
Abstract
Background:
We conducted a multicentre feasibility study for single agent long-term S-1 chemotherapy following docetaxel plus cisplatin in patients with curatively resected stage II–IIIA non-small cell lung cancer.
Methods:
Patients received three cycles of docetaxel (60 mg m−2) plus cisplatin (80 mg m−2) and then received S-1 (40 mg m−2 twice daily) for 14 consecutive days with a 1-week rest for >6 months (maximum, 1 year). The primary end point was feasibility, which was defined as the proportion of patients who completed eight or more cycles of S-1 chemotherapy. If the lower 95% confidence interval (CI) of this proportion was 50% or more, then the treatment was considered as feasible. The sample size was set at 125 patients.
Results:
One hundred and thirty-one patients were enrolled, of whom 129 patients were eligible and assessable. In all, 109 patients (84.5%) completed 3 cycles of docetaxel plus cisplatin and 66 patients (51.2%, 95% CI: 42.5–59.8) completed 8 or more cycles of S-1 treatment. Grade 3/4 toxicities during the S-1 chemotherapy included anaemia (7.3%), neutropaenia (3.7%), and anorexia (3.7%).
Conclusion:
The toxicity level was acceptable, although the results did not meet our criterion for feasibility. Modification of the treatment schedule for S-1 chemotherapy might improve the treatment compliance.
Similar content being viewed by others

Main
Primary surgery is the standard of care for resectable clinical stage I or II non-small cell lung cancer (NSCLC). The 5-year survival rate for patients with clinical stage IB and stage II surgically resected NSCLC was ∼66% and 50%, respectively. The majority of patients with recurrences have distant metastases, indicating that systemic micrometastases are common in patients with completely resected NSCLC. To control distant micrometastasis and to improve patients’ survival, adjuvant chemotherapy has been examined in patients with completely resected NSCLC of pathological stage I–III. Several randomised studies and meta-analyses have demonstrated that cisplatin-based adjuvant chemotherapy improved the overall survival (OS) in patients with pathological stage IB to III NSCLC (Arriagada et al, 2004; Hotta et al, 2004; Winton et al, 2005; Douillard et al, 2006; Pignon et al, 2006). However, the absolute increase in survival was only 4% at 5 years. Thus, new treatment strategies or drugs are needed to improve the clinical outcome in patients with resectable NSCLC.
A randomised phase III study demonstrated that adjuvant chemotherapy with uracil-tegafur (UFT) improved survival among patients with completely resected pathological stage I adenocarcinoma of the lung. The 5-year OS was 88% in the UFT group and 85% in the control group (hazard ratio 0.71, 95% confidence interval (CI) 0.52–0.98) (Kato et al, 2004). S-1 is an oral anticancer agent comprises tegafur, gimeracil (an inhibitor of dihydropyrimidine dehydrogenase, which degrades fluorouracil), and oteracil (which inhibits the phosphorylation of fluorouracil in the gastrointestinal tract, thereby reducing the gastrointestinal toxicity of fluorouracil) in a molar ratio of 1 : 0.4 : 1 (Shirasaka et al, 1996). S-1 is approved for the treatment of NSCLC as well as gastric, colorectal, head and neck, breast, pancreatic, and biliary tract cancer in Japan. In a phase II trial, S-1 monotherapy produced a response rate of 22% as a first-line treatment in patients with advanced NSCLC (Kawahara et al, 2001). S-1 is believed to have a stronger antitumour activity against NSCLC than UFT, since UFT monotherapy produced a response rate of only 6% in another phase II study (Keicho et al, 1986). A randomised phase III trial demonstrated that S-1 plus carboplatin (CBDCA) was non-inferior in terms of OS, compared with paclitaxel plus CBDCA, in patients with advanced NSCLC (Okamoto et al, 2010). Another randomised phase III trial also demonstrated that S-1 plus CDDP was non-inferior in terms of OS, compared with docetaxel plus CDDP, in patients with advanced NSCLC (Katakami et al, 2012). Previous phase II trials demonstrated that S-1 monotherapy produced a response rate of 7–14% as a second-line treatment for advanced NSCLC (Totani et al, 2009; Govindan et al, 2011; Shiroyama et al, 2011).
Recent phase III trials have demonstrated that switch maintenance chemotherapy consisting of pemetrexed or erlotinib prolonged the OS of patients with advanced NSCLC who showed no signs of progression after four cycles of platinum-based chemotherapy (Ciuleanu et al, 2009; Cappuzzo et al, 2010). Continuation maintenance with pemetrexed also prolonged the OS in patients with non-squamous NSCLC in another randomised trial (Paz-Ares et al, 2012a, 2012b). Maintenance chemotherapy has thus received considerable attention.
