took a week. Grade-3 QTc prolongation and grade-4 thrombocytopenia were both seen within the first 2 weeks of therapy (C1D8 or D15), and did not recur after dose adjustments. Fifteen patients (32%) had at least one dose reduction: 7 of 9 patients (78%) treated at 40 mg and 8 of 38 (21%) treated at 20 mg thrice a week. Eleven of these patients (23%) did not require further dose reduction during the study. There were no treatment-related deaths. Overall, there was no correlation between the occurrence of grade 3–4 toxicity and efficacy; in particular, the median PFS of patients treated at 40 mg thrice a week was 1.5 months (range 1.0–4.8 months).
Discussion
This is, to the best of our knowledge, the first study investigating the activity of an HDAC inhibitor as a single agent in patients with advanced STS. Although preclinical studies indicated interesting activity in several sarcoma models (Kutko et al, 2003; Ito et al, 2004; Sakimura et al, 2005; Hrzenjak et al, 2006; Sonnemann et al, 2007; Hrzenjak et al, 2008; Lubieniecka et al, 2008; Owen et al, 2008; Dickinson et al, 2009), our study failed to meet its primary endpoint, suggesting that activity in unselected patients with advanced STS may be limited. Whether the significant dose reduction required for tolerability (from 40 to 20 mg thrice a week) had any impact on efficacy remains unclear. All patients treated at 40 mg thrice a week (n=9) progressed within 5 months of starting therapy, and most of them (6 of 9) at or before the first assessment (6 weeks). Six patients in the whole study population (13%) were free of progression at 6 months, which, therefore, suggest potential activity in subsets of patients. This is particularly relevant, as all patients were required to have (and actually had) RECIST-defined disease progression within 6 months before enrolment. Consequently, the 6-month progression-free rate fell in the rates of active agents according to the EORTC standards, whereas this was not the case at 3 months. However, because of the small number of patient, this percentage, although numerically close to the 14% threshold described as compatible with an ‘active agent’ by Van Glabbeke et al (2002), is not statistically different from the 8% rate given for ‘inactive agents’. Of the 6 patients who had prolonged SD, 4 had non-translocation-related sarcomas (1 SFT, 1 of 11 LPS and 2 of 10 LMS) and only 2 had translocation-related sarcoma (2 among 3 ESS). Preclinical studies have shown that ESS may be sensitive to HDAC inhibition, and our data are therefore consistent with these preclinical findings. The same is not true, however, for all translocation-related sarcomas. In this study, the activity of panobinostat was not significantly different between translocation-related and non-translocation-related sarcomas, although this study did not have the power to detect differences that could have been clinically significant between these two groups (Table 2). Furthermore, although several preclinical studies suggested activity of HDAC inhibitors against synovial sarcoma (Ito et al, 2005; Lubieniecka et al, 2008; Su et al, 2010), we saw no responses among the six patients with synovial sarcoma, and four of these patients had PFS less or equal to 2 months, whereas the two other patients had PFS of 2.4 and 5.5 months, respectively. The reasons for the discrepancy between preclinical data and our clinical results are unclear. Su et al (2010, 2012) reported similarly high activity with romidepsin (FK228/debsipeptide) and several hydroxamic-class HDAC inhibitors (MS-275, PXD101, SAHA and SB939) in synovial sarcoma models. On the otherhand, our patients who were heavily pretreated may have acquired different genetic events, rendering them less sensitive to HDAC inhibition. Other possible explanation may lie in the difficulties seen in dose escalating HDAC inhibitors due to significant side effects (QTc prolongation and haematological toxicities), especially in pretreated patients. Other histological subtypes where panobinostat showed some activity include well-differenciated LPS, LMS, SFT and ESS. Well-differenciated LPS can often behave indolently; therefore, the value of prolonged SD in this disease can be questioned. The same could be commented about SFTs (Levard et al, 2013), although this patient had very prolonged disease stabilisation (more than 18 months). Leiomyosarcoma are often aggressive, and disease stabilisation in this setting is clinically meaningful. Lee et al have also reported activity of vorinostat as a single agent in a patient with LMS, therefore, supporting further evaluation in a subtype-specific study. Finally, prolonged SD in ESS is also of limited value, as these tumours are considered low-grade, chemosensitive tumours (Reichardt, 2012). On the basis of these observations, further development of panobinostat in advanced STS does not seem warranted in unselected population of patients but is worth further exploring in LMS, ESS and SFT. In another phase II study, the combination of vorinostat (another pan-HDAC inhibitor) and bortezomib failed to demonstrate significant activity in a similar setting for STS (Attia et al, 2011).
