
Abstract
Background:
Fluoropyrimidine drugs are extensively used for the treatment of solid cancers. However, adverse drug reactions are a major clinical problem, often necessitating treatment discontinuation. The aim of this study was to identify pharmacogenetic markers predicting fluoropyrimidine toxicity.
Methods:
Toxicity in the first four cycles of 5-fluorouracil or capecitabine-based chemotherapy were recorded for a series of 430 patients. The association between demographic variables, DPYD, DPYS, TYMS, MTHFR, CDA genotypes, and toxicity were analysed using logistic regression models.
Results:
Four DPYD sequence variants (c.1905+1G>A, c.2846A>T, c.1601G>A and c.1679T>G) were found in 6% of the cohort and were significantly associated with grade 3–4 toxicity (P<0.0001). The TYMS 3′-untranslated region del/del genotype substantially increased the risk of severe toxicity (P=0.0123, odds ratio (OR)=3.08, 95% confidence interval (CI): 1.38–6.87). For patients treated with capecitabine, a MTHFR c.1298CC homozygous variant genotype predicted hand–foot syndrome (P=4.1 × 10−6, OR=9.99, 95% CI: 3.84–27.8). The linked CDA c.−92A>G and CDA c.−451C>T variants predicted grade 2–4 diarrhoea (P=0.0055, OR=2.3, 95% CI: 1.3–4.2 and P=0.0082, OR=2.3, 95% CI: 1.3–4.2, respectively).
Conclusion:
We have identified a panel of clinically useful pharmacogenetic markers predicting toxicity to fluoropyrimidine therapy. Dose reduction should be considered in patients carrying these sequence variants.
Similar content being viewed by others

Main
The fluoropyrimidine drug 5-fluorouracil (5-FU) and the prodrug capecitabine have been extensively used for almost 5 decades (Ezzeldin and Diasio, 2004) either as monotherapy or in combination therapy for a variety of solid cancers including gastrointestinal tract and breast. However, adverse drug reactions are a major clinical problem, often necessitating dose reduction and treatment discontinuation. Diarrhoea, mucositis, myelosuppression and hand–foot syndrome are the most frequent and troublesome side effects. A meta-analysis of 1219 colorectal cancer (CRC) patients receiving 5-FU reported that grade 3–4 toxicity was encountered in 31–34% of patients, with 0.5% mortality (Cancer M-AGI, 1998). A significant proportion of these adverse drug reactions are likely to be the result of inter-individual genetic variation. Identification of the genetic factors underlying such variation would provide a basis for individualised patient dosing strategies and a significant advance on current empirical dosing based solely on body surface area. To date, no robust pharmacogenetic markers of fluoropyrimidine toxicity have been validated for use as a standard of care for the routine management of patients with cancer (Ezzeldin and Diasio, 2008).
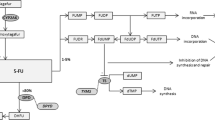
The metabolic pathways by which 5-FU and the prodrug capecitabine are converted to active nucleotide analogues are well described (Thorn et al, 2011) and have led to a number of candidate gene-based pharmacogenetic studies (Figure 1). As most of the administered 5-FU dose (80–90%) is degraded via dihydropyrimidine dehydrogenase (DPD, encoded by the DPYD gene), several studies have highlighted the role of DPD deficiency in the development of severe 5-FU related toxicity. These have been extensively reviewed by Amstutz et al (2011). Significant associations between variation in the DPYS gene encoding dihydropyrimidinase, the next step in fluoropyrimidine degradation and toxicity to 5-FU, have also been reported (Hamajima et al, 1998; van Kuilenburg et al, 2003).

Capecitabine and 5-FU metabolism. Enzymes: Carboxyl esterase (CES), deoxycytidine kinase (dCK), deoxycytidine monophosphate deaminase (dCMDA), cytidine deaminase (CDA), thymidine phosphorylase (TP), uridine phosphorylase (UP), dihydropyrimidine dehydrogenase (DPYD), dihydropyrimidinase (DPYS), orotate phosphoribosyltransferase (OPRT), uridine kinase (UK), uridine monophosphate kinase (UMPK), uridine diphosphate kinase (UDPK), ribonucleotide reductase (RNR), thymidine kinase (TK), thymidine synthase (TS), deoxyuridine triphosphatase (DUT), methylene tetrahydrofolate reductase (MTHFR). Metabolites: deoxyfluorocytidine riboside (5′-dFCR), deoxyfluorocytidine monophosphate (5′-dFCMP), deoxyfluorouridine monophosphate (5-FdUMP), deoxyfluorouracil (5′-dFUR), fluorouracil (5-FU), fluorouridine (5-FUridine), fluorouracil monophosphate (5-FUMP), fluorouracil di, tri-phosphate (5-FUDP, 5-FUTP), deoxyfluorouracil di, tri-phosphate (5-FdUDP, 5-FdUTP), deoxyuridine mono, tri –phosphate (dUMP, dUTP), deoxycytidine mono, tri-phosphate (dTMP, dTTP), 5,10-methylenetetrahydrofolate (5,10-MTHF), 5-methyltetrahydrofolate (5-MeTHF), dihydrofolate (DHF), dihydrofluorouracil (DHFU), beta-fluoroureido propionic acid (β-FUPA).
Genetic polymorphisms in the promoter and 3′-untranslated regions (3′-UTR) of the TYMS gene are known to influence TS expression and have been associated with both toxicity and an improved clinical response (Horie et al, 1995; Kawakami and Watanabe, 2003; Lecomte et al, 2004; Mandola et al, 2004). Inhibition of TS requires binding of both 5,10-methylenetetrahydrofolate (5,10-MTHF) and 5-FdUMP. Methylene tetrahydrofolate reductase (MTHFR) catalyses the conversion of 5,10-MTHF to 5-methyltetrahydrofolate (5-MTHF). Studies correlating the MTHFR 677TT and MTHFR 1298CC variant homozygous genotypes with toxicity and response have been contradictory (De Mattia and Toffoli, 2009).
Capecitabine, a prodrug of 5-FU, is first hydrolysed by carboxyl esterases in the liver to form 5-fluorodeoxycytidine, which is then deaminated by cytidine deaminase (CDA) in liver and neoplastic tissue to form 5-fluorodeoxyuridine, which in turn is converted to 5-FU by thymidine phosphorylase (TP) (Hameed and Cassidy, 2011). Decreased CDA activity is predicted to lead to the accumulation of potentially toxic fluoro-cytidine metabolites. Variation in CDA expression has been linked to polymorphism in the CDA promoter region (Fitzgerald et al, 2006) and has been suggested to impact on both gemcitabine (Gilbert et al, 2006) and capecitabine metabolism (Mercier et al, 2009; Caronia et al, 2011).