The Thoracic Oncology Research Group (TORG) conducted a randomised phase II study comparing docetaxel (DOC) plus CDDP with paclitaxel (PTX) plus CBDCA as an adjuvant chemotherapy in patients with completely resected stage IB to IIIA NSCLC (TORG 0503). This study showed that DOC plus CDDP had a promising activity with a favourable 2-year recurrence-free survival (RFS) rate (74.1% vs 72.5%, respectively) (Ohira et al, 2011). Taking these rationales into consideration, we conducted a feasibility study for adjuvant chemotherapy consisting of DOC plus CDDP followed by single agent long-term S-1 chemotherapy in patients with completely resected NSCLC (TORG 0809).
Patients and methods
Patient population
Patients were required to have completely resected stage II or IIIA (according to the Union Internationale Contre le Cancer (UICC) fifth TNM edition) NSCLC, an age of 20–74 years, and an ECOG performance status (PS) of 0 or 1. Other criteria included a PaO2 at room air ⩾70 torr or an SpO2 at room air ⩾95%, and adequate organ function (i.e., total bilirubin ⩽1.2 mg dl−1, AST and ALT ⩽100 IU l−1, serum creatinine ⩽1.2 mg dl−1, creatinine clearance ⩾60 ml min−1, leukocyte count ⩾4000 per mm3 and ⩽12 000 per mm3, neutrophil count ⩾2000 per mm3, haemoglobin ⩾10.0 g dl−1, and platelets ⩾100 000 per mm3). Patients were required to start the protocol treatment within 10 weeks after surgical resection.
Key exclusion criteria were a lack of recovery from surgical complications; active infection; interstitial pneumonia as determined using computed tomography (CT) of the chest; acute cardiac infarction within 6 months; uncontrolled heart disease, liver dysfunction, or diabetes mellitus; grade 2 or worse peripheral neuropathy; active concomitant malignancy; pregnancy or breastfeeding; a history of hypersensitivity to drugs including polysorbate-80; and the concurrent use of flucytosine. Patients who had undergone a pneumonectomy were also excluded. All the patients were required to provide written informed consent.
Treatment plan
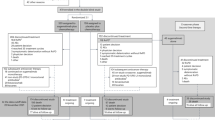
The treatment schema is shown in Figure 1. Treatment was started within 1 week after enrolment in the study. Patients received adjuvant chemotherapy with DOC (60 mg m−2, day 1) and CDDP (80 mg m−2, day 1) every 3–4 weeks for up to three cycles. After the completion of adjuvant chemotherapy with DOC plus CDDP, if the leukocyte count was ⩾3000 per mm3, the neutrophil count was ⩾1500 per mm3, the platelet count was ⩾100 000 per mm3, the AST and/or ALT level was ⩽100 IU l−1, the total bilirubin level was ⩽1.5 mg dl−1, the serum creatinine level was <1.5 mg dl−1, and all other non-haematological toxicities were grade 1 or better with the exception of alopecia, body weight loss, and hyponatraemia, then the patients were treated with oral S-1 at a dose of 40 mg m−2 twice daily for 14 consecutive days, followed by a 1-week rest. The actual dose of S-1 was selected as follows: patients with a body surface area (BSA) of <1.25 m2 received 80 mg daily; those with a BSA of 1.25 m2 or more but <1.5 m2 received 100 mg daily; and those with a BSA of 1.5 m2 or more received 120 mg daily. If the serum creatinine level was 1.2 mg dl−1 or more but <1.5 mg dl−1 before the initiation of S-1 chemotherapy, then the S-1 dose was reduced to a lower level. This 3-week cycle was repeated for 6 months (maximum, 1 year) if neither unacceptable toxicity nor tumour recurrence was observed. In the event of a leukocyte count of <2000 per mm3, a platelet count of <75 000 per mm3, an AST and/or ALT level of ⩾100 IU l−1, a total bilirubin level of ⩾2.5 mg dl−1, a serum creatinine level of ⩾1.5 mg dl−1, appetite loss, diarrhoea, mucositis, nausea/vomiting of grade 2 or worse despite appropriate antiemetic therapy, and/or other grade 2 non-haematological toxicities other than body weight loss, alopecia, or hyponatraemia, the daily dose of S-1 was reduced from 120 to 100 mg, 100 to 80 mg, or 80 to 50 mg in the next cycle. If the patients experienced the above-mentioned toxicities after the dose reduction, then their daily dose of S-1 was reduced from 100 to 80 mg, or 80 to 50 mg. If a patient with a BSA of <1.25 m2 experienced the above toxicities at 50 mg, then the S-1 chemotherapy was terminated. If the adjuvant chemotherapy of DOC+CDDP was terminated after one or two cycles, then a shift to S-1 chemotherapy was allowed. However, these patients were not considered to have completed the protocol treatment.