Another finding is that panobinostat was overall poorly tolerated and was about one-third of patients who required at least one dose reduction, even after the protocol was amended to reduce the starting dose by half. Thrombocytopenia was the most common drug-related grade 3–4 AE and has been linked to the inhibition of pro-platelets from megakaryocytes by HDAC inhibitors (Bishton et al, 2011). Grade 3–4 fatigue, lymphopenia and anaemia were also noted in more than 10% of patients. Grade-3 QTc interval prolongation, another common toxicity of HDAC inhibitors, was noted in four patients. Most of these AEs occurred early during treatment, typically within the first 2 weeks of treatment, suggesting that more intensive monitoring (clinical, laboratory and ECG) is necessary during the first cycle, but may be lightened afterwards. The majority of patients (88%) treated with the initial dose of 40 mg thrice a week and 42% of patients treated at 20 mg thrice a week required a dose interruption or dose reduction. This is in fact in line with data from the phase II study of panobinostat in patients with Hodgkin’s lymphoma, where 67% of patients required a dose reduction (Younes et al, 2012).
In conclusion, results from this single-arm phase II study suggest that panobinostat has only limited activity in patients with unselected STS. However, long-term (>6 months) SD was noted in 13% of patients, some of which had SD for longer than a year, including some patients with rare histologies, such as ESS and SFTs. Conducting specific trials in these rare histotypes may be worthwhile but will likely require international collaboration. Efforts to identify predictive biomarkers may allow patients’ selection for this type of therapy.
Change history
20 August 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Attia S, Mahoney MR, Okuno SH, Adkins D, Ahuja HG, Ducker TP, Maples WJ, Ochs L, Wentworth-Hartung NL, Erlichman C, Bailey HH (2011) A phase II consortium trial of vorinostat and bortezomib for advanced soft tissue sarcomas. J Clin Oncol 29: 10075.
Bishton MJ, Harrison SJ, Martin BP, McLaughlin N, James C, Josefsson EC, Henley KJ, Kile BT, Prince HM, Johnstone RW (2011) Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood 117: 3658–3668.
Blay JY, Van GM, Verweij J, van Oosterom AT, Le CA, Oosterhuis JW, Judson I, Nielsen OS (2003) Advanced soft-tissue sarcoma: a disease that is potentially curable for a subset of patients treated with chemotherapy. Eur J Cancer 39: 64–69.
Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5: 769–784.
Claus R, Lubbert M (2003) Epigenetic targets in hematopoietic malignancies. Oncogene 22: 6489–6496.
Dickinson M, Ritchie D, DeAngelo DJ, Spencer A, Ottmann OG, Fischer T, Bhalla KN, Liu A, Parker K, Scott JW, Bishton M, Prince HM (2009) Preliminary evidence of disease response to the pan deacetylase inhibitor panobinostat (LBH589) in refractory Hodgkin Lymphoma. Br J Haematol 147: 97–101.
Ellis L, Pan Y, Smyth GK, George DJ, McCormack C, Williams-Truax R, Mita M, Beck J, Burris H, Ryan G, Atadja P, Butterfoss D, Dugan M, Culver K, Johnstone RW, Prince HM (2008) Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res 14: 4500–4510.
Hrzenjak A, Kremser ML, Strohmeier B, Moinfar F, Zatloukal K, Denk H (2008) SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J Pathol 216: 495–504.
Hrzenjak A, Moinfar F, Kremser ML, Strohmeier B, Staber PB, Zatloukal K, Denk H (2006) Valproate inhibition of histone deacetylase 2 affects differentiation and decreases proliferation of endometrial stromal sarcoma cells. Mol Cancer Ther 5: 2203–2210.
Italiano A, Mathoulin-Pelissier S, Cesne AL, Terrier P, Bonvalot S, Collin F, Michels JJ, Blay JY, Coindre JM, Bui B (2010) Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer 117: 1049–1054.
Ito T, Ouchida M, Ito S, Jitsumori Y, Morimoto Y, Ozaki T, Kawai A, Inoue H, Shimizu K (2004) SYT, a partner of SYT-SSX oncoprotein in synovial sarcomas, interacts with mSin3A, a component of histone deacetylase complex. Lab Invest 84: 1484–1490.
Ito T, Ouchida M, Morimoto Y, Yoshida A, Jitsumori Y, Ozaki T, Sonobe H, Inoue H, Shimizu K (2005) Significant growth suppression of synovial sarcomas by the histone deacetylase inhibitor FK228 in vitro and in vivo. Cancer Lett 224: 311–319.
Kutko MC, Glick RD, Butler LM, Coffey DC, Rifkind RA, Marks PA, Richon VM, LaQuaglia MP (2003) Histone deacetylase inhibitors induce growth suppression and cell death in human rhabdomyosarcoma in vitro. Clin Cancer Res 9: 5749–5755.
Le Cesne A, Judson I, Crowther D, Rodenhuis S, Keizer HJ, van HQ, Blay JY, Frisch J, Van GM, Hermans C, Van OA, Tursz T, Verweij J (2000) Randomized phase III study comparing conventional-dose doxorubicin plus ifosfamide versus high-dose doxorubicin plus ifosfamide plus recombinant human granulocyte-macrophage colony-stimulating factor in advanced soft tissue sarcomas: A trial of the European Organization for Research and Treatment of Cancer/Soft Tissue and Bone Sarcoma Group. J Clin Oncol 18: 2676–2684.
