
Abstract
Background:
Cytosine arabinoside-based chemotherapy coupled with anthracycline is currently the first-line treatment for acute myeloid leukaemia (AML), but diverse responses to the regimen constitute obstacles to successful treatment. Therefore, outcome prediction to chemotherapy at diagnosis is believed to be a critical consideration.
Methods:
The mRNA expression of 12 genes closely involved in the actions of cytosine arabinoside and anthracycline was evaluated by real-time reverse transcriptase PCR (RT–PCR), in 54 diagnostic bone marrow specimens of M2-subtype AML.
Results:
Low expression levels of ribonucleotide reductase M2 (RRM2) and high expression levels of topoisomerase 2 beta (TOP2B) were correlated with longer survival in a univariate analysis. Another interesting finding is that high ratios of TOP2B/RRM2 and TOP2B/TOP2 alpha (TOP2A) in a combined analysis were also shown to have a prognostic impact for longer survival with improved accuracy. Among the four markers, when adjusted for the influence of other clinical factors in multivariate analysis, the TOP2B/TOP2A ratio was significantly correlated with treatment outcomes; patients with high ratios trended toward longer disease-free survival (HR, 0.24; P=0.002) and overall survival (HR, 0.29; P=0.005).
Conclusion:
Genes with distinct expression profiles such as TOP2B/TOP2A expression ratio at diagnosis can be employed for outcome prediction after the treatment with standard regimens in AML patients with M2 subtype.
Similar content being viewed by others


Main
Acute myeloid leukaemia (AML) is a heterogeneous haematologic malignancy and is the most common type of the condition, accounting for up to 60% of leukaemia cases. The French–American–British classification subdivides AML into nine subtypes (M0-M7 and M4Eo), which can be distinguished by their lineages and the degree of differentiation of myeloid cells (Löwenberg et al, 1999). Among these, the M2 subtype is the top-ranked disease (∼30%) in terms of incidence.
Nucleoside analogues or combination with anthracyclines have been used for the majority of AML patients and are generally regarded as the most effective therapies (Wiernik et al, 1992). Although approximately two-thirds of the patients, at least, achieve complete remission after receiving intensive induction regimens, most of these patients suffer from relapsed diseases. Thus, a failure of first-line chemotherapy in these patients can also attenuate the antitumour effect of subsequent chemotherapeutics, owing to substantial multi-drug resistance acquisition (Greenberg et al, 2004). At diagnosis, therefore, it is believed that the accurate response prediction against initial regimens is an extremely important consideration, and can provide another option for AML treatment.
Once cytosine arabinoside, a pyrimidine analogue, enters into the cells through specialised transporters such as human equilibrative nucleoside transporters (hENT) and concentrative nucleoside transporters (hCNT), it is finally converted into an active drug by deoxycytidine kinase (dCK) (Stegmann et al, 1995; Hubeek et al, 2005). Recent studies using clinical samples have demonstrated that high levels of dCK and hCNT3 expression were correlated with favourable responses among patients with AML and CLL, respectively, who received nucleoside drug-based chemotherapy (Mackey et al, 2005; Yamauchi et al, 2009). Conversely, a reversal of drug metabolism by ecto-5′-nucleotidase (CD73), 5′,3′-nucleotidase (NT5C) and cytidine deaminase (CDA) has been regarded as indicators of poor outcome in AML treatment (Ujházy et al, 1996; Galmarini et al, 2004; Mahlknecht et al, 2009). Anthracyclines such as idarubicin and daunorubicin, supplemental drugs used in the treatment of AML, exert cytotoxic effects by intercalating into DNA strand and inhibiting topoisomerase activities in cancer cells (Carella et al, 1990; Richardson and Johnson, 1997). Although there have been attempts to elucidate the relationship between topoisomerase II expression and drug response in clinical AML samples (Kaufmann et al, 1994; McKenna et al, 1994; Wang et al, 2009), it still remains unsettled. Another drug resistance mechanism in the chemotherapy containing nucleotide analogues is the reduction of intracellular drug accumulation by ATP-binding cassette (ABC) transporters (Pogliani et al, 2001; Steinbach et al, 2006). These findings imply that the genes encoding for molecules associated with action mechanism and delivery of the induction regimens are potential predictive parameters of clinical outcomes in the standard chemotherapy for AML.
