
Abstract
Background:
Combination of S-1, an oral fluorouracil derivative, plus docetaxel against non-small cell lung cancer (NSCLC) showed promising efficacy but clinically problematic emesis. A phase I/II study utilising a new schedule for this combination was conducted.
Methods:
A biweekly regimen of docetaxel on day 1 with oral S-1 on days 1–7 was administered to previously treated NSCLC patients. Doses of docetaxel/S-1 were escalated to 30/80, 35/80, and 40/80 mg m−2, respectively, and its efficacy was investigated at the recommended dose below maximum tolerated dose (MTD).
Results:
In phase I study employing 13 patients, dose-limiting toxicities were febrile neutropenia and treatment delay, with the respective MTDs for docetaxel 40 mg m−2/S-1 80 mg m−2. In the phase II study, 34 patients were treated with docetaxel 35 mg m−2/S-1 80 mg m−2 for a median cycle of 6. The response and disease control rates were 34.3% (95% confidence interval (CI), 18.6–50.0%) and 62.9% (95% CI, 46.8–72.9%), respectively. Median progression-free survival was 150.5 days. Haematologic grade 4 toxicities were observed in neutropenia (11.8%) and thrombocytopenia (2.9%). Regarding non-haematologic toxicities, including emesis, there were no grade 3/4 side effects.
Conclusion:
Combination of 1-week administration of S-1 with biweekly docetaxel is safe and active for NSCLC.
Similar content being viewed by others

Main
For patients with advanced or metastatic non-small cell lung cancer (NSCLC), platinum-based chemotherapy is the mainstay of first-line treatment on the basis of the moderate improvement in survival and quality of life (Sandler et al, 2007; Scagliotti et al, 2008; Azzoli et al, 2009; Reck et al, 2009). Recently, first-line therapy with a tyrosine kinase inhibitor (TKI) was employed in the case of individuals with advanced NSCLC harbouring EGFR mutations (Maemondo et al, 2010; Mitsudomi et al, 2010; Zhou et al, 2011; Rosell et al, 2011); however, most of patients with mutated tumours were eventually treated with platinum-based chemotherapy after progression on TKI treatment. After failure of platinum-based regimens in NSCLC patients with or without EGFR mutations, current options for second-line treatment include single-agent therapy with docetaxel, pemetrexed, or erlotinib (Gridelli et al, 2008). However, the efficacy of such treatments have remained largely unsatisfactory, with reported response rates in unselected NSCLC patients that had been treated previously being 6.7–10.8% with docetaxel (Shepherd et al, 2000), 9.1% with pemetrexed (Hanna et al, 2004), and 8.9% with erlotinib (Shepherd et al, 2005). Furthermore, the progression-free survival (PFS) rates of these patients were less than 4 months and 1-year survival rates were 25–37% (Shepherd et al, 2000; Fossella et al, 2000; Hanna et al, 2004; Shepherd et al, 2005; Kim et al, 2008). Therefore, further research should focus on the development of regimens that are more effective against previously treated advanced NSCLC.
S-1 (Taiho Pharmaceutical Co., Ltd, Tokyo, Japan) is an oral fluoropyrimidine agent comprising the 5-fuorouracil (5-FU) prodrug, tegafur, along with two enzyme inhibitors of 5-chloro-2,4-dihydroxypyrimidine (CDHP) and potassium oxonate, in a molar ratio of 1 : 0.4 : 1. The CDHP enhances the serum levels of 5-FU by competitive inhibition of dihydropyrimidine dehydrogenase (DPD), an enzyme responsible for 5-FU catabolism. Potassium oxonate is a reversible competitive inhibitor of orotate phosphoribosyl transferase (OPRT), a phosphoenzyme for 5-FU, and functions to reduce the gastrointestinal toxicity of 5-FU (Wada et al, 2006). These mechanisms mean that oral S-1 administration can generate a higher concentration of 5-FU than protracted intravenous injection of 5-FU alone. S-1 monotherapy has achieved an overall response rate (ORR) of 22% and median survival time (MST) of 11 months when applied to previously untreated patients with advanced NSCLC (Kawahara et al, 2001).