Treatment schema for this study.
Safety assessment and follow-up
For the toxicity assessment, blood samples were obtained before the start of each cycle. A chest X-ray examination was performed monthly throughout the study period. Toxicities were graded according to the Common Terminology Criteria for Adverse Events (CTCAE), version 3.0. A CT examination of the chest was performed at 1, 2, 3, and 5 years after the initiation of the protocol treatment.
Study design and statistical analysis
This trial was designed as a multicentre, prospective, single-arm, feasibility study and the study protocol was approved by the institutional review board of each participating institution. All the study data were managed by the TORG0809 data centre at Kitasato University Research Center for Clinical Pharmacology.
The primary end point of this study was feasibility, which was defined as the proportion of patients who had completed eight or more cycles of S-1 chemotherapy. If the lower 95% CI of this proportion was 50% or more, then the treatment was considered as feasible. If a patient received 75% or more of S-1 in a cycle, that is, 21 times per cycle, this patient was considered to have completed the treatment cycle. If 72 out of 120 patients (60%) completed the protocol treatment, then the 95% CI of the proportion of the treatment completion was 51.2–68.8%. Considering the possibility of ineligible patients, the sample size was set at 125 patients.
The secondary end points included adverse events, OS, RFS, and recurrence pattern. Because of the short follow-up period, we will report the OS and RFS data elsewhere. We plan to analyse the OS and RFS at 5 years after the last enrolment, as described in the study protocol. The statistical analysis was performed using SAS version 9.2 (SAS Institute, Cary, NC, USA).
This study was registered with the UMIN Clinical Trials Registry (number UMIN000001779).
Results
Patient population
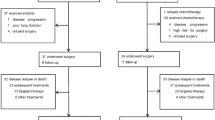
A total of 131 patients were enrolled in this study between January 2009 and November 2010 from 20 institutions in Japan. One patient did not receive any protocol treatment at the patient’s request. Another patient was enrolled as p-stage IIIA according to the UICC 7th edition; however, the p-stage corresponded to IIIB according to the UICC 5th edition, making this patient ineligible. This patient received three cycles of docetaxel plus cisplatin and two cycles of S-1 chemotherapy, and she was included in the safety analysis. A total of 129 patients were eligible (Figure 2). The patient characteristics are listed in Table 1. Sixty-four percent of the patients were male; the median age was 63 years. Seventy-eight percent of the patients had an adenocarcinoma histology.

CONSORT diagram.
Treatment delivery and protocol compliance
Overall, 114 patients received two cycles or more of DOC+CDDP. Of these, 67 patients (58.8%) required a dose reduction of DOC or CDDP. The most common reason for the dose reduction of DOC and CDDP was grade 4 neutropaenia (n=63), followed by a fever of 38.0 °C or higher (n=16). The dose of CDDP was reduced because of anorexia, nausea, and/or vomiting of grade 2 or worse for more than a week (n=16) and an elevated serum creatinine level of 1.5 mg dl−1 or more (n=6).
In total, 109 patients (84.5%) completed three cycles of adjuvant chemotherapy consisting of DOC+CDDP (Table 2). The main reasons for the discontinuation of the adjuvant chemotherapy were toxicity (n=15) and patient refusal because of toxicity (n=7) (Table 3). One patient terminated the DOC+CDDP treatment after one cycle and completed eight cycles of S-1 chemotherapy. Another patient terminated the DOC+CDDP treatment after two cycles and received three cycles of S-1 chemotherapy.
One hundred and eight patients received S-1 chemotherapy. Of these, 34 patients (31.5%) required the interruption of S-1 during a treatment cycle. Thirty-one patients (28.7%) required a dose reduction of S-1. The majority of the reasons for the interruption or dose reduction of S-1 were appetite loss, diarrhoea, mucositis, or nausea/vomiting of grade 2 or worse (n=27), followed by other non-haematologic toxicities of grade 2 or worse (n=20).