In this study, in an effort to address the clinical significance of 12 candidate genes that perform critical functions in the actions of cytosine arabinoside and anthracycline, we evaluated their mRNA levels in bone marrow (BM) samples from 54 diagnostic AML patients with the M2 subtype. The findings of correlation analyses of gene profiles with clinical outcomes may provide us with valuable insights into possible strategies to improve treatment outcomes in AML therapy.
Materials and methods
Clinical samples
BM aspirates of 54 adults newly diagnosed with the M2 subtype of AML and 4 healthy donors were collected from Catholic University Seoul St. Mary’s Hospital and Chonnam National University Hwasun Hospital (Table 1). Written informed consents for the cryopreservation and use for further research of all BM samples were obtained from the patients. The Institutional Review Board of Catholic University Seoul St Mary’s Hospital and Chonnam National University Hwasun Hospital approved all research on the human subjects participating in this study. The patient population consisted of 28 males and 26 females with a median age of 42.5 years (range, 20–72). Induction chemotherapies were executed by using a combination of 1-β-D-arabinofuranosyl cytosine (Ara-C, N=14) or N4-behenoyl-1-β-D-arabinofuranosyl cytosine (BH-AC, N=35) combined with idarubicin. Four patients received Ara-C alone and one did Ara-C with etoposide. Among these, 33 patients (61.1%) received haematopoietic stem cell transplantation (HSCT) after induction and consolidation chemotherapies. A total of 16 patients (29.6%) evidenced no cytogenetic changes, and the others (70.4%) exhibited cytogenetical abnormalities including t(8;21). Additionally, normal BM samples from four healthy donors were used as references throughout the experiments and data analyses.
RNA preparation and real-time PCR
All of the BM mononuclear cells (MNCs) from patients and from healthy donors were kept at −80 °C until use. Total RNA extracted using a Qiagen RNA isolation kit (Hilden, Germany) was converted to cDNA with a reverse transcription kit (Invitrogen, Carlsbad, CA, USA). The cDNA was mixed with SYBR Green PCR Master Mix (PE Applied Biosystems, Foster City, CA, USA) and the specific primers (Supplemental Table S1) for 12 genes including CD73, NT5C, CDA, dCK, hCNT3, hENT1, ribonucleotide reductase M1 (RRM1), RRM2, topoisomerase 2 alpha (TOP2A), TOP2 beta (TOP2B), ABC subfamily A member 3 (ABCA3) and ABCB1. The candidate genes and GAPDH were amplified in triplicate using an ABI Prism 7900 Sequence Detection System (PE Applied Biosystems) under the following conditions: 40 cycles of 95 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s. To normalise the raw data, ΔCT was generated by subtracting the CT value for GAPDH from the CT of 12 genes in an individual patient sample. The results were subsequently adjusted to the average gene levels in four healthy donors (control ΔCT−patient ΔCT=ΔΔCT). The results were expressed in arbitrary quantitative PCR units (2−(ΔΔCT)). In some cases, these data were applied to gene-clustering analysis to explore whether there was any unique pattern between the individual gene expression.
Correlation analysis of gene expression with clinical outcomes
The mean expression values for each gene in four healthy donors were set as the cutoff point 1.0 and the gene levels in patient samples were expressed as relative values compared with the mean values of each gene from healthy donors. We then dichotomised total study population of 54 members into two groups for 12 individual genes by the cutoff value. Populations with expression level lower and higher than 1.0 were named the low and high groups, respectively. We defined DFS and OS as the time from first remission to relapse or death and the time of diagnosis to death or date of last follow-up, respectively. Using both populations clustered according to the levels of each gene and their clinical information, Kaplan–Meier estimate constructed survival curves for DFS and OS. Differences in the clinical outcomes and statistical significance between both groups were then determined using a univariate analysis with log-rank test. In combined analysis using the expression ratios of the two genes, especially, the patients were grouped by the median value of the gene ratio. Cox proportional hazards model was employed to investigate the influence of clinical factors against the prognostic gene markers. The details of this are provided in the figure legends. All statistic analyses were done by MS Office Excel program and SPSS 12.0 software (Chicago, IL, USA). A level of P<0.05 was regarded as statistically significant in this study.
Cell lines and maintenance
Human leukaemia cell OCI/AML-2 (AML-2) was cultured in alpha-MEM media (Gibco, Grand Island, NY, USA) supplemented with 10% FBS and antibiotics. We generated and maintained AML-2/IDAC cells double-resistant to Ara-C and idarubicin as previously described (Song et al, 2009a).