In preclinical studies, docetaxel has shown no cross-resistance in 5-FU-resistant human tumour cell lines (Hill et al, 1994). In addition, the combination of S-1 and docetaxel has been reported to have synergistic effects in vitro (Wada et al, 2006). The combination of S-1 and docetaxel holds particularly great promise, because both drugs display substantial anti-tumour activity as single agents and they have different mechanisms of action and toxicity profiles (Shepherd et al, 2000; Fossella et al, 2000; Furuse et al, 2001; Kubota et al, 2008; Maruyama et al, 2008). Recent preclinical studies have shown that S-1 and docetaxel have synergistic effects in human cancer xenografts (Takahashi et al, 2005; Suto et al, 2006; Wada et al, 2006). Docetaxel can modulate the expression and activity of DPD and OPRT. Moreover, a low level of DPD activity and a high level of OPRT activity can enhance the anti-tumour effect of 5-FU and S-1.
Several phase II studies found that the combination of docetaxel and S-1 is effective in patients with NSCLC (Atagi et al, 2008; Yanagihara et al, 2010). These trials employed a combination of docetaxel with oral S-1 for more than 14 consecutive days. However, the administration of oral S-1 for more than 14 consecutive days was subsequently withdrawn because of modest, but continuous, emesis. No trials have evaluated the combination of docetaxel and S-1 treatment for 1 week in patients with previously treated advanced NSCLC. Thus, we conducted a phase I study to find the maximum tolerated doses (MTDs) of biweekly administered docetaxel with oral S-1 for 1 week, and a phase II study to evaluate the efficacy and toxicity of this combination strategy.
Patients and methods
Eligibility criteria
Patients meeting all of the following criteria were enrolled: (1) histologically or cytologically confirmed NSCLC; (2) stages IIIB–IV that is inadequate for curative-intent thoracic radiotherapy, or relapse after surgery; (3) measurable lesions using Response Evaluation Criteria In Solid Tumours (RECIST); (4) previously received cytotoxic chemotherapy; (5) at least 4 weeks had to have elapsed from completion of prior chemotherapy; (6) Eastern Cooperative Oncology Group performance status of 0–2; (7) age 20 years or more; (8) adequate organ functions (white blood cell count of 4000–12 000 per μl, neutrophil count of 2000 per μl or more, platelet count of 100 000 per μl or more, haemoglobin level of 9 g dl−1 or more, levels of aspartate aminotransferase and alanine aminotransferase less than or equal to 2.5-fold of the upper limits of the normal ranges, total serum bilirubin level less than 1.5 mg dl−1, total serum creatinine level less than the upper limit of the normal range, creatinine clearance rate of 50 ml min−1 or more); (9) life expectancy exceeding 3 months; (10) capable of oral intake; and (11) written informed consent provided.
Drug administration
S-1 was given orally twice daily after a meal for 7 consecutive days, followed by 7 days of rest (Figure 1). The dose of S-1 (80 mg m−2 per day) was simplified according to body surface area (BSA) as follows: 80 mg per day for patients with a BSA of less than 1.25 m2, 100 mg per day for those with a BSA of 1.25–1.5 m2, and 120 mg per day for those with a BSA greater than 1.5 m2. Docetaxel (escalating dose: 30 mg m−2 in level 1, 35 mg m−2 in level 2, and 40 mg m−2 in level 3) was diluted in 500 ml of normal saline and given as an intravenous drip infusion over 60 min on day 1. Anti-emetic treatment comprised of glucocorticoids and 5HT3 antagonist was usually given. This regimen was repeated every 2 weeks for at least two courses and until progression, unless predefined dose reduction or stopping criteria were encountered.

Therapeutic experiments for combination therapy of S-1 and docetaxel. Two previous studies have reported regimens of (A) docetaxel 40 mg m−2 on day 1 with S-1 80 mg m−2 for 2 weeks plus a 1-week rest, and (B) docetaxel 35 mg m−2 on day 1 and 15 with S-1 80 mg m−2 for 2 weeks plus a 2-week rest in patients with previously treated advanced NSCLC. The schedule employed in this study was (C) docetaxel 30–40 mg m−2 on day 1 with S-1 80 mg m−2 for 1 week, in which patients were treated by an anti-emetic treatment, plus a 1-week rest.
In phase I study, dose-limiting toxicity (DLT) was defined as grade 3 or 4 non-haematological toxicities (excluding nausea or vomiting, appetite loss, fatigue), grade 4 haematological toxicities, or any adverse event requiring a break in administration of S-1 for more than 4 days during two courses, or any adverse event requiring delayed administration of docetaxel for more than 8 days during the first three courses. The MTD was estimated based on the analysis of DLT as follows. If DLT occurrence was three out of three, this level was defined as the MTD. If DLT occurrence ranged from one out of three to two out of three, then three patients were added and the same dose level was repeated. If DLT occurrence was three out of six or more, this level was defined as MTD. If this level was non-MTD, then the dose would be escalated to the next level. Recommended dose (RD) was defined as one level lower than the MTD; moreover, RD needed a feasibility of more than four courses. In phase II study, docetaxel at RD and S-1 were administered similarly to the phase I study. Post-treatment was withheld until evident disease progression, followed by no restriction afterward.