In this series of 430 patients treated with fluoropyrimidine-based chemotherapy for predominantly colorectal malignancy, we identify a panel of pharmacogenetic markers predicting toxicity.
Materials and methods
Patients and clinical data
A series of 430 patients were recruited from oncology clinics forming part of a regional cancer network in South East London, UK. Ethical approval was obtained from St Thomas’ Hospital Research Ethics Committee (07/H0802/143) and written consent was provided by all patients. For inclusion in the study, patients had to fulfil the following criteria: (1) World Health Organisation performance status <2; (2) life expectancy ⩾3 months; (3) any previous chemotherapy completed ⩾6 months ago; and (4) adequate haematological and cardiac status. Although the study was retrospective, clinical outcome data were obtained from standardised oncology outcome records completed at each clinic visit. Pre-treatment evaluation included a complete physical examination and recording of the following information: (1) baseline patient demographics (age, sex and ethnicity) and medical history; (2) diagnosis of tumour and staging (tumour, node, metastasis system); (3) current chemotherapy regimen (drug, dosing regimen) and (4) baseline blood analyses. Patients were assessed for treatment tolerance and had full blood count, renal function and liver function monitored before each chemotherapy cycle. All chemotherapy related toxicity in the first four cycles of treatment was recorded according to the National Cancer Institute Common Toxicity Criteria version 3. Patient outcome data were not disclosed to investigators undertaking the genetic analysis.
Laboratory methods
DNA was extracted from EDTA whole blood using the QIAamp DNA Mini Kit (Qiagen Ltd, Crawley, UK).
Single-nucleotide polymorphisms were genotyped by TaqMan assay (Applied Biosystems, Warrington, UK) following the manufacturer’s instructions using an Agilent Mx3005P RT–PCR instrument (Agilent Technologies, Edinburgh, UK) and are shown in Table 1. The nine candidate DPYD sequence variants genotyped were identified from the literature and from previous research in our laboratory (Loganayagam et al, 2010). Two common MTHFR deficiency associated sequence variants c.677C>T and c.1298A>C were genotyped. Coding region non-synonymous and non-coding region variants in CDA and DPYS were identified from the online single-nucleotide polymorphism (SNP) registry (http://www.ncbi.nlm.nih.gov/SNP) and included variants reported in the literature to be associated with response to fluoropyrimidine therapy or in the case of CDA, to modulate mRNA expression.
The TYMS 5′-UTR region containing the 28 bp 2R/3R tandem repeat polymorphism was amplified using primers forward 5′-CTC CGT TCT GTG CCA CAC C-3′ and reverse 5′-GTC TGT AAG GCG AGG AGG AC-3′, designed using the web-based tool primer3 (http://frodo.wi.mit.edu/) and synthesised by MWG Biotech, (Ebersberg Germany). PCR products were amplified using Hot Start DNA Polymerase (Rovalab, Teltow, Germany). PCR conditions were 1 min denaturation at 94 °C then 35 cycles of 30 s denaturation at 94 °C, 30 s annealing at 48 °C and 30 s extension at 72 °C and a last cycle consisting of 5 min extension at 72 °C. PCR products were purified using QIAquick PCR purification kit (Qiagen Ltd). Dye-terminator cycle sequencing was performed using the BigDye terminator v3.1 cycle sequencing kit (Applied Biosystems). Excess dye terminators were removed using Agencourt CleanSeq (Beckman Coulter Limited, High Wycombe, UK). Samples were run on an ABI PRISM 3130xl Genetic Analyser (Applied Biosystems). Sequences were analysed by Mutation Surveyor Local v3.20 (Softgenetics LLC, State College, PA, USA). The TYMS 5′-UTR polymorphisms were classified by both the number of repeats and the number of functional upstream stimulatory factor (USF)-binding sites (depending on G>C or C>G SNPs within the tandem repeat). The homozygous double-repeat variant genotype was designated 2R/2R, the heterozygous genotype was designated as 2R/3R, and the homozygous triple repeat as 3R/3R. Variants with higher number of repeats were grouped into 3R for statistical analysis.
TYMS c.1494del6b 3′-UTR polymorphism was genotyped by amplifying the sample twice. Both amplifications shared the same forward primer 5′-ATTACAACAGGTCGTACAATTATGGC-3′, and differed in the reverse primer. The positive 5′-CTTTATTATAGCAACATATAAAACAACTATAACT-3′ primer was designed to anneal when the 6 bp sequence was present, and the negative 5′-TTTATTATAGCAACATATAAAACAACTATA AAGT-3′ reverse primer annealed specifically to the DNA sequence when the 6 bp fragment was absent. The fragments amplified with each set of primers were ∼673 bp long. Each sample was run in consecutive lanes on a 2% standard agarose gel and scored for the presence or absence of the 673 bp band. A PCR blank and genotype controls were included in each series of PCR reactions.
Statistical analysis
Differences in the demographic and clinical characteristics of patients with and without toxicity were tested using t-tests (continuous variables) or Fisher’s exact test (discrete variables). A baseline clinical model for toxicity was determined using logistic regression and backward stepwise selection used to identify relevant clinical characteristics to be included in the model. For the genetic association testing, sex, age and ethnicity (coded as European vs non-European ancestry) were included in all models as standard covariates, together with GFR.
The frequency of each SNP was compared with the published frequencies for Caucasian individuals in dbSNP and all SNPs were tested for departure from Hardy–Weinberg equilibrium. Association was tested for two toxicity outcome measures: diarrhoea, neutropenia and mucositis in the first four cycles of treatment were dichotomised as either mild to moderate (grade 0–2) or severe (grade 3–4). In capecitabine patients, hand–foot syndrome was dichotomised as grades 0–1 and 2–3. For statistical analysis of rare variants in a gene, carriers of at least one rare variant were identified, and Fisher’s exact test was used to test for association across pooled variants with toxicity. For common variants, two analysis strategies were performed. First, a gene-based logistic regression analysis was performed across all common variants in the gene to assess cumulative evidence for association with toxicity. Second, analysis of each variant was performed using an allele-based Fisher’s exact test, and with a logistic regression model, assuming an additive genetic model within and between variants, and including baseline clinical covariates. A gene-based logistic regression model was used to predict toxicity effects (0–2 vs 3–4) with all variants genotyped within the gene, together with explanatory variables of sex, age at initiation, GFR and ethnicity (Caucasian, non-Caucasian) included in all models. Separate models were fitted for each gene. In the initial analysis, genotypes were included as an additive model (0, 1, 2). Dominant and recessive models were tested to follow-up our significant results and when testing prior hypotheses from other studies. The number of repeats (in the TYMS promoter enhancer region (TSER) was analysed as a continuous variable.