Methylation-specific PCR (MSP) analysis
Cell lines were rinsed twice in PBS, and their gDNA were isolated using AccuPrep Genomic DNA Extraction kit (Bioneer, Daejeon, Korea). Sodium bisulfite-converted DNA was then obtained from 0.5 μg of the gDNA using an EZ DNA Methylation kit (Zymo Research, Irvine, CA, USA). Total DNA of 15 ng were used for MSP corresponsive to CpG islands. The primer pairs were designed using MethPrimer software (Li and Dahiya, 2002), and are as follows: RRM2, Fw: 5′-ttagtttggtcgataaggagaatac-3′ and Re: 5′-taaaaaaacaatacgactttaacgc-3′ for methylation (305 to 566 from transcription start site, TSS), and Fw: 5′-tagtttggttgataaggagaatatgg-3′ and Re: 5′-aaaaaaacaatacaactttaacacc-3′ for unmethylation (306 to 565 from TSS); TOP2B, Fw: 5′-gatagtatggcgttgattgatagc-3′ and Re: 5′-tacttcaaaaacaaccttaacctcg-3′ for methylation (−679 to −476 from TSS), and Fw: 5′-tggatagtatggtgttgattgatagt-3′ and Re: 5′-cttcaaaaacaaccttaacctcact-3′ for unmethylation (−681 to −78 from TSS); TOP2A, Fw: 5′-ggttggagtgtagtggagtaattac-3′ and Re: 5′-aacacattaaaaaaccgaaacga-3′ for methylation (−792 to −64 from TSS), and Fw: 5′-aggttggagtgtagtggagtaattat-3′ and Re: 5′-caacacattaaaaaaccaaaacaaa-3′ for unmethylation (−793 to −63 from TSS). The products were analysed by agarose gel electrophoresis.
Semi-quantitative RT–PCR analysis
Total RNA from cell lines was converted to cDNA using a CycleScript Reverse Transcriptase kit (Bioneer). After mixing the cDNA with specific primers for RRM2, TOP2A and TOP2B, PCR was carried out. GAPDH was used as an internal control. The product band intensity was estimated using Image J software (http://rsbweb.nih.gov/ij/) and normalised using GAPDH. The quantitative results were then displayed in terms of relative expression to the wild-type control cells. The result analysis was performed using Student’s t-test.
Results
Patient population and clinical features
We obtained 54 BM MNCs from patients with the M2 subtype of AML at diagnosis; their details are described in Table 1. A total of 16 patients exhibited normal karyotype and 24 patients had t(8;21). Each of the others evidenced cytogenetically distinct abnormalities. For remission induction, a majority of the patients received standard chemotherapy with Ara-C (N=14) or BH-AC (N=35) coupled with idarubicin. Four of the remaining patients were treated with Ara-C alone, and one received a regimen of Ara-C plus etoposide. After induction and subsequent consolidation chemotherapies, 33 patients (61.1%) received HSCT. Median DFS and OS periods for all populations were 521 (95% CI, 99–943) and 737 (95% CI, 222–1252) days, respectively. In a univariate analysis, we noted that HSCT resulted in prolonged patient survival (P<0.05). Patients with t(8;21) also evidenced better responses than the population with normal or other cytogenetics. The correlation between other clinical features (age, sex, RI regimen and blast % in BM) and treatment outcomes did not reach statistical significance at the specified level (Table 2).
Transcriptional levels in AML samples and their correlation with clinical outcomes
Expression profiles for 12 genes were analysed in a total of 54 samples to identify a prediction marker for treatment outcome at diagnosis. Relative expression levels from real-time PCR analysis of 12 genes were displayed in box plots (Figure 1B). In order to characterise the relationship of the gene expression pattern with clinical outcomes, we clustered the patients into two groups according to the mRNA levels of each of the genes relative to the levels of healthy control subjects. The prognostic impacts of individual genes between the low and high groups were evaluated via a univariate analysis by Kaplan–Meier estimation and log-rank test, and summarised (Table 2). In this analysis, we noted that the high RRM2-level group (N=18) evidenced 2.85-fold (95% CI, 1.22–6.62; P=0.002) and 2.69-fold (95% CI, 1.17–6.20; P=0.005) increases in the hazard ratio (HR) for DFS and OS, respectively, relative to the low group (Figure 2). High TOP2B at the transcriptional level was also associated with longer DFS (P=0.023) and OS (P=0.024) in the AML patients. Because the two clinical factors, cytogenetics abnormality and SCBT, already evidenced an association with treatment outcome, we investigated whether there was a correlation between both factors and biological markers (Supplemental Table S2). In the study, no significant correlation of gene markers with cytogenetics was observed, and HSCT only showed a weak correlation with TOP2A (correlation coefficient, 0.296).