Evaluation during chemotherapy
Chest CT scan was examined before the treatment, and thereafter, it was re-examined to investigate tumour response by the RECIST criteria at appropriate times. Tumour response in every patient was evaluated by an external reviewer according to the RECIST criteria, and was classified into four categories: complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD). A minimum of a 6-week interval from the start of therapy was required for establishing SD. As of PFS, chest X ray and chest CT scan were repeated at least every 4 and 8 weeks, respectively, until disease progression. And other radiographic modalities were also performed when there was a possibility of distant metastasis.
Symptoms, physical examination, complete blood counts, and serum chemistries were monitored weekly during chemotherapy. Toxicity was evaluated for every course according to the common terminology criteria for adverse events (CTCAE) version 3.0.
Dose-reduction and -termination criteria of chemotherapy
The next course of treatment was started when the neutrophil count returned to 1500 per μl, the platelet count returned to 100 000 per μl, and the non-haematologic toxicity recovered to grade 0 or 1. For patients who experienced febrile neutropenia, grade 4 thrombocytopenia, or grade 3 or 4 non-haematologic toxicity, the dose of docetaxel was reduced by 5 mg m−2 and the dose of S-1 was also reduced by 20 mg per day of the initial dose. For patients who still experienced the same toxicity after the dose reduction, docetaxel was reduced by 5 mg m−2 and S-1 was reduced by 20 mg per day of the reduced dose, and this could be done up to two times. If recovery from such toxicities at a reduced dose was confirmed, administration at the reduced dose was continued. Patients who still experienced the same toxicity after the dose reduction were withdrawn from the study treatment.
Statistical and ethical considerations
The primary endpoint of the phase II study was to evaluate tumour response, and the secondary endpoints were to evaluate PFS, overall survival (OS), and safety. We chose a 25% response rate as a desirable target level and a 10% response rate as an undesirable target level. The study design had a power of >80% to detect a response with an error of <5%. It was necessary to enrol a minimum of 33 patients. According to this, we aimed for 35 patients to take non-evaluable patients into consideration. Exact confidence interval (CI) for ORR was based on the binomial distribution. Overall survival was calculated from the date of registration until death from any cause, whereas PFS was defined as being until disease progression or death from any cause. The OS and PFS were analysed using the Kaplan–Meier method. All statistical tests were one sided, and a P-value of less than 0.05 was considered statistically significant.
Results
Patient characteristics
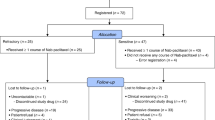
The present study was conducted after the protocol had been approved by the Institutional Review Board of each participating institutions. From February 2007 to April 2009, 13 patients in the phase I portion were enrolled from 2 facilities. From October 2009 to November 2010, 35 patients in phase II portion were enrolled from 5 facilities. Patient characteristics are listed in Table 1. All the patients had stage IV or relapsed disease, and most of them had PS 0 and adenocarcinoma histology. Sensitive EGFR mutations were observed in seven patients who had progress disease after EGFR–TKI treatment. In the phase I portion, there was one unfit patient in level 2, as described below. In the phase II portion, one patient had withdrawn informed consent immediately before treatment. All of the patients had progressed after at least one previous platinum-based regimen, such as cisplatin plus pemetrexed (8 patients), vinorelbin (5 patients) or irinotecan (1 patient), and carboplatin plus paclitaxel (8 patients), pemetrexed (11 patients), or gemcitabine (6 patients). Twenty-two of the 34 patients were given this treatment as a second-line therapy.
Phase I study results
Toxicities encountered during the phase I study are summarised in Table 2. Three patients were enrolled in level 1, and none experienced DLT. Three patients were enrolled in level 2, and one patient experienced DLT due to grade 3 headache. Consequently, three more patients were added as mentioned in the protocol. One of the three patients, when entering this study, displayed silent brain metastasis; however, this became symptomatic (secondary epilepsy) when the study protocol was stopped. Thus, one more patient was added. Finally, there was one patient who experienced DLT in level 2. In level 3, three patients were enrolled; one experienced DLT due to febrile neutropenia and two experienced DLT due to delay of administering docetaxel. The MTD among 13 patients was 80 mg m−2 S-1 with 40 mg m−2 docetaxel. S-1 compliance in every course for all patients in levels 1–3 was 100%. The RD for the phase II study was determined as 80 mg m−2 S-1 and 35 mg m−2 docetaxel (level 2).