Models were tested against the baseline model with only clinical factors included (sex, age, European ancestry and GFR). Akaike’s information criterion was used to determine the SNPs to be included in the backward stepwise regression, and a likelihood ratio test was used to compare the final model with the baseline clinical model. A global model of all genetic variants was also analysed, to test for risk effects across genes for all patients and for capecitabine patients and the predictive ability of models with DPYD rare variants was compared with a model including all variants and clinical predictors. Predictive ability was assessed by the area under the curve (AUC) of the receiver-operating characteristic curve. Statistical analysis was performed using R v2.7.0.
A Bonferroni correction for multiple testing of 5 genes (DPYD, DYPS, TYMS, MTHFR, CDA) was applied, giving a significance threshold 0.01 (=0.05/5) for association P-values. Applying this across the common variants tested (n=12) would give a Bonferroni-corrected P-value of 0.0042.
Results
Of the 430 patients recruited to the study, 186 (43%) were treated with 5-FU-based chemotherapy and 244 (57%) were on capecitabine-based therapy. Demographics and clinical characteristics are shown in Table 2. The majority of patients (85%) were treated for CRC. Of the 430 patients, only 19 patients (4%) had less than a full initial fluoropyrimidine dose, receiving a 75% dose owing to poor renal function.
The fluoropyrimidine dose was reduced to 75% of the initial dose in 126/430 patients, 104 of whom were dose reduced due to grade 3–4 toxicity (Table 3). Grade 3–4 diarrhoea, mucositis or neutropenia tended to occur in a polyvisceral syndrome in the first four cycles of treatment. There were no reported cases of mortality secondary to toxicity. Treatment was stopped in 31 out of 430 patients due to grade 3–4 toxicity and in 7 patients due to either disease progression (n=2), persistent line sepsis (n=1), frailty (n=2) or patient choice (n=2). The characteristics of patients with and without grade 3–4 toxicity (diarrhoea, mucositis or neutropenia) are shown in Table 4. In agreement with previous studies, patients with grade 3–4 toxicity were older than those with grade 1–2 toxicity (P=0.025, Student’s t-test) and the average GFR values were significantly lower in the patients with grade 3–4 toxicity (P=0.0014, Student’s t-test). There was no difference in toxicity according to gender (P=0.426), by Caucasian/non-Caucasian ethnicity (P=0.351), tumour stage (P=0.8618) or by the type of fluoropyrimidine drug (P=1.000). In capecitabine-treated patients, hand–foot syndrome was assessed on a severity scale 0–3, with 55 of 244 patients (23%) having severity score of 2–3.
Pharmacogenetic markers for fluoropyrimidine toxicity and clinical response
The allele frequencies in the study cohort are shown in Table 1, and were similar to those reported in online databases. All genotypes were in Hardy–Weinberg equilibrium. The genotyping success rate for all polymorphisms studied was >99%.
Four rare DPYD variants predict grade 3–4 toxicity
The study cohort was genotyped for nine DPYD sequence variants of which six variants were polymorphic in the cohort. Four rare DPYD sequence variants (c.1905+1G>A, c.2846A>T, c.1601G>A and c.1679T>G) known to be associated with DPD deficiency were present in 24 patients in heterozygous or compound heterozygous genotypes. All 24 patients experienced grade 3–4 diarrhoea, mucositis and/or neutropaenia in the first four cycles of chemotherapy (Table 5), P<10−16, logistic regression. Two patients were compound heterozygous for the variants c.1601G>A/c.1905+1G>A and c.1601G>A/c.2846A>T, and both suffered grade 4 toxicity within the first two cycles, requiring hospital admission for 16 and 19 days, respectively. Of the three patients with heterozygous c.1905+1G>A genotypes, two patients experienced toxicity in the first two cycles of therapy, and continued therapy after 25 and 50% dose reductions. The third patient experienced grade 3–4 toxicities in cycles 3–4 and discontinued therapy. Ten of fourteen patients heterozygous for the c.1601G>A variant experienced 3–4 toxicity during the first two cycles of therapy. Eight patients tolerated a 25% dose reduction, two discontinued therapy, the remaining patient was not dose adjusted and suffered grade 4 toxicity in the subsequent cycle. Four patients experienced toxicity in cycle 3 and three tolerated a 25% dose reduction with one patient withdrawing from therapy. Three of four patients with heterozygous c.2846A>T genotypes experienced early grade 3–4 toxicity, one of whom discontinued and two continued therapy after 25% dose reductions. Toxicity was delayed to cycle 3 in the fourth patient and this patient tolerated a 25% dose reduction. The single patient with a heterozygous c.1679T>G variant genotype tolerated a 25% dose reduction after experiencing toxicity in cycles 3–4. The sensitivity, specificity, positive and negative-predictive values for the DPYD variants and toxicity are shown in Table 6. When all four variants were considered, the positive-predictive value for toxicity was 98% with a specificity of 100%.
Overall, the 104 patients experiencing grade 3–4 toxicity were admitted to hospital for a total of 423 days compared with 65 days for the 326 patients with grade 0–2 toxicity. Although the 24 patients carrying a c.1601G>A, c.1679T>G, c.1905+1G>A or c.2846A>T sequence variant comprised just 6% of the cohort, admissions for these patients accounted for 171/488 admission days or 35% of the total.
The DPYD intronic variant rs75017182 (c.1129-5923C>G) has been reported to be associated with mis-splicing of an intronic sequence into DPYD mRNA and 5-FU toxicity (van Kuilenburg et al, 2010). In our study, only heterozygous patients were found (n=15), none of which carried one of the rare variants analysed above. This variant, rs75017182, was not associated with grade 3–4 diarrhoea, mucositis or neutropaenia overall (P=0.2210). When the 24 patients with a deleterious DPYD genotype were excluded, the association between a heterozygous rs75017182 genotype and toxicity strengthened, but did not reach significance (P=0.088). The positive-predictive value for this variant was just 40% (Table 6). The common variant DPYD c.496A>G (p.Met166Val) was not associated with toxicity (P=0.3957).