Levard A, Derbel O, Meeus P, Ranchere D, Ray-Coquard I, Blay JY, Cassier PA (2013) Outcome of patients with advanced solitary fibrous tumors: the Centre Leon Berard experience. BMC Cancer 13: 109.
Lubieniecka JM, de Bruijn DR, Su L, van Dijk AH, Subramanian S, van de RM, Poulin N, van Kessel AG, Nielsen TO (2008) Histone deacetylase inhibitors reverse SS18-SSX-mediated polycomb silencing of the tumor suppressor early growth response 1 in synovial sarcoma. Cancer Res 68: 4303–4310.
Owen LA, Kowalewski AA, Lessnick SL (2008) EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing's sarcoma. PLoS One 3: e1965.
Prince HM, George D, Patnaik A, Mita M, Dugan M, Butterfoss D, Masson E, Culver KW, Burris HA, Beck J (2007) Phase I study of oral LBH589, a novel deacetylase (DAC) inhibitor in advanced solid tumors and non-hodgkin's lymphoma. J Clin Oncol 25: 3500.
Reichardt P (2012) The treatment of uterine sarcomas. Ann Oncol 23 (Suppl 10): x151–x157.
Sakimura R, Tanaka K, Nakatani F, Matsunobu T, Li X, Hanada M, Okada T, Nakamura T, Matsumoto Y, Iwamoto Y (2005) Antitumor effects of histone deacetylase inhibitor on Ewing's family tumors. Int J Cancer 116: 784–792.
Sonnemann J, Dreyer L, Hartwig M, Palani CD, Hong le TT, Klier U, Broker B, Volker U, Beck JF (2007) Histone deacetylase inhibitors induce cell death and enhance the apoptosis-inducing activity of TRAIL in Ewing's sarcoma cells. J Cancer Res Clin Oncol 133: 847–858.
Su L, Cheng H, Sampaio AV, Nielsen TO, Underhill TM (2010) EGR1 reactivation by histone deacetylase inhibitors promotes synovial sarcoma cell death through the PTEN tumor suppressor. Oncogene 29: 4352–4361.
Su L, Sampaio AV, Jones KB, Pacheco M, Goytain A, Lin S, Poulin N, Yi L, Rossi FM, Kast J, Capecchi MR, Underhill TM, Nielsen TO (2012) Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell 21: 333–347.
Van Glabbeke M, Verweij J, Judson I, Nielsen OS (2002) Progression-free rate as the principal end-point for phase II trials in soft-tissue sarcomas. Eur J Cancer 38: 543–549.
Younes A, Sureda A, Ben-Yehuda D, Zinzani PL, Ong TC, Prince HM, Harrison SJ, Kirschbaum M, Johnston P, Gallagher J, Le CC, Shen A, Engert A (2012) Panobinostat in patients with relapsed/refractory Hodgkin’s lymphoma after autologous stem-cell transplantation: results of a phase II study. J Clin Oncol 30: 2197–2203.
Acknowledgements
We thank members of the independent safety monitoring committee Dr Nicolas Isambert (Medical oncologist), Dr Olivier Collard (Medical oncologist) and Dr Xavier Paoletti (Biostatistician). We also thank radiologists Dr Marie Peix and Dr Bertrand Richioud. The trial registration number is Trial registration: NCT01136499. This study was funded by an unrestricted research grant from Novartis Pharma, which also provided the study drug.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The following authors have received honoraria from Novartis PAC, J-YB, AL, IR-C, AL, FD, CC. J-YB has received honoraria, research grant, travel grant from Novartis, MSD and GSK. All other authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Cassier, P., Lefranc, A., Y Amela, E. et al. A phase II trial of panobinostat in patients with advanced pretreated soft tissue sarcoma. A study from the French Sarcoma Group. Br J Cancer 109, 909–914 (2013). https://doi.org/10.1038/bjc.2013.442
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.442
Keywords
This article is cited by
-
Establishment and functional testing of a novel ex vivo extraskeletal osteosarcoma cell model (USZ20-ESOS1)
Human Cell (2023)
-
Relapsed Synovial Sarcoma: Treatment Options
Current Treatment Options in Oncology (2023)
-
Molecular mechanisms underpinning sarcomas and implications for current and future therapy
Signal Transduction and Targeted Therapy (2021)
-
MicroRNA-mRNA networks define translatable molecular outcome phenotypes in osteosarcoma
Scientific Reports (2020)
-
Assessment of Synergistic Contribution of Histone Deacetylases in Prognosis and Therapeutic Management of Sarcoma
Molecular Diagnosis & Therapy (2020)