Treatment response and survival in the phase II study
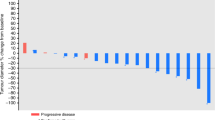
A total of 261 courses of chemotherapy were given in phase II. The average and median number of courses given per patient was 7.6 and 6 (range 2–18), respectively. Results of tumour response in the phase I or II study are shown in Table 3. Of the 34 patients in the phase II study, none (0%) achieved CR, 12 (34.3%) achieved PR, 10 (28.6%) had SD, 11 (31.4%) had PD, and 2 (5.7%) were not evaluable, resulting in an ORR of 34.3% (95% CI, 18.6–50.0%). The disease-control rate was 62.9% (95% CI, 46.8–78.9%). As shown in Figure 2, the median PFS was 150.5 days (95% CI, 96–227 days) and the median OS time was 503 days (95% CI, 446–606 days).

The figure shows progression-free survival (PFS) and overall survival (OS) from the start of this treatment, estimated by the Kaplan–Meier method. The median PFS time was 150.5 days (range 96–227 days). The median OS time was 503 days (range 446–606 days).
Toxicity in the phase II study
All adverse events are shown in Table 4. The most frequent grade 3 or 4 haematological toxicities were neutropenia (41.2%), febrile neutropenia (2.9%), and thrombocytopenia (2.9%). No patient received platelet transfusions. There was no non-haematological grade 3 or 4 toxicity. Nine patients experienced grade 1 nausea, but there were no incidences of grade 2 nausea. Although four patients had grade 2 pulmonary toxicity due to drug-induced pneumonitis (11.8%), these patients recovered after receiving corticosteroids and discontinued this treatment. There was no death or irreversible toxicity in this study that was considered to be related to treatment. Doses of docetaxel administration were reduced in two patients because of grade 4 thrombocytopenia, lasting a week, and febrile neutropenia. In addition, doses of S-1 were not reduced in any patients. The next course of chemotherapy was delayed in 5 (14.7%) of the 34 patients, mainly because of neutropenia, thrombocytopenia, or increased aspartate aminotransferase or alanine aminotransferase levels.
Discussion
Many reports have been published investigating combination chemotherapy using two non-platinum agents for recurrent NSCLC, with the objective of improving outcomes further. However, there have been relatively higher or intolerable toxicities observed in this context (Pectasides et al, 2001; Spiridonidis et al, 2001; Nelli et al, 2004; Takeda et al, 2009). In the present study, we evaluated the efficacy and safety of a new scheduled combination of S-1 and docetaxel, each of which have shown promising efficacy in the treatment of advanced or metastatic NSCLC.
Our schedule using this combination chemotherapy conferred efficacy with an ORR of 34.3% and a median PFS time of 150 days. The ORR observed in this study was slightly higher than expected. Furthermore, the survival benefits as second- or third-line therapy that were observed compare favourably with other chemotherapy regimens, such as monotherapy with docetaxel, pemetrexed, or erlotinib (Hanna et al, 2004; Shepherd et al, 2005). Two previous studies have reported that docetaxel at a dose of 40 mg m−2 on day 1 and S-1 at a dose of 80 mg m−2 per day on days 1–14 every 3 weeks (Figure 1A) in patients with previously treated advanced NSCLC achieved ORRs of 24.1 and 18.4% with median PFS times of 3.9 months and 4.4 months, respectively. Moreover, their MSTs and 1-year survival rates were 11.8 months and 16.1 months, and 42 and 60%, respectively (Atagi et al, 2008; Yanagihara et al, 2010). In addition, a previous study has reported that the combination of 80 mg m−2 S-1 per day for 14 consecutive days and 35 mg m−2 docetaxel on days 1 and 15 every 4 weeks (Figure 1B) in patients with previously treated advanced NSCLC achieved ORRs of 16.3% with median PFS time of 3 months, MST of 9 months, and 1-year survival rates of 42%, respectively (Oki et al, 2011). Our response rates and median PFS time were slightly higher than those of studies reported by Atagi et al, 2008, Yanagihara et al, 2010, and Oki et al, 2011.