DPYS, MTHFR and TYMS polymorphisms
The association between polymorphisms in DPYS, TYMS and MTHFR and toxicity is shown in Table 7. Owing to the highly significant association between the four rare DPYD sequence variants (c.1905+1G>A, c.2846A>T, c.1601G>A or c.1679T>G) and grade 3 and 4 toxicity, the 24 patients with variant DPYD genotypes were excluded from subsequent analysis. Of the four DPYS coding sequence variants tested, two were not polymorphic in the cohort (rs34895123 (c.937T>A, p.Asn313Tyr) and rs61758444 (c.1423C>T p.Arg475Ter)). The rare coding region variant rs36027551 (DPYS c.541C>T, p.Arg181Trp) was polymorphic but was not significantly associated with toxicity (5 carriers observed; 1 carrier experienced toxicity; P=1.00). The common intron 1 sequence variant rs2669429 (c.265-58T>C) previously reported to protect against side effects (Fidlerova et al, 2010) was not significantly associated with protection from diarrhoea, mucositis and/or neutropaenia (Table 7; P=0.513).
The five TYMS-coding region sequence variants reported in SNP databases and listed in Table 1 were not polymorphic in the cohort. Sequencing of the TYMS promoter region revealed considerable variation, with the number of repeats varying from 2 to 4. No association between repeat number and toxicity was seen in the either logistic regression model (P=0.080) or Fisher’s exact test, classifying ⩽2 variants and >2 variants (P=0.140). The number of functional USF-binding sites present in the repeat region, which is a function of additional single-nucleotide substitutions in the repeat region, did not predict toxicity. The gene-based analysis of TYMS variants showed some evidence of association (P=0.031), which would not withstand correction for multiple testing of genes. Further analyses of these variants by genotype did not strengthen the association.
Given the reported influence of the TYMS 3′-UTR c.1494del6b variant on mRNA stability, further exploration suggested a recessive effect for the deletion with Fisher’s exact test showing a significant association between the rare homozygous del/del genotype and diarrhoea, neutropaenia and mucositis (P=0.0123, OR=3.08, 95% confidence interval (CI): 1.38–6.87).
The MTHFR 677C>T and 1298A>C variants were not significantly associated with grade 3–4 diarrhoea, mucositis and neutropenia in an analysis including both 5-FU and capecitabine-treated patients (Table 7).
Full modelling analysis of all genotyped variants showed the best fitting model with rare DPYD variants, the number of functional USF-binding sites in the 5′-UTR of TYMS, and the TYMS 3′-UTR deletion (full results shown in Supplementary Materials). However, adding the common variants provided little increase in predication compared with a model with the clinical variables only, with AUC increasing from 0.72–0.74 (Figure 2).

Receiver-operating characteristic curves for the prediction models with all variables (rare DPYD variants, common variants, clinical variables) for all patients ( A ) and the capecitabine cohort ( B).
Subgroup analysis: Capecitabine Cohort
Three sequence variants in the CDA promoter region, c.−92A>G (rs602950), c.−451C>T (rs532545) and c.−943insC (rs3215400) have been associated with increased CDA expression in vitro. After excluding 12 capecitabine patients with known DPYD mutations, the logistic regression model showed evidence of an association between the presence of the CDA c.−92A>G variant and diarrhoea, mucositis and neutropaenia (P=0.052), although this was not significant using Fisher’s exact test (P=0.38). Further analysis of five toxicity phenotypes in a stepwise logistic regression including only the c.−92A>G polymorphism, revealed an association between a heterozygous or homozygous c.−92A>G genotype and the development of grade 2–4 diarrhoea (P=0.002) and dehydration (P=0.042) in the first four cycles of chemotherapy (Table 8). The c.−92G variant allele showed an additive effect with each c−92G allele present increasing the risk of diarrhoea two-fold (Fisher’s exact test, P=0.0055, OR=2.3, 95% CI: 1.3–4.2). Similarly, the CDA c.−451C>T variant was also associated with the development of grade 2–4 diarrhoea in the first four cycles of chemotherapy (P=0.0082, OR=2.3, 95% CI: 1.3–4.2). There is a strong linkage disequilibrium between these two variants (r2=0.95) and only 2.2% of haplotypes showed discordant alleles. The promoter variant c.−943insC was not associated with toxicity nor were any of the three CDA variants associated with hand–foot syndrome. A global model of all variants for toxicity showed the best fitting model with rare DPYD variants and the CDA c.−92A>G genotype. However, as with the full cohort, adding the common variants provided little increase in prediction, with AUC increasing only from 0.74 to 0.75 with the addition of common SNPs to a model with DYPD rare variants and clinical variables (Figure 2).
Unexpectedly, a subgroup analysis restricted to patients treated with capecitabine revealed a significant association between MTHFR677T and MTHFR1298C variant genotypes and hand–foot syndrome (P=0.0046; logistic regression model). The best fitting model was a recessive model for MTHFR 1298CC genotypes, which significantly increased the risk of hand–foot syndrome (logistic regression, P=4.1 × 10−6, OR=9.99, 95% CI: 3.84–27.8).
Discussion
We have previously reported that three DPYD variants (c.1905+1G>A, c.1679T>G (p.I560S) and c.2846A>T (p.D949V)) are prevalent in the UK population and are significant associated with grade 3–4 toxicity (Loganayagam et al, 2010). In the well-powered study reported here, we have confirmed these findings, and we have identified a fourth DPYD variant c.1601G>A (p.S534N) significantly associated with severe fluoropyrimidine toxicity. A total of 24 patients were heterozygous or compound heterozygous for these variants and all 24 patients experienced severe diarrhoea, mucositis and/or neutropenia in the first four cycles of therapy. In line with previous reports, DPD deficiency accounted for 23% of cases with grade 3–4 toxicity (Morel et al, 2006; Loganayagam et al, 2010). Patients with a variant DPYD allele comprised 6% of the cohort but accounted for a disproportionate 35% of all hospital admission days.