One hundred and six patients (82.2%) completed three cycles of DOC+CDDP and subsequently switched to S-1 chemotherapy. Of these, 31 patients terminated the S-1 chemotherapy after receiving 3 or fewer cycles. A total of 66 patients (51.2%; 95% CI, 42.5–59.8%) completed 8 cycles or more of S-1 treatment (Table 2). The lower limit of the 95% CI for the completion rate was 42.5%, which was less than our previously defined criterion for treatment feasibility. The reasons for the discontinuation of the S-1 chemotherapy included toxicity (n=17), patient refusal because of toxicity (n=15), and recurrence (n=6) (Table 3).
Safety and toxicity
The most common grade 3 or 4 toxicity experienced during the DOC+CDDP treatment was neutropaenia (78.5%) (Table 4). Ten patients (7.7%) developed febrile neutropaenia; however, all these patients recovered after receiving appropriate antibiotic therapy. Two patients experienced grade 3 or 4 allergic reactions to DOC during the first cycle, resulting in treatment termination.
Grade 3 or 4 toxicities during the S-1 chemotherapy included anaemia (7.3%), neutropaenia (3.7%), anorexia (3.7%), dyspnoea (1.8%), and infection with neutropaenia of grade 0–2 (1.8%). Febrile neutropaenia was not observed. One treatment-related death occurred during the study. This patient was a 63-year-old man. After two cycles of S-1 chemotherapy, he developed grade 3 fatigue. On day 36 of the second cycle of S-1, grade 3 dyspnoea was observed, and his SpO2 was 92% in room air. A CT scan of the chest revealed bilateral diffuse ground-glass opacities. Prednisolone (80 mg day−1; 1 mg kg−1 per day) was administered, and an improvement in the opacities was observed. The prednisolone was tapered to 30 mg day−1 for 6 weeks; however, multiple cavity lesions were visible on a chest CT image obtained 2 months after the initiation of the steroid therapy. Multiple abscesses at the neck, axilla, chest, and femur were noted, and the patient developed hypotension. Nocardia was isolated in blood and abscess samples, with a diagnosis of disseminated nocardiosis. Sulfamethoxazole/trimethoprim and antibiotics were administered and artificial ventilation therapy was performed. The patient was taken off the respirator once, but the pneumonitis recurred and disseminated intravascular coagulation also developed, leading to death.
Discussion
This feasibility study was designed to evaluate the tolerability, safety, and efficacy of single agent long-term administration of S-1 chemotherapy following three cycles of DOC plus CDDP in patients with completely resected stage II or IIIA NSCLC. Fifty-one percent of the patients (95% CI, 42.5–59.8%) completed three cycles of DOC plus CDDP and eight cycles or more of S-1 chemotherapy. The lower limit of the CI for this proportion was lower than the predefined criterion of 50%. Grade 3–4 haematologic toxicities were observed in 7.3% of patients, while grade 3–4 non-haematologic toxicities were observed in only 4%. However, grade 1–2 anorexia and/or fatigue were common, with rates of ∼50–60%. S-1 was administered for 2 weeks with a 1-week rest. The long duration of S-1 administration might have been responsible for the low-grade but extended non-haematologic toxicities and might have been too intensive for patients especially after platinum-doublet chemotherapy. In a previous phase III study of adjuvant chemotherapy for gastric cancer with single agent of S-1, 78% of patients received S-1 for at least 6 months (Sakuramoto et al, 2007). Adjuvant chemotherapy of DOC+CDDP probably affected the compliance of S-1 chemotherapy negatively in our study. A modification of the treatment schedule for S-1 chemotherapy, such as a 2-week rest period rather than a 1-week rest period, might improve treatment compliance.