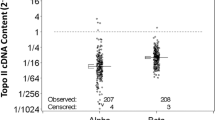
Distribution of mRNA expression levels for 12 genes in 54 patients. (A) Schematic flow of therapeutic actions of nucleosides and anthracycline. Effector molecules encoded by the genes analysed in this study are marked in bold. (B) The expression of 12 candidate genes evaluated by quantitative RT–PCR. Details regarding the processes of raw data are described in the ‘Materials and Methods’ section. The final data values were expressed on log10-scale x-axis. The solid line in box plots (ranging from 25th to 75th percentile) and the circular bullets indicate the median and outlier values, respectively.

Kaplan–Meier evaluation of the prognostic impact of RRM2, TOP2B and TOP2A for clinical outcomes. According to the expression levels of RRM2, TOP2B and TOP2A, a total of 54 patients were divided into low and high groups. The disease-free survival (A) and overall survival (B) curves were generated by Kaplan–Meier survival analyses. Statistical difference (s.d.), HR and P-value were calculated via log-rank test.
Next, gene-clustering analysis was conducted in order to evaluate the correlations among expression patterns of each specific gene. The results visualised a conflict of mRNA expression between TOP2B and RRM2 genes. In addition, TOP2B evidenced a noticeable trend of counter-expression against its 170 kDa isoform-encoding TOP2A gene in the AML samples (Figure 3A). Therefore, we attempted to divide the 54 patients into two groups according to a reference TOP2B/RRM2 ratio value of 1.0 (range, 0.015–19.021). Kaplan–Meier estimation curves for DFS and OS showed meaningful differences between both groups (Figure 3B). The HRs of the high group against the low group were 0.40 (95% CI, 0.19–0.85; P=0.010) and 0.41 (95% CI, 0.19–0.85; P=0.014) for DFS and OS, respectively, in a univariate analyses conducted using log-rank tests. In a similar manner, the total patient sample was again dichotomised into two groups by a reference TOP2B/TOP2A value of 1.0 (range, 0.005–9.027), allowing both groups to be clearly distinguished in Kaplan–Meier Survival estimations (Figure 3C). The differences between the two groups subdivided by TOP2A alone did not reach the threshold for statistical significance (Table 2). When the mRNA level for TOP2A was combined with that of TOP2B, however, the univariate analysis generated great impact and reliable significance for good response with a lowered HR of <0.3 (95% CI, 0.14–0.62 for DFS and 0.13–0.60 for OS; P<0.001 for both).

Prediction of disease-free survival and overall survival using TOP2B/RRM2 or TOP2B/TOP2A ratios. (A) A 54-patient population (rows) was rearranged in accordance with the gene expression profiles of RRM2, TOP2B and TOP2A genes (columns) via gene-clustering analysis. (B, C) The patients were divided into low and high groups according to the reference value, 1.0, of TOP2B/RRM2 (B) or TOP2B/TOP2A (C) ratio. Kaplan–Meier estimation created the survival curves for disease-free survival and overall survival. Statistical difference (s.d.), HR and P-value for survival curves were calculated via log-rank test.
We further conducted multivariate analyses to explore the influence of clinical factors in regard to the prognostic properties of four gene markers, which were identified in the univariate analyses (Table 3). A Cox proportional hazards model adjusted for age, HSCT and cytogenetic abnormalities indicated that a high TOP2B/TOP2A ratio was an independent favourable prognostic factor for both DFS (HR, 4.17; P=0.002) and OS (HR, 3.4; P=0.005), as shown in Figure 4, whereas the predictive likelihoods of the others fizzled out in this analysis. Cytogenetic status, among the clinical features applied, was identified as an independent risk factor for DFS (P=0.020), but not for OS.

Survival curves modified by the Cox proportional hazards model. A total of 54 patients were divided into low and high groups according to a reference TOP2B/TOP2A ratio of 1.0. The curves for disease-free survival (A) and overall survival (B) were rearranged from a multivariate analysis by Cox proportional hazards model via the stepwise method.