Dihydropyrimidine dehydrogenase deficiency is recognised as a significant cause of grade 3–4 toxicity (Amstutz et al, 2011) and the four DPYD variants we found to be associated with severe toxicity have been previously described in the literature as variants associated with decreased DPD enzyme activity and a high risk of severe 5-FU toxicity (van Kuilenburg et al, 2002; Seck et al, 2005; Morel et al, 2006; Loganayagam et al, 2010). The majority of patients 18/24 (75%) carrying a DPYD variant experienced severe toxicity in the first two cycles of therapy, with the remaining 6 patients experiencing severe toxicity in cycle 3 or 4. Although different in design, our study is broadly in line with the findings of Deenen et al (2011) who reported that not all patients carrying a DPYD variant experienced severe toxicity in the first two cycles of therapy, specifically, we and Deenen et al (2011), found that all patients heterozygous for the 1905+1G>A intron 14 splice site variant experienced severe toxicity, contrasting markedly with the findings of those of Schwab et al (2008) who reported toxicity in just 6/13 patients with the DPYD intron 14 splice variant. For the c.2846A>T variant we found 3/4 patients with a heterozygous genotype experienced severe toxicity in the first two cycles of therapy, not dissimilar to the 62% of patients experiencing toxicity within the first two cycles reported in the Deenen et al (2011) study and the 3/5 patients reported by Schwab et al (2008). The c.1601G>A variant is the most frequent toxicity-associated variant identified in our study, with 14 patients carrying the variant allele in a heterozygous genotype of whom 10 (71%) experienced early severe toxicity. These results differ markedly from those of Schwab et al (2008) where 2/7 cases carrying this variant experienced toxicity. Similarly, Amstutz et al (2009) reported that 1/5 patients heterozygous for this variant experienced toxicity. Interestingly, although Deenen et al (2011) did not find a significant association between the c.1601G>A variant and early toxicity, this study did report the specificity for this variant for any toxicity as 94% with a positive-predictive values of 83%, comparable to the specificity of 100% and positive-predictive value of >99% found in our study (Table 6). There is additional evidence that the c.1601G>A (p.S534N) variant impacts on DPD activity (Seck et al, 2005) and the variant has been associated with severe toxicity in a series of smaller studies (Collie-Duguid et al, 2000; Gross et al, 2003). The single patient with a heterozygous c.1679T>G genotype experienced late toxicity after cycle 2.
We were unable to replicate the significant association between the deep intronic DPYD variant rs75017182 (c.1129-5923C>G), which is in disequilibrium with the synonymous exonic variant c.1236G>A, and toxicity. The intronic variant is reported to result in the insertion of 44 base pair sequence derived from intron 10 into DPYD mRNA and was found to be significantly (P=0.033) overrepresented in a retrospective cohort of 203 cancer patients (van Kuilenburg et al, 2010) and in a c.1236G>A haplotype, with severe diarrhoea (Deenen et al, 2011). Similarly we found that the c.496A>G (p.Met166Val) variant was not significantly associated with toxicity, contrary to the report of Gross et al (2008).
Small case studies have suggested that deficiency of dihydropyrimidinase, the second enzyme in 5-FU catabolism, results in severe 5-FU toxicity (Hamajima et al, 1998; van Kuilenburg et al, 2003). However, in our study, only one out of the three DPYS coding SNPs genotyped was polymorphic in the cohort and no significant associations were found. The intron 1 sequence variant rs2669429 (c.265-58T>C), previously reported to protect against side effects (Fidlerova et al, 2010), was not significantly associated with protection from diarrhoea, mucositis and/or neutropaenia.
Thymidylate synthase is a key enzyme in thymidine nucleotide biosynthesis, and is the main intracellular target of the active 5-FU metabolite, FdUMP, which forms a ternary complex with TS and 5,10-MTHF (Pinedo and Peters, 1988). Variable numbers (2–9) of a 28-bp tandem repeat sequence (VNTR) are present in the 5′-UTR in the TSER with the most frequent being 2R and 3R repeats. The 2R repeat contains one USF-binding site and two are found in the 3R. Functional studies have shown a stepwise increase in TS transcription with an increasing number of tandem repeats (Horie et al, 1995). Additional polymorphic variation within repeats also alters USF-binding sequences and influences TYMS transcription (Kawakami and Watanabe, 2003; Mandola et al, 2003; Lincz et al, 2007). We found no significant associations between the number of TYMS 5′-UTR region repeats, or the number of functional USF-binding sites and toxicity. The findings from our study differ from a large prospective study where the high expression genotypes 2R/3R or 3R/3R were significantly associated with a lower risk of toxicity, (Schwab et al, 2008). Other studies have produced contradictory results (Largillier et al, 2006; Schwab et al, 2008; Sharma et al, 2008; Gusella et al, 2009). We would agree with a recent meta-analysis concluding that although variation in the TYMS 5′-UTR region may be associated with adverse reactions, the effect is likely to be small and testing would not be clinically useful (Jennings et al, 2012).
The 6-bp deletion in the 3′-UTR region of the TYMS gene (TYMS c.1494del6b) has been reported to modulate gene regulation at a post-transcriptional level through decreased mRNA stability (Mandola et al, 2004). We found a significant association between the c.1494del6bp variant and toxicity. The presence of a homozygous del/del genotype approximately doubled the risk of grade 3–4 toxicity. However, other studies have failed to show an association between a homozygous del/del genotype and severe toxicity (Sharma et al, 2008; Braun et al, 2009; Lurje et al, 2009). With a specificity for the homozygous genotype and toxicity of 54% and a positive-predictive value of just 25%, we suggest that testing for this variant would not be clinically useful.
The MTHFR 677C>T and 1298A>C variant genotypes were not significantly associated with severe 5-FU toxicity overall. This is in agreement with previous published studies (Cohen et al, 2003; Ruzzo et al, 2007, 2008; Schwab et al, 2008) but contradicts the findings of others, which have reported significant associations between variant MTHFR genotypes and clinical outcomes (Sharma et al, 2008; Afzal et al, 2009; Gusella et al, 2009; Castillo-Fernandez et al, 2010).
In a subgroup analysis, we then analysed the 244 patients treated with the fluoropyrimidine prodrug capecitabine. The MTHFR c.1298A>C polymorphism was significantly associated with grade 2–3 hand–foot syndrome, which occurred in 22% of patients. Patients with the MTHFR 1298CC homozygous variant genotype were ten times more likely to develop hand–foot syndrome. Methylene tetrahydrofolate reductase catalyses the irreversible conversion of 5,10-MTHF to 5-MTHF. As 5,10-MTHF inhibits TS activity in conjunction with 5-FdUMP, reduced MTHFR activity, which is associated with increased levels of 5,10-MTHF, theoretically leads to more effective TS inhibition. The association between the MTHFR 1298CC genotype and hand–foot syndrome would be consistent with a localised increase in the conversion of capeticabine to active metabolites and increased inhibition of TS. Interestingly, TP, which catalyses the conversion of the intermediate 5-fluorodeoxyuridine to 5-FU, is highly expressed in the skin, and activity of this enzyme has been suggested to be a mechanism whereby a localised increase in the production of 5-FU leads to hand–foot syndrome (Milano et al, 2008). Variation in CDA has also been previously reported to be associated with hand–foot syndrome. Caronia et al (2011) found a significant association between the CDA c.−451C>T variant allele and grade 3 hand–foot syndrome, and a protective effect for the CDA rs315400insC variant (Caronia et al, 2011), although the latter variant was not significantly associated with toxicity in an earlier study (Ribelles et al, 2008). We were, however, unable to replicate these associations.