Efficacious treatment for advanced stage disease has been introduced and investigated in an adjuvant setting, such as bevacizumab plus platinum-doublet chemotherapy in patients with non-squamous cell carcinoma or erlotinib in patients with a mutated epidermal growth factor receptor gene. Recent phase III trials have demonstrated that switch maintenance chemotherapy consisting of pemetrexed or erlotinib, which were efficacious for second-line chemotherapy, prolonged the OS in patients with advanced NSCLC (Ciuleanu et al, 2009; Cappuzzo et al, 2010). Switch maintenance chemotherapy can be recognised as an early second-line chemotherapy. The purpose of adjuvant chemotherapy is to control micrometastasis and to prevent recurrence. Switch maintenance chemotherapy is considered to enhance the efficacy of adjuvant chemotherapy. Previous phase II trials have demonstrated that S-1 monotherapy produced a response rate of 7–14%, a median progression-free survival (PFS) of 3–4 months, and a median OS of 7–16 months as a second-line treatment for advanced NSCLC (Totani et al, 2009; Govindan et al, 2011; Shiroyama et al, 2011). Pemetrexed is effective against non-squamous NSCLC; on the other hand, S-1 is effective against both non-squamous and squamous NSCLC. A randomised trial comparing S-1 and docetaxel as a second- or third-line chemotherapy is now underway in Asia. Switch maintenance chemotherapy using S-1 is also being evaluated as a first-line chemotherapy for patients with advanced NSCLC in a phase II study (UMIN000003676). If promising RFS or OS data in this trial are obtained, then a prospective randomised trial will be warranted to compare adjuvant chemotherapy with or without single agent long-term administration of S-1 chemotherapy.
A recent phase III trial has also demonstrated that continuation maintenance chemotherapy consisting of pemetrexed prolonged the OS and PFS in patients with advanced non-squamous NSCLC. However, concurrent chemoradiotherapy consisting of pemetrexed plus CDDP followed by four cycles of pemetrexed did not improve OS over concurrent chemoradiotherapy consisting of etoposide plus CDDP in patients with stage III non-squamous NSCLC (PROCLAIM study). Up to four cycles of pemetrexed in the PROCLAIM study, comparable to S-1 chemotherapy in our study, might be unable to enhance curative treatment effect. We might have to distinguish strategy for stage IV disease from that for curative situations in completely resected stage II/III disease.
Combination chemotherapy consisting of DOC plus CDDP is a standard regimen for the treatment of patients with advanced NSCLC. A randomised trial demonstrated that DOC+CDDP resulted in a more favourable response rate and OS than vinorelbine (VNR) plus CDDP in chemo-naïve patients with advanced NSCLC. The median OS period was 11.3 months for patients treated with DOC plus CDDP and 10.1 months for patients treated with VNR plus CDDP. The hazard ratio was 1.183 (97.2% CI, 0.989–1.416) (Fossella et al, 2003). A higher incidence of grade 3–4 anaemia, nausea, and vomiting was observed in VNR+CDDP arm, compared with DOC+CDDP arm. Febrile neutropaenia occurred in <5% of patients in both regimens. Furthermore, the single agent DOC had a more favourable OS period than the single agent VNR in both first-line and second-line settings in patients with advanced NSCLC (Fossella et al, 2000). TORG0503 study demonstrated that >90% of patients completed three planned cycles of adjuvant chemotherapy in both DOC+CDDP and PTX+CBDCA arms. On the other hand, the most common regimen for adjuvant chemotherapy for pathological stage II or III NSCLC is VNR+CDDP, because most randomised trials, which resulted in positive results, adopted VNR+CDDP. Considering the promising results of clinical trials for advanced NSCLC, it might be reasonable to select DOC+CDDP as an adjuvant chemotherapy in patients with completely resected stage II or III NSCLC. Indeed, DOC+CDDP has been selected as one of the standard adjuvant chemotherapy regimens in ECOG1505 study, which is a randomised phase III trial of adjuvant chemotherapy with or without bevacizumab in patients with completed resected early-stage NSCLC (Wakelee et al, 2011). However, 7.7% of patients experienced grade 3 febrile neutropaenia during the chemotherapy of DOC+CDDP in our study. Relatively high incidence of febrile neutropaenia could not support the use of adjuvant chemotherapy with DOC+CDDP as a new alternative. Four cycles of VNR+CDDP followed by long-term administration of S-1 might be a better strategy in a future study.
The treatment cycle for DOC plus CDDP was set at three because the actual median numbers of cycles delivered in previous phase III studies of adjuvant chemotherapy were three or four (Winton et al, 2005; Douillard et al, 2006), and a randomised study demonstrated that four cycles or more of platinum-based chemotherapy did not improve the OS in patients with advanced NSCLC (Smith et al, 2001). In the TORG0503 study, the number of treatment cycles for DOC plus CDDP or for PTX plus CBDCA as an adjuvant chemotherapy was also set at three, and a favourable 2-year RFS rate was observed (Ohira et al, 2011).