The majority of non-haematologic toxicities were mild and tolerable without grade 3 or 4 non-haematologic toxicity. These toxicity results are lower than those observed in phase I or II studies described above in patients with NSCLC (Atagi et al, 2008; Yanagihara et al, 2010; Oki et al, 2011). Especially, in terms of emesis, about one-fourth of the patients felt grade 1 nausea for a short duration, but there were no patients that exhibited grade 2 nausea in our study. The incidence (26.4%) of emesis in our study was lower than studies reported by Yanagihara et al, 2010 (45%) and ; Oki et al, 2011 (44.8%). Emesis by our treatment might be weak compared with these previous regimens (Yanagihara et al, 2010; Oki et al, 2011). A characteristic point of our treatment is the short duration of oral S-1, which is treated by anti-emetic therapy with 5HT3 antagonist and steroids to counteract docetaxel-induced emesis. Results of the median number of courses in our regimen (6 courses: range 2–18) tended to be higher compared with those in the phase II study reported by Atagi et al, 2008 (3 courses: range 1–8), Oki et al, 2011 (2 courses: range 1–6), and Yanagihara et al, 2010 (6 courses: range 1–8). Our increased feasibility and dosing might have an impact on the better response rates, longer PFS, and longer OS observed, as compared with that previously documented (Atagi et al, 2008; Yanagihara et al, 2010; Oki et al, 2011).
Caucasians have lower tolerance to oral S-1 than the Japanese; namely, 80 mg m2 S-1 per day has been given in Japan, whereas 50 mg m−2 S-1 per day has been clinically used in the West (Koizumi et al, 2003; Ajani et al, 2010). Interest in S-1 in the West has been considerable, but the well-tolerated doses used in Japan resulted in considerable emetic toxicity in Caucasians (Ajani et al, 2005). This difference in tolerance is probably due to differences in the efficiency of the cytochrome P-450 enzyme system determined by polymorphisms in different populations. There have been many reports of ethnic differences in the safety and efficacy profile of S-1 and docetaxel (Yamaguchi et al, 2006; Yoshida et al, 2006; Atagi et al, 2008; Yanagihara et al, 2010; Ajani et al, 2010), and it has been shown that the CYP2A6*9 genetic polymorphism is a potential predictive marker, for efficacy and toxicity, in patients receiving the combination of S-1 and docetaxel for metastatic gastric carcinoma (Park et al, 2007). In the development of a S-1/docetaxel combination therapy in the United States and Europe, further optimisation of the dose of each agent may be required to account for these differences. Our short duration of administering oral S-1 might overcome the low tolerance to S-1 in the West.
There are three limitations in this study. First, of the total 34 patients in the phase II study, sensitive EGFR mutations were detected in 4 patients who had disease progression after gefitinib or erlotinib treatment, and there were 14 patients with unknown status of EGFR mutations. Sixteen patients (47%) had post-study treatments, and EGFR–TKI was administrated in 32.3% of patients. Thus, the use of EGFR–TKI in post study might contribute to the extension of OS. Second, although the sample sizes in phase I and II studies were different and rather small, the response rates in phase I and II were different, meaning that a further confirmatory study should be considered. Third, 4 patients had grade 2 pulmonary toxicity due to drug-induced interstitial lung disease (ILD; 11.8%) in this study. The toxicity of drug-induced ILD in previous studies of S-1 alone, S-1 plus docetaxel, and S-1 plus gemcitabin have been reported 3.3, 3.5–5, and 9%, respectively (Wada et al, 2006; Atagi et al, 2008; Yanagihara et al, 2010; Takiguchi et al, 2011). We speculated that, comparing with the previous studies, drug-induced ILD might occur in our present study due to longer term of the treatment and biweekly dosing of docetaxel. But limited number of the patients in phase II portion produces a wide range of 95% CI (4–28%), indicating a further accumulation of the patients treated by this regimen.
Conclusion
A new combination of 1-week administration of S-1 with biweekly docetaxel is safe and active for advanced NSCLC after failure of prior chemotherapy. A phase III trial comparing docetaxel with or without S-1 would warrant further investigation.