The CDA promoter region variants, c.−92 A>G (rs602950) and c.−451C>T (rs532545), have been associated with increased CDA expression in vitro (Fitzgerald et al, 2006) and we predicted that these variants would influence capecitabine metabolism and hence clinical outcome to therapy. In our study, the CDA promoter region variants, c.−92 A>G and c.−451C>T, were associated with grade 2–4 diarrhoea, and capecitabine-treated patients with variant genotypes twice as likely to develop diarrhoea in the first four cycles of chemotherapy. However, the positive-predictive value of 40% is relatively low and further studies are needed to determine whether testing for this variant is clinically useful.
In conclusion, our results, and those of others, provide convincing evidence that patients with the four variant DPYD genotypes c.1905+1G>A, c.1601G>A, c.1679T>G and c.2846A>T will experience grade 3–4 toxicity. The combined positive predicted value for the four variants is >99% with a negative-predictive value of 80%. We suggest there is sufficient evidence to justify testing for these variants before the start of therapy and dose reducing patients with variant genotypes. For the c.1905+1G>A variant, we agree with previous reports suggesting that the severity of side effects will be reduced by a dose reduction of 50–60% before the start of therapy (Deenen et al, 2011; Yang et al, 2011; van Kuilenburg et al, 2012), while patients carrying the other variant DPYD genotypes may benefit from a lesser dose reduction of 25%.
Diarrhoea and hand–foot syndrome have been reported to be amongst the most frequent toxicities requiring dose reduction patients on capecitabine therapy (Walko and Lindley, 2005). We have identified two markers in the CDA gene, which are significantly associated with grade 2–4 diarrhoea. There is, however, currently no evidence supporting a dose reduction strategy in patients with variant CDA genotypes. At the very least, these markers may be used by clinicians to anticipate severe diarrhoea and to prescribe appropriate doses of anti-diarrhoeal agents. We have also identified a homozygous MTHFR 1298CC variant genotype as a pharmacogenetic marker MTHFR for hand–foot syndrome in patients treated with capecitabine. Avoidance of hand–foot syndrome would add substantially to quality of life. Further studies are needed to determine whether capecitabine dose reduction would compromise clinical efficacy.
Finally, the majority of cases of severe toxicity remain unexplained. Pharmacogenetic variation in genes involved in the metabolism of co-therapies are likely to explain a subset of these adverse events.
Change history
25 June 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Afzal S, Jensen SA, Vainer B, Vogel U, Matsen JP, Sorensen JB, Andersen PK, Poulsen HE (2009) MTHFR polymorphisms and 5-FU-based adjuvant chemotherapy in colorectal cancer. Ann Oncol 20 (10): 1660–1666.
Amstutz U, Farese S, Aebi S, Largiader CR (2009) Dihydropyrimidine dehydrogenase gene variation and severe 5-fluorouracil toxicity: a haplotype assessment. Pharmacogenomics 10 (6): 931–944.
Amstutz U, Froehlich TK, Largiader CR (2011) Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5-fluorouracil toxicity. Pharmacogenomics 12 (9): 1321–1336.
Braun MS, Richman SD, Thompson L, Daly CL, Meade AM, Adlard JW, Allan JM, Parmar MK, Quirke P, Seymour MT (2009) Association of molecular markers with toxicity outcomes in a randomized trial of chemotherapy for advanced colorectal cancer: the FOCUS trial. J Clin Oncol 27 (33): 5519–5528.
Cancer M-AGI (1998) Toxicity of fluorouracil in patients with advanced colorectal cancer: effect of administration schedule and prognostic factors. J Clin Oncol 16: 3537–3541.
Caronia D, Martin M, Sastre J, de la Torre J, Garcia-Saenz JA, Alonso MR, Moreno LT, Pita G, Diaz-Rubio E, Benitez J, Gonzalez-Neira A (2011) A polymorphism in the cytidine deaminase promoter predicts severe capecitabine-induced hand-foot syndrome. Clin Cancer Res 17 (7): 2006–2013.
Castillo-Fernandez O, Santibanez M, Bauza A, Calderillo G, Castro C, Herrera R, Serrano A, Arrieta O, Herrera LA (2010) Methylenetetrahydrofolate reductase polymorphism (677 C>T) predicts long time to progression in metastatic colon cancer treated with 5-fluorouracil and folinic acid. Arch Med Res 41 (6): 430–435.
Cohen V, Panet-Raymond V, Sabbaghian N, Morin I, Batist G, Rozen R (2003) Methylenetetrahydrofolate reductase polymorphism in advanced colorectal cancer: a novel genomic predictor of clinical response to fluoropyrimidine-based chemotherapy. Clin Cancer Res 9 (5): 1611–1615.
Collie-Duguid ES, Etienne MC, Milano G, McLeod HL (2000) Known variant DPYD alleles do not explain DPD deficiency in cancer patients. Pharmacogenetics 10 (3): 217–223.
De Mattia E, Toffoli G (2009) C677T and A1298C MTHFR polymorphisms, a challenge for antifolate and fluoropyrimidine-based therapy personalisation. Eur J Cancer 45 (8): 1333–1351.
Deenen MJ, Tol J, Burylo AM, Doodeman VD, de Boer A, Vincent A, Guchelaar HJ, Smits PH, Beijnen JH, Punt CJ, Schellens JH, Cats A (2011) Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin Cancer Res 17 (10): 3455–3468.
Ezzeldin H, Diasio R (2004) Dihydropyrimidine dehydrogenase deficiency, a pharmacogenetic syndrome associated with potentially life-threatening toxicity following 5-fluorouracil administration. Clin Colorectal Cancer 4 (3): 181–189.
Ezzeldin HH, Diasio RB (2008) Predicting fluorouracil toxicity: can we finally do it? J Clin Oncol 26 (13): 2080–2082.