A previous randomised phase II study demonstrated that adjuvant chemotherapy with pemetrexed plus CDDP was safe and feasible with less toxicity and superior dose delivery compared with VNR+CDDP (Kreuter et al, 2013). Pemetrexed plus CDDP is considered as suitable for adjuvant chemotherapy because of relatively less toxic and promising antitumour activity in patients with non-squamous NSCLC. A randomised phase III study is underway comparing pemetrexed plus CDDP and VNR+CDDP in patients with completely resected stage II–IIIA non-squamous NSCLC in Japan. However, it is difficult to conduct a randomised phase III study of adjuvant chemotherapy in patients with NSCLC, because large sample size and long-term follow-up are needed. Therefore, a randomised phase II study containing control arm should be taken into consideration to select appropriate experimental treatment.
Aprepitant, a standard antiemetic drug for cisplatin therapy, was approved in December 2009 in Japan. As a result, ∼20 patients did not receive aprepitant. If aprepitant had been available for all the enrolled patients, then the treatment compliance might have improved. Furthermore, 2 out of the 129 patients experienced grade 3 or 4 allergic reactions to DOC during the first cycle, resulting in treatment termination. Premedication for DOC+CDDP included dexamethasone only on day 1 in this study. The administration of dexamethasone on the day before the initiation of DOC+CDDP and an antihistamine on day 1 might be recommended in future clinical trials to prevent anaphylaxis in response to DOC.
In conclusion, the toxicity level of S-1 chemotherapy was acceptable, although the treatment completion rate did not meet our criterion for feasibility. A modification of the treatment schedule for S-1 chemotherapy, such as a 2-week rest period rather than a 1-week rest period, might improve treatment compliance. After referring to the results for OS and RFS, we would like to plan a randomised trial to investigate whether platinum-based chemotherapy followed by single agent long-term administration of S-1 chemotherapy improves survival in patients with completely resected stage II or III NSCLC.
Change history
06 August 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Arriagada R, Bergman B, Dunant A, Le Chevalier T, Pignon JP, Vansteenkiste J (2004) Cisplatin-based adjuvant chemotherapy in patients with completely resected non-small-cell lung cancer. N Engl J Med 350 (4): 351–360.
Cappuzzo F, Ciuleanu T, Stelmakh L, Cicenas S, Szczesna A, Juhasz E, Esteban E, Molinier O, Brugger W, Melezinek I, Klingelschmitt G, Klughammer B, Giaccone G (2010) Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: a multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol 11 (6): 521–529.
Ciuleanu T, Brodowicz T, Zielinski C, Kim JH, Krzakowski M, Laack E, Wu YL, Bover I, Begbie S, Tzekova V, Cucevic B, Pereira JR, Yang SH, Madhavan J, Sugarman KP, Peterson P, John WJ, Krejcy K, Belani CP (2009) Maintenance pemetrexed plus best supportive care versus placebo plus best supportive care for non-small-cell lung cancer: a randomised, double-blind, phase 3 study. Lancet 374 (9699): 1432–1440.
Douillard JY, Rosell R, De Lena M, Carpagnano F, Ramlau R, Gonzales-Larriba JL, Grodzki T, Pereira JR, Le Groumellec A, Lorusso V, Clary C, Torres AJ, Dahabreh J, Souquet PJ, Astudillo J, Fournel P, Artal-Cortes A, Jassem J, Koubkova L, His P, Riggi M, Hurteloup P (2006) Adjuvant vinorelbine plus cisplatin versus observation in patients with completely resected stage IB-IIIA non-small-cell lung cancer (Adjuvant Navelbine International Trialist Association [ANITA]): a randomised controlled trial. Lancet Oncol 7 (9): 719–727.
Fossella F, Pereira JR, von Pawel J, Pluzanska A, Gorbounova V, Kaukel E, Mattson KV, Ramlau R, Szczesna A, Fidias P, Millward M, Belani CP (2003) Randomized, multinational, phase III study of docetaxel plus platinum combinations versus vinorelbine plus cisplatin for advanced non-small-cell lung cancer: the TAX 326 study group. J Clin Oncol 21 (16): 3016–3024.
Fossella FV, DeVore R, Kerr RN, Crawford J, Natale RR, Dunphy F, Kalman L, Miller V, Lee JS, Moore M, Gandara D, Karp D, Vokes E, Kris M, Kim Y, Gamza F, Hammershaimb L (2000) Randomized phase III trial of docetaxel versus vinorelbine or ifosfamide in patients with advanced non-small-cell lung cancer previously treated with platinum-containing chemotherapy regimens. The TAX 320 Non-Small Cell Lung Cancer Study Group. J Clin Oncol 18 (12): 2354–2362.