Expression of RRM2, TOP2B and TOP2A in drug-sensitive and -resistant AML cell lines
In an effort to confirm whether the interesting expressions in the other candidates as well as the marker genes in the BM blasts of patients was also observed in drug-resistant cell models, we conducted real-time PCR analysis in Ara-C and idarubicin-resistant AML-2 cell lines. The drug-resistant AML-2/IDAC cells evidenced higher transcriptional levels of RRM2 and TOP2A than were observed in parental AML-2 cells, whereas TOP2B was downregulated in AML-2/IDAC cells. These are quite consistent with the results observed in AML patients (Figure 5A). Additionally, an unusual downexpression of dCK was showed in AML-2/IDAC cells, reconfirming our previous finding that the extremely decreased expression of dCK gene is a main reason of Ara-C resistance in this cell model (Song et al, 2009b). When verified by semi-quantitative PCR and gel electrophoresis, the expression patterns of RRM2, TOP2B and TOP2A genes in the cell lines were in an agreement with those from real-time PCR (Figure 5B). Next, we evaluated the DNA methylation status of the genes for RRM2, TOP2B and TOP2A in order to determine whether the differently observed mRNA expressions were modulated by some epigenetic modification inherent to drug-resistant AML cells. As anticipated, we observed that CpG island methylation for RRM2 and TOP2A was slightly reduced in the AML-2/IDAC cells, but CpG island methylation for TOP2B was higher, relative to that observed in the wild-type cells (Figure 5C). These results were quite similar to the observations noted with the clinical specimens, specifically that most patients with poor prognoses evidenced higher RRM2 and TOP2A, but lower TOP2B.

Expression and methylation analyses for the marker genes in drug-sensitive and -resistant AML cell lines. (A) mRNA extracted from AML-2 and AML-2/IDAC cells were applied to semi-quantitative RT–PCR for 12 genes. The results were obtained from the two independent experiments and expressed as relative values against mRNA levels in AML-2 cells. N.D, not determined. (B) mRNA extracted from AML-2 (WT) and AML-2/IDAC (IDAC) cells were also used for semi-quantitative RT–PCR for RRM2, TOP2B and TOP2A. GAPDH was used as an internal control. Using densitometric analysis, the results were expressed as the means±s.d. from three independent experiments. *P<0.05. (C) gDNA from WT and IDAC cells were applied to MSP analyses for unmethylated (UM) or methylated (M) sequences of RRM2, TOP2B and TOP2A. The PCR products were analysed via agarose gel electrophoresis. Densitometric analysis was conducted, and the results were expressed as mean ratios±s.d. between M and UM bands from at least two independent experiments. *P<0.05.
Discussion
Discovering a novel marker that can be applied to the categorisation between patient populations with diverse outcomes against routine induction regimens at diagnosis is very important. Additionally, a more specialised study employing a patient subset with a specific subtype is clearly necessary, owing to the high heterogeneity of AML. In this study, we searched key determinants allowing for the prediction of treatment outcomes in AML patients. The results from univariate analysis showed that TOP2B/RRM2 and TOP2B/TOP2A ratios, as well as individual RRM2 and TOP2B expressions, are statistically reliable predictors for DFS and OS. When applied to multivariate analysis including other clinical factors, as described in Table 3, we concluded that a segregation of patients based on the ratios of the two TOP2 isoforms was the optimal means of predicting treatment outcomes; this also implies an independence of the marker as a prognostic determinant against chemotherapy.