Fidlerova J, Kleiblova P, Bilek M, Kormunda S, Formankova Z, Novotny J, Kleibl Z (2010) Contribution of dihydropyrimidinase gene alterations to the development of serious toxicity in fluoropyrimidine-treated cancer patients. Cancer Chemother Pharmacol 65 (4): 661–669.
Fitzgerald SM, Goyal RK, Osborne WR, Roy JD, Wilson JW, Ferrell RE (2006) Identification of functional single nucleotide polymorphism haplotypes in the cytidine deaminase promoter. Hum Genet 119 (3): 276–283.
Gilbert JA, Salavaggione OE, Ji Y, Pelleymounter LL, Eckloff BW, Wieben ED, Ames MM, Weinshilboum RM (2006) Gemcitabine pharmacogenomics: cytidine deaminase and deoxycytidylate deaminase gene resequencing and functional genomics. Clin Cancer Res 12 (6): 1794–1803.
Gross E, Busse B, Riemenschneider M, Neubauer S, Seck K, Klein HG, Kiechle M, Lordick F, Meindl A (2008) Strong association of a common dihydropyrimidine dehydrogenase gene polymorphism with fluoropyrimidine-related toxicity in cancer patients. PLoS One 3 (12): e4003.
Gross E, Ullrich T, Seck K, Mueller V, de Wit M, von Schilling C, Meindl A, Schmitt M, Kiechle M (2003) Detailed analysis of five mutations in dihydropyrimidine dehydrogenase detected in cancer patients with 5-fluorouracil-related side effects. Hum Mutat 22 (6): 498.
Gusella M, Frigo AC, Bolzonella C, Marinelli R, Barile C, Bononi A, Crepaldi G, Menon D, Stievano L, Toso S, Pasini F, Ferrazzi E, Padrini R (2009) Predictors of survival and toxicity in patients on adjuvant therapy with 5-fluorouracil for colorectal cancer. Br J Cancer 100 (10): 1549–1557.
Hamajima N, Kouwaki M, Vreken P, Matsuda K, Sumi S, Imaeda M, Ohba S, Kidouchi K, Nonaka M, Sasaki M, Tamaki N, Endo Y, De Abreu R, Rotteveel J, van Kuilenburg A, van Gennip A, Togari H, Wada Y (1998) Dihydropyrimidinase deficiency: structural organization, chromosomal localization, and mutation analysis of the human dihydropyrimidinase gene. Am J Hum Genet 63 (3): 717–726.
Hameed H, Cassidy J (2011) Use of capecitabine in management of early colon cancer. Cancer Manag Res 3: 295–299.
Horie N, Aiba H, Oguro K, Hojo H, Takeishi K (1995) Functional analysis and DNA polymorphism of the tandemly repeated sequences in the 5′-terminal regulatory region of the human gene for thymidylate synthase. Cell Struct Funct 20 (3): 191–197.
Jennings BA, Kwok CS, Willis G, Matthews V, Wawruch P, Loke YK (2012) Functional polymorphisms of folate metabolism and response to chemotherapy for colorectal cancer, a systematic review and meta-analysis. Pharmacogenet Genomics 22 (4): 290–304.
Kawakami K, Watanabe G (2003) Identification and functional analysis of single nucleotide polymorphism in the tandem repeat sequence of thymidylate synthase gene. Cancer Res 63 (18): 6004–6007.
Largillier R, Etienne-Grimaldi MC, Formento JL, Ciccolini J, Nebbia JF, Ginot A, Francoual M, Renee N, Ferrero JM, Foa C, Namer M, Lacarelle B, Milano G (2006) Pharmacogenetics of capecitabine in advanced breast cancer patients. Clin Cancer Res 12 (18): 5496–5502.
Lecomte T, Ferraz JM, Zinzindohoue F, Loriot MA, Tregouet DA, Landi B, Berger A, Cugnenc PH, Jian R, Beaune P, Laurent-Puig P (2004) Thymidylate synthase gene polymorphism predicts toxicity in colorectal cancer patients receiving 5-fluorouracil-based chemotherapy. Clin Cancer Res 10 (17): 5880–5888.
Lincz LF, Scorgie FE, Garg MB, Ackland SP (2007) Identification of a novel single nucleotide polymorphism in the first tandem repeat sequence of the thymidylate synthase 2R allele. Int J Cancer 120 (9): 1930–1934.
Loganayagam A, Arenas-Hernandez M, Fairbanks L, Ross P, Sanderson JD, Marinaki AM (2010) The contribution of deleterious DPYD gene sequence variants to fluoropyrimidine toxicity in British cancer patients. Cancer Chemother Pharmacol 65 (2): 403–406.
Lurje G, Manegold PC, Ning Y, Pohl A, Zhang W, Lenz HJ (2009) Thymidylate synthase gene variations: predictive and prognostic markers. Mol Cancer Ther 8 (5): 1000–1007.
Mandola MV, Stoehlmacher J, Muller-Weeks S, Cesarone G, Yu MC, Lenz HJ, Ladner RD (2003) A novel single nucleotide polymorphism within the 5' tandem repeat polymorphism of the thymidylate synthase gene abolishes USF-1 binding and alters transcriptional activity. Cancer Res 63 (11): 2898–2904.
Mandola MV, Stoehlmacher J, Zhang W, Groshen S, Yu MC, Iqbal S, Lenz HJ, Ladner RD (2004) A 6 bp polymorphism in the thymidylate synthase gene causes message instability and is associated with decreased intratumoral TS mRNA levels. Pharmacogenetics 14 (5): 319–327.
Mercier C, Dupuis C, Blesius A, Fanciullino R, Yang CG, Padovani L, Giacometti S, Frances N, Iliadis A, Duffaud F, Ciccolini J (2009) Early severe toxicities after capecitabine intake: possible implication of a cytidine deaminase extensive metabolizer profile. Cancer Chemother Pharmacol 63 (6): 1177–1180.
Milano G, Etienne-Grimaldi MC, Mari M, Lassalle S, Formento JL, Francoual M, Lacour JP, Hofman P (2008) Candidate mechanisms for capecitabine-related hand-foot syndrome. Br J Clin Pharmacol 66 (1): 88–95.
Morel A, Boisdron-Celle M, Fey L, Soulie P, Craipeau MC, Traore S, Gamelin E (2006) Clinical relevance of different dihydropyrimidine dehydrogenase gene single nucleotide polymorphisms on 5-fluorouracil tolerance. Mol Cancer Ther 5 (11): 2895–2904.
Pinedo HM, Peters GF (1988) Fluorouracil: biochemistry and pharmacology. J Clin Oncol 6 (10): 1653–1664.