Govindan R, Morgensztern D, Kommor MD, Herbst RS, Schaefer P, Gandhi J, Saito K, Zergebel C, Schiller J (2011) Phase II trial of S-1 as second-line therapy in patients with advanced non-small cell lung cancer. J Thorac Oncol 6 (4): 790–795.
Hotta K, Matsuo K, Ueoka H, Kiura K, Tabata M, Tanimoto M (2004) Role of adjuvant chemotherapy in patients with resected non-small-cell lung cancer: reappraisal with a meta-analysis of randomized controlled trials. J Clin Oncol 22 (19): 3860–3867.
Katakami N, Gemma A, Sakai H, Kubota K, Nishio M, Inoue A, Okamoto H, Isobe H, Kunitoh H, Takiguchi Y, Kobayashi K, Nakamura Y, Ohmatsu H, Sugawara S, Minato K, Fukuda M, Yokoyama A, Takeuchi M, Michimae H, Kudoh S (2012) Randomized phase III trial of S-1 plus cisplatin versus docetaxel plus cisplatin for advanced non-small-cell lung cancer (TCOG0701). J Clin Oncol 30 (suppl): abstr 7515.
Kato H, Ichinose Y, Ohta M, Hata E, Tsubota N, Tada H, Watanabe Y, Wada H, Tsuboi M, Hamajima N, Ohta M (2004) A randomized trial of adjuvant chemotherapy with uracil-tegafur for adenocarcinoma of the lung. N Engl J Med 350 (17): 1713–1721.
Kawahara M, Furuse K, Segawa Y, Yoshimori K, Matsui K, Kudoh S, Hasegawa K, Niitani H (2001) Phase II study of S-1, a novel oral fluorouracil, in advanced non-small-cell lung cancer. Br J Cancer 85 (7): 939–943.
Keicho N, Saijo N, Shinkai T, Eguchi K, Sasaki Y, Tamura T, Sakurai M, Sano T, Hoshi A (1986) Phase II study of UFT in patients with advanced non-small cell lung cancer. Jpn J Clin Oncol 16 (2): 143–146.
Kreuter M, Vansteenkiste J, Fischer JR, Eberhardt W, Zabeck H, Kollmeier J, Serke M, Frickhofen N, Reck M, Engel-Riedel W, Neumann S, Thomeer M, Schumann C, De Leyn P, Graeter T, Stamatis G, Zuna I, Griesinger F, Thomas M (2013) Randomized phase 2 trial on refinement of early-stage NSCLC adjuvant chemotherapy with cisplatin and pemetrexed versus cisplatin and vinorelbine: the TREAT study. Ann Oncol 24 (4): 986–992.
Ohira T, Kubota K, Seto T, Kunitoh H, Shimada N, Ikeda N, Tsuboi M, Okamoto H, Masuda N, Maruyama R, Shibuya M (2011) A randomized phase II trial of adjuvant chemotherapy with docetaxel plus cisplatin versus paclitaxel plus carboplatin in patients with completely resected non-small cell lung cancer: TORG 0503. J Thorac Oncol 6 (suppl): S1555–S1556.
Okamoto I, Yoshioka H, Morita S, Ando M, Takeda K, Seto T, Yamamoto N, Saka H, Asami K, Hirashima T, Kudoh S, Satouchi M, Ikeda N, Iwamoto Y, Sawa T, Miyazaki M, Tamura K, Kurata T, Fukuoka M, Nakagawa K (2010) Phase III trial comparing oral S-1 plus carboplatin with paclitaxel plus carboplatin in chemotherapy-naive patients with advanced non-small-cell lung cancer: results of a west Japan oncology group study. J Clin Oncol 28 (36): 5240–5246.
Paz-Ares L, de Marinis F, Dediu M, Thomas M, Pujol JL, Bidoli P, Molinier O, Sahoo TP, Laack E, Reck M, Corral J, Melemed S, John W, Chouaki N, Zimmermann AH, Visseren-Grul C, Gridelli C (2012a) Maintenance therapy with pemetrexed plus best supportive care versus placebo plus best supportive care after induction therapy with pemetrexed plus cisplatin for advanced non-squamous non-small-cell lung cancer (PARAMOUNT): a double-blind, phase 3, randomised controlled trial. Lancet Oncol 13 (3): 247–255.