Change history
11 October 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ajani JA, Faust J, Ikeda K, Yao JC, Anbe H, Carr KL, Houghton M, Urrea P (2005) Phase I pharmacokinetic study of S-1 plus cisplatin in patients with advanced gastric carcinoma. J Clin Oncol 23: 6957–6965
Ajani JA, Rodriguez W, Bodoky G, Moiseyenko V, Lichinitser M, Gorbunova V, Vynnychenko I, Garin A, Lang I, Falcon S (2010) Multicenter phase III comparison of cisplatin/S-1 with cisplatin/infusional fluorouracil in advanced gastric or gastroesophageal adenocarcinoma study: the FLAGS trial. J Clin Oncol 28: 1547–1553
Atagi S, Kawahara M, Kusunoki Y, Takada M, Kawaguchi T, Okishio K, Kubo A, Uehira K, Yumine K, Tomizawa Y, Saito R, Fukai S, Komatsu H (2008) Phase I/II study of docetaxel and S-1 in patients with previously treated non-small cell lung cancer. J Thorac Oncol 3: 1012–1017
Azzoli CG, Baker S, Temin S, Pao W, Aliff T, Brahmer J, Johnson DH, Laskin JL, Masters G, Milton D, Nordquist L, Pfister DG, Piantadosi S, Schiller JH, Smith R, Smith TJ, Strawn JR, Trent D, Giaccone G (2009) American Society of Clinical Oncology Clinical Practice Guideline update on chemotherapy for stage IV non-small-cell lung cancer. J Clin Oncol 27: 6251–6266
Fossella FV, DeVore R, Kerr RN, Crawford J, Natale RR, Dunphy F, Kalman L, Miller V, Lee JS, Moore M, Gandara D, Karp D, Vokes E, Kris M, Kim Y, Gamza F, Hammershaimb L (2000) Randomized phase III trial of docetaxel versus vinorelbine or ifosfamide in patients with advanced non-small-cell lung cancer previously treated with platinum-containing chemotherapy regimens. J Clin Oncol 18: 2354–2362
Furuse K, Kawahara M, Hasegawa K, Kudoh S, Takada M, Sugiura T, Ichinose Y, Fukuoka M, Ohashi Y, Niitani H, S-1 Cooperative Study Group (Lung Cancer Working Group) (2001) Early phase II study of S-1, a new oral fluoropyrimidine, for advanced non-small cell lung cancer. Int J Clin Oncol 6: 236–241
Gridelli C, Ardizzoni A, Ciardiello F, Hanna N, Heymach JV, Perrone F, Rosell R, Shepherd FA, Thatcher N, Vansteenkiste J, De Petris L, Di Maio M, De Marinis F (2008) Second-line treatment of advanced non-small cell lung cancer. J Thorac Oncol 3: 430–440
Hanna N, Shepherd FA, Fossella FV, Pereira JR, De Marinis F, von Pawel J, Gatzemeier U, Tsao TC, Pless M, Muller T, Lim HL, Desch C, Szondy K, Gervais R, Shaharyar Manegold C, Paul S, Paoletti P, Einhorn L, Bunn PA (2004) Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol 22: 1589–1597
Hill BT, Whelan RD, Shellard SA, McClean S, Hosking LK (1994) Differential cytotoxic effects of docetaxel in a range of mammalian tumor cell lines and certain drug resistant sublines in vitro. Invest New Drugs 12: 169–182
Kawahara M, Furuse K, Segawa Y, Yoshimori K, Matsui K, Kudoh S, Hasegawa K, Niitani H, S-1 Cooperative Study Group (Lung Cancer Working Group) (2001) Phase II study of S-1, a novel oral fluorouracil, in advanced non-small-cell lung cancer. Br J Cancer 85: 939–943
Kim ES, Hirsh V, Mok T, Socinski MA, Gervais R, Wu YL, Li LY, Watkins CL, Sellers MV, Lowe ES, Sun Y, Liao ML, Osterlind K, Reck M, Armour AA, Shepherd FA, Lippman SM, Douillard JY (2008) Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomized phase III trial. Lancet 372: 1809–1818
Koizumi W, Tanabe S, Saigenji K, Ohtsu A, Boku N, Nagashima F, Shirao K, Matsumura Y, Gotoh M (2003) Phase I/II study of S-1 combined with cisplatin in patients with advanced gastric cancer. Br J Cancer 89: 2207–2212
Kubota K, Kawahara M, Ogawara M, Nishiwaki Y, Komuta K, Minato K, Fujita Y, Teramukai S, Fukushima M, Furuse K, Japan Multi-National Trial Organisation (2008) Vinorelbine plus gemcitabine followed by docetaxel versus carboplatin plus paclitaxel in patients with advanced non-small-cell lung cancer: a randomised open-label, phase III study. Lancet Oncol 9: 1135–1142
Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, Ando M, Miyazawa H, Tanaka T, Saijo Y, Hagiwara K, Morita S, Nukiwa T, North-East Japan Study Group (2010) Gefitinib or chemotherapy for non-small cell lung cancer with mutated EGRF. N Engl J Med 362: 2380–2388
Maruyama R, Nishiwaki Y, Tamura T, Yamamoto N, Tsuboi M, Nakagawa K, Shinkai T, Negoro S, Imamura F, Eguchi K, Takeda K, Inoue A, Tomii K, Harada M, Masuda N, Jiang H, Itoh Y, Ichinose Y, Saijo N, Fukuoka M (2008) Phase III study V-15–32 of gefitinib versus docetaxel in previously treated Japanese patients with non-small-cell lung cancer. J Clin Oncol 26: 4244–4252
Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, Asami K, Katakami N, Takada M, Yoshioka H, Shibata K, Kudoh S, Shimizu E, Saito H, Toyooka S, Nakagawa K, Fukuoka M, West Japan Oncology Group (2010) Gefitinib versus cisplatin plus docetaxel in patients with non-small cell lung cancer harbouring mutations of epidermal growth factor receptor (WJTOG3405). Lancet Oncol 11 (2): 121–128
Nelli F, Naso G, De Pasquale Ceratti A, Saltarelli R, Dauria G, Lugini A, Ferraldeschi R, Picone V, Moscetti L, Cortesi E (2004) Weekly vinorelbine and docetaxel as second-line chemotherapy for pretreated non-small cell lung cancer patients: a phase I-II trial. J Chemother 16: 392–399
Oki Y, Hirose T, Yamaoka T, Kusumoto S, Shirai T, Sugiyama T, Okuda K, Nakashima M, Murata Y, Ohmori T, Adachi M (2011) Phase II study of S-1, a novel oral fluoropyrimidine, and biweekly administration of docetaxel for previously treated advanced non-small-cell lung cancer. Cancer Chemother Pharmacol 67 (4): 791–797
Park SR, Park MS, Park YL, Kim NK, Lee JS, Kim HK, Lim HS (2007) CYP2A6 genetic polymorphism as a predictive marker for clinical outcomes in patients with metastatic gastric carcinoma treated with S-1 plus docetaxel. 2007 ASCO Annual Meeting Proceedings. J Clin Oncol 25: 230s (abstr 4633)
Pectasides D, Kalofonos HP, Samantas E, Nicolaides C, Papacostas P, Onyenadum A, Visvikis A, Skarlos D, Fountzilas G (2001) An out-patient second-line chemotherapy with gemcitabine and vinorelbine in patients with non-small cell lung cancer previously treated with cisplatin-based chemotherapy. A phase II study of the Hellenic Co-operative Oncology Group. Anticancer Res 21: 3005–3010
Rosell R, Gervais R, Vergnenegre A, Massuti B, Felip E, Cardenal F, Garcia Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Di Seri M, Garrido Lopez P, Insa A, De Marinis F, Corre R, Carreras M, Carcereny E, Taron M, Paz-Ares LG, Spanish Lung Cancer Group (2011) Erlotinib versus chemotherapy (CT) in advanced non-small cell lung cancer (NSCLC) patients (p) with epidermal growth factor (EGFR) mutations. 2011 ASCO Annual Meeting Proceeding J Clin Oncol 29 (suppl abstr 7503))
Reck M, von Pawel J, Zatloukal P, Ramlau R, Gorbounova V, Hirsh V, Leighl N, Mezger J, Archer V, Moore N, Manegold C (2009) Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for non-squamous non-small-cell lung cancer: AVAil. J Clin Oncol 27: 1227–1234
Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH (2007) Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 355 (24): 2542–2550
Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C, Serwatowski P, Gatzemeier U, Digumarti R, Zukin M, Lee JS, Mellemgaard A, Park K, Patil S, Rolski J, Goksel T, de Marinis F, Simms L, Sugarman KP, Gandara D (2008) Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol 26: 3543–3551
Shepherd FA, Dancey J, Ramlau R, Mattson K, Gralla R, O'Rourke M, Levitan N, Gressot L, Vincent M, Burkes R, Coughlin S, Kim Y, Berille J (2000) Prospective randomized trial of docetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy. J Clin Oncol 18: 2095–2103
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabárbara P, Seymour L, National Cancer Institute of Canada Clinical Trials Group (2005) Erlotinib in previously treated non-small cell lung cancer. N Engl J Med 353: 123–132