Ribelles N, Lopez-Siles J, Sanchez A, Gonzalez E, Sanchez MJ, Carabantes F, Sanchez-Rovira P, Marquez A, Duenas R, Sevilla I, Alba E (2008) A carboxylesterase 2 gene polymorphism as predictor of capecitabine on response and time to progression. Curr Drug Metab 9 (4): 336–343.
Ruzzo A, Graziano F, Loupakis F, Rulli E, Canestrari E, Santini D, Catalano V, Ficarelli R, Maltese P, Bisonni R, Masi G, Schiavon G, Giordani P, Giustini L, Falcone A, Tonini G, Silva R, Mattioli R, Floriani I, Magnani M (2007) Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFOX-4 chemotherapy. J Clin Oncol 25 (10): 1247–1254.
Ruzzo A, Graziano F, Loupakis F, Santini D, Catalano V, Bisonni R, Ficarelli R, Fontana A, Andreoni F, Falcone A, Canestrari E, Tonini G, Mari D, Lippe P, Pizzagalli F, Schiavon G, Alessandroni P, Giustini L, Maltese P, Testa E, Menichetti ET, Magnani M (2008) Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFIRI chemotherapy. Pharmacogenomics J 8 (4): 278–288.
Schwab M, Zanger UM, Marx C, Schaeffeler E, Klein K, Dippon J, Kerb R, Blievernicht J, Fischer J, Hofmann U, Bokemeyer C, Eichelbaum M (2008) Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: a prospective clinical trial by the German 5-FU Toxicity Study Group. J Clin Oncol 26 (13): 2131–2138.
Seck K, Riemer S, Kates R, Ullrich T, Lutz V, Harbeck N, Schmitt M, Kiechle M, Diasio R, Gross E (2005) Analysis of the DPYD gene implicated in 5-fluorouracil catabolism in a cohort of Caucasian individuals. Clin Cancer Res 11 (16): 5886–5892.
Sharma R, Hoskins JM, Rivory LP, Zucknick M, London R, Liddle C, Clarke SJ (2008) Thymidylate synthase and methylenetetrahydrofolate reductase gene polymorphisms and toxicity to capecitabine in advanced colorectal cancer patients. Clin Cancer Res 14 (3): 817–825.
Thorn CF, Marsh S, Carrillo MW, McLeod HL, Klein TE, Altman RB (2011) PharmGKB summary: fluoropyrimidine pathways. Pharmacogenet Genomics 21 (4): 237–242.
van Kuilenburg AB, Dobritzsch D, Meinsma R, Haasjes J, Waterham HR, Nowaczyk MJ, Maropoulos GD, Hein G, Kalhoff H, Kirk JM, Baaske H, Aukett A, Duley JA, Ward KP, Lindqvist Y, van Gennip AH (2002) Novel disease-causing mutations in the dihydropyrimidine dehydrogenase gene interpreted by analysis of the three-dimensional protein structure. Biochem J 364 (Pt 1): 157–163.
van Kuilenburg AB, Hausler P, Schalhorn A, Tanck MW, Proost JH, Terborg C, Behnke D, Schwabe W, Jabschinsky K, Maring JG (2012) Evaluation of 5-fluorouracil pharmacokinetics in cancer patients with a c.1905+1G>A mutation in DPYD by means of a Bayesian limited sampling strategy. Clin Pharmacokinet 51 (3): 163–174.
van Kuilenburg AB, Meijer J, Mul AN, Meinsma R, Schmid V, Dobritzsch D, Hennekam RC, Mannens MM, Kiechle M, Etienne-Grimaldi MC, Klumpen HJ, Maring JG, Derleyn VA, Maartense E, Milano G, Vijzelaar R, Gross E (2010) Intragenic deletions and a deep intronic mutation affecting pre-mRNA splicing in the dihydropyrimidine dehydrogenase gene as novel mechanisms causing 5-fluorouracil toxicity. Hum Genet 128 (5): 529–538.
van Kuilenburg AB, Meinsma R, Zonnenberg BA, Zoetekouw L, Baas F, Matsuda K, Tamaki N, van Gennip AH (2003) Dihydropyrimidinase deficiency and severe 5-fluorouracil toxicity. Clin Cancer Res 9 (12): 4363–4367.
Walko CM, Lindley C (2005) Capecitabine: a review. Clin Ther 27 (1): 23–44.
Yang CG, Ciccolini J, Blesius A, Dahan L, Bagarry-Liegey D, Brunet C, Varoquaux A, Frances N, Marouani H, Giovanni A, Ferri-Dessens RM, Chefrour M, Favre R, Duffaud F, Seitz JF, Zanaret M, Lacarelle B, Mercier C (2011) DPD-based adaptive dosing of 5-FU in patients with head and neck cancer: impact on treatment efficacy and toxicity. Cancer Chemother Pharmacol 67 (1): 49–56.
Acknowledgements
We acknowledge funding from Purine Metabolic Patient Association (PUMPA), CANHELP and Guy’s and St Thomas’ Charity. We acknowledge support from the National Institutes of Health Research Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Loganayagam, A., Arenas Hernandez, M., Corrigan, A. et al. Pharmacogenetic variants in the DPYD, TYMS, CDA and MTHFR genes are clinically significant predictors of fluoropyrimidine toxicity. Br J Cancer 108, 2505–2515 (2013). https://doi.org/10.1038/bjc.2013.262
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.262
Keywords
This article is cited by
-
Dihydropyrimidine dehydrogenase gene variants for predicting grade 4-5 fluoropyrimidine-induced toxicity: FUSAFE individual patient data meta-analysis
British Journal of Cancer (2024)
-
Impact of pharmacogenomic DPYD variant guided dosing on toxicity in patients receiving fluoropyrimidines for gastrointestinal cancers in a high-volume tertiary centre
BMC Cancer (2023)
-
Fluoropyrimidine-associated toxicity and DPYD variants c.85T>C, c.496A>G, and c.1236G>A: impact of haplotype
Cancer Chemotherapy and Pharmacology (2023)
-
Role of Genetic Polymorphisms in Drug-Metabolizing Enzyme-Mediated Toxicity and Pharmacokinetic Resistance to Anti-Cancer Agents: A Review on the Pharmacogenomics Aspect
Clinical Pharmacokinetics (2022)
-
The prevalence and clinical relevance of 2R/2R TYMS genotype in patients with gastrointestinal malignancies treated with fluoropyrimidine-based chemotherapy regimens
The Pharmacogenomics Journal (2021)