Paz-Ares L, De Marinis F, Dediu M, Thomas M, Pujol JL, Bidoli P, Molinier O, Sahoo TP, Laack E, Reck M, Jaime JC, Melemed S, John W, Chouaki N, Zimmermann AH, Visseren-Grul C, Gridelli C (2012b) PARAMOUNT: Final overall survival results of the phase III study of maintenance pemetrexed plus best supportive care versus placebo plus best supportive care immediately following induction treatment with pemetrexed plus cisplatin for advanced nonsquamous non-small cell lung cancer. J Clin Oncol 30 (suppl): abstr LBA7507.
Pignon JP, Tribodet H, Scagliotti GV, Douillard JY, Shepherd F, Le Chevalier T (2006) Lung adjuvant cisplatin evaluation (LACE): A pooled analysis of five randomized clinical trials including 4,584 patients. J Clin Oncol 24 (18S Part I): 366s.
Sakuramoto S, Sasako M, Yamaguchi T, Kinoshita T, Fujii M, Nashimoto A, Furukawa H, Nakajima T, Ohashi Y, Imamura H, Higashino M, Yamamura Y, Kurita A, Arai K (2007) Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 357 (18): 1810–1820.
Shirasaka T, Shimamato Y, Ohshimo H, Yamaguchi M, Kato T, Yonekura K, Fukushima M (1996) Development of a novel form of an oral 5-fluorouracil derivative (S-1) directed to the potentiation of the tumor selective cytotoxicity of 5-fluorouracil by two biochemical modulators. Anticancer Drugs 7 (5): 548–557.
Shiroyama T, Komuta K, Imamura F, Hirashima T, Kijima T, Tachibana I, Kawase I (2011) Phase II study of S-1 monotherapy in platinum-refractory, advanced non-small cell lung cancer. Lung Cancer 74 (1): 85–88.
Smith IE, O'Brien ME, Talbot DC, Nicolson MC, Mansi JL, Hickish TF, Norton A, Ashley S (2001) Duration of chemotherapy in advanced non-small-cell lung cancer: a randomized trial of three versus six courses of mitomycin, vinblastine, and cisplatin. J Clin Oncol 19 (5): 1336–1343.
Totani Y, Saito Y, Hayashi M, Tada T, Kohashi Y, Mieno Y, Kato A, Imizu H, Yoneda Y, Hoshino T, Uchiyama Y, Takeuchi Y, Okazawa M, Sakakibara H (2009) A phase II study of S-1 monotherapy as second-line treatment for advanced non-small cell lung cancer. Cancer Chemother Pharmacol 64 (6): 1181–1185.
Wakelee HA, Dahlberg SE, Keller SM, Gandara DR, Graziano SL, Leighl NB, Adjei AA, Schiller JH (2011) Interim report of on-study demographics and toxicity from E1505, a phase III randomized trial of adjuvant chemotherapy with or without bevacizumab for completely resected early-stage non-small cell lung cancer. J Clin Oncol 29 (15S): 456s.
Winton T, Livingston R, Johnson D, Rigas J, Johnston M, Butts C, Cormier Y, Goss G, Inculet R, Vallieres E, Fry W, Bethune D, Ayoub J, Ding K, Seymour L, Graham B, Tsao MS, Gandara D, Kesler K, Demmy T, Shepherd F (2005) Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. N Engl J Med 352 (25): 2589–2597.
Acknowledgements
This work was supported by Taiho Pharmaceutical Co., Ltd. Funding was provided by Taiho. We are indebted to Ms Miki Fukutani and Ms Yoshiko Kanazu for data management (Department of Biostatistics and Pharmaceutical Medicine, School of Pharmaceutical Sciences, Kitasato University School of Medicine, Tokyo), and Dr Teruhiko Koike (Niigata Cancer Center Hospital, Niigata), Dr Masanori Tsuchida (Niigata University Medical and Dental Hospital, Niigata), Dr Motohiro Yamashita (National Hospital Organization Shikoku Cancer Center, Matsuyama), Dr Osamu Kawashima (National Hospital Organization Nishigunma National Hospital, Shibukawa), and Dr Kazuma Kishi (Toranomon Hospital, Tokyo) for their contributions to this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Niho, S., Ikeda, N., Michimae, H. et al. Feasibility trial for adjuvant chemotherapy with docetaxel plus cisplatin followed by single agent long-term administration of S-1 chemotherapy in patients with completely resected non-small cell lung cancer: Thoracic Oncology Research Group Study 0809. Br J Cancer 109, 545–551 (2013). https://doi.org/10.1038/bjc.2013.378
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.378