Spiridonidis CH, Laufman LR, Carman L, Moore T, Blair S, Jones J, George C, Patel T, Roach R, Rupert R, Zangmeister J, Colborn D, Kuebler JP (2001) Secondline chemotherapy for non-small-cell lung cancer with monthly docetaxel and weekly gemcitabine: a phase II trial. Ann Oncol 12: 89–94
Suto A, Kubota T, Fukushima M, Ikeda T, Takeshita T, Ohmiya H, Kitajima M (2006) Antitumor effect of combination of S-1 and docetaxel on the human breast cancer xenograft transplanted into SCID mice. Oncol Rep 15: 1517–1522
Takahashi I, Emi Y, Kakeji Y, Uchida J, Fukushima M, Maehara Y (2005) Increased antitumor activity in combined treatment TS-1 and docetaxel. A preclinical study using gastric cancer xenografts. Oncology 68: 130–137
Takeda K, Negoro S, Tamura T, Nishiwaki Y, Kudoh S, Yokota S, Matsui K, Semba H, Nakagawa K, Takada Y, Ando M, Shibata T, Saijo N (2009) Phase III trial of docetaxel plus gemcitabine versus docetaxel in second-line treatment for non-small-cell lung cancer: results of a Japan Clinical Oncology Group trial (JCOG0104). Ann Oncol 20: 835–884
Takiguchi Y, Seto T, Ichinose Y, Nogami N, Sinkai Okamoto H, Minato K, Seki N, Eguchi K, Kishi K, Nichikawa M, Watanabe K (2011) Long-term administration of second-line chemotherapy with S-1 and gemcitabine or platinum-resistant non-small cell cancer. J Thorac Oncol 6: 156–160
Wada Y, Yoshida K, Suzuki T, Mizuiri H, Konishi K, Ukon K, Tanabe K, Sakata Y, Fukushima M (2006) Synergistic effects of docetaxel and S-1 by modulating the expression of metabolic enzymes of 5-Xuorouracil in human gastric cancer cell lines. Int J Cancer 119: 783–791
Yamaguchi K, Shimamura T, Hyodo I, Koizumi W, Doi T, Narahara H, Komatsu Y, Kato T, Saitoh S, Akiya T, Munakata M, Miyata Y, Maeda Y, Takiuchi H, Nakano S, Esaki T, Kinjo F, Sakata Y (2006) Phase I/II study of docetaxel and S-1 in patients with advanced gastric cancer. Br J Cancer 94: 1803–1808
Yanagihara K, Yoshimura K, Niimi M, Yasuda H, Sasaki T, Nishimura T, Ishiguro T, Matsumoto S, Kitano T, Kanai M, Misawa A, Tada H, Teramukai S, Mio T, Fukushima M (2010) Phase II study of S-1 and docetaxel for previously treated patients with locally advanced or metastatic non-small cell lung cancer. Cancer Chemother Pharmacol 66: 913–918
Yoshida K, Ninomiya M, Takakura N, Hirabayashi N, Takiyama W, Sato Y, Todo S, Terashima M, Gotoh M, Sakamoto J, Nishiyama M (2006) Phase II study of docetaxel and S-1 combination therapy for advanced or recurrent gastric cancer. Clin Cancer Res 12: 3402–3407
Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, Hu C, Luo Y, Chen L, Ye M, Huang J, Zhi X, Zhang Y, Xiu Q, Ma J, Zhang L, You C (2011) Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small cell lung cancer (OPTIMAL, CTONG-0802). Lancet Oncol 12 (8): 735–742
Acknowledgements
This work was supported by the Kanto Respiratory Disease Study Group. This trial was designed and conducted independently of any profit organisation.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
The content of this study was presented during a poster section at the 14th World Conference on Lung Cancer (WCLC) 2011.
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Komiyama, K., Kobayashi, K., Minezaki, S. et al. Phase I/II trial of a biweekly combination of S-1 plus docetaxel in patients with previously treated non-small cell lung cancer (KRSG-0601). Br J Cancer 107, 1474–1480 (2012). https://doi.org/10.1038/bjc.2012.437
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.437
Keywords
This article is cited by
-
A multi-institution phase II study of gemcitabine/cisplatin/S-1 (GCS) combination chemotherapy for patients with advanced biliary tract cancer (KHBO 1002)
Cancer Chemotherapy and Pharmacology (2015)