The most interesting finding in this study is that it may prove a possibility to predict treatment responses by evaluating TOP2B and TOP2A expression levels, showing the conflicting expression trends between the two genes. Although we anticipated finding overexpression of both genes in the patient group with the shorter survival rate, the majority of cases (43 of 54 patients, 79.6%) evidenced a differential pattern, with more than a 1.5-fold difference in mRNA expression levels noted between the two genes. Thus, we concluded that the prediction of DFS rate via the ratio of TOP2B/TOP2A as opposed to that of TOP2B alone is more accurate, and maximises the difference between the two patient populations (12.9 vs 5.2) in the univariate analysis. The TOP2 isoforms have been identified as essential nuclear enzymes, which potentially encourage the rapid replication of tumour cells by modifying DNA structures (Vos et al, 2011), but their expression and roles in sensitivity against anthracycline-containing regimens are controversial as ever. Most studies have focused on TOP2A and reported involvement of tumour chemoresistance to DNA-damaging agents in AML as well as other tumours (Chekerov et al, 2006; Chen et al, 2009; Coss et al, 2009). Some groups suggested no significant correlation between topoisomerase levels and treatment outcome at diagnosis (Kaufmann et al, 1994; McKenna et al, 1994). Another recent study indicated that a break of double-strand DNA by TOP2B, as opposed to that by TOP2A, is more closely associated with the development of secondary malignancy after treatment with TOP2-targeting drugs such as etoposide and doxorubicin (Azarova et al, 2007). With regard to the case of cancer cells that escape from or resist anticancer drugs, one possibility is that reduced topoisomerase II levels occasionally allow the cells to survive from cytotoxic attack by maintaining their quiescence (Friche et al, 1991; Chen et al, 2011). Apart from relationship studies between topoisomerase expression and drug response, it has been reported that TOP2A activity but not TOP2B in AML blast was decreased by treating with daunorubicin and idarubicin, indicating that a primary target of anthracycline is TOP2A (Gieseler et al, 1996). Microarray-based approach of diagnostic AML with normal cytogenetics showed that TOP2B expression was much higher in patients showing continuous CR than in refractory or relapsed patients after CR, although the researchers did not evidence a direct correlation between TOP2B expression and sensitivity to the induction chemotherapy (Vey et al, 2004). Therefore, our results imply the existence of a mechanism whereby high TOP2B assists rapid proliferation of leukaemia cells and induces a high degree of susceptibility to chemotherapy, thereby indicating that TOP2B as well as TOP2A manage in drug sensitivity against anthracyclines by distinct independent ways. Another possible explanation is that TOP2B/TOP2A ratio might be not relevant to a susceptibility against antitumour agents but to a biological feature of AML with good or poor outcome, empathising the value of the parameter as a predictive marker only. However, the functional and differential roles of both topoisomerases remain to be clearly elucidated. The reason for the differential regulation of their expression in AML cells will require elucidation in future studies.
Previous in vivo and in vitro studies have demonstrated that nucleoside drug metabolism-related genes may have an important role in responses against the drugs and can be used as a prognostic indicator not only in leukaemias. One study using samples of Ara-C-treated AML patients indicated that high dCK and low NT5C expression were noted in the group with longer survival rates (Galmarini et al, 2003). Additionally, the roles of dCK, CDA and hENT1 were addressed in several studies employing in vitro leukaemia cell models (Ge et al, 2004; Cai et al, 2008; Song et al, 2009b). In our study, despite its identification as a pivotal key enzyme, dCK was not statistically correlated with DFS (P=0.965) or OS (P=0.852), according to univariate analyses. We also detected no meaningful significance in survival analyses using other candidates. The discrepancy between our results and those of previous studies may be attributable to the specialised role of dCK in the activity of anticancer drugs, and to the fact that other studies have employed differently subtyped AML populations. Indeed, to screen outcome predictors, other research groups have employed mixed populations of AML patients treated with non-unified regimens, whereas in this study we used more focused populations in the aspect of disease type and chemotherapy regimen. Additionally, our previous in vitro study showing that dCK performed a critical function in Ara-C resistance, but not in idarubicin resistance, of AML cells bolsters this discrepant observation (Song et al, 2009b).
We also attempted to address the regulation mechanisms underlying the differential expression of marker genes between AML patients with good or poor outcomes. DNA methylation is a well-established mechanism of gene regulation in AML (Figueroa et al, 2010). One study using 42 AML samples suggested that a poor prognosis by overexpression of ABCB1 was correlated with hypomethylation in its promoter region (Nakayama et al, 1998). In order to confirm any correlation between methylation status of the selected genes and drug resistance, we employed a drug-resistant in vitro leukaemia model. As described in a previous report, an AML-2/IDAC (double-resistant to Ara-C and idarubicin) cell line was originated by way of a chronic drug exposure of the drug-sensitive parental AML-2 cells (Song et al, 2009a). We hypothesised that RRM2 and TOP2A expression would be downregulated in drug-resistant cells, whereas TOP2B expression would be upregulated. As anticipated, altered gene expressions were noted in AML-2/IDAC cells, as compared with the AML-2 cells (Figures 5A and B). We also determined that the differential expression of the genes can be regulated by the DNA methylation status in the CpG islands (Figure 5C). Although AML-2 cells are not fully representative of the clinical AML blasts used in this study because both resistant cell lines were already exposed to the drugs, these results are reflective of a connection between the RRM2, TOP2A and TOP2B genes with the drug-resistant phenotype in AML cells. Additionally, this cell line may prove useful as a good model for in vitro attempts to define the contribution of the markers in response to treatment.
In summary, the results of this study demonstrated that the expression ratio of the TOP2B and TOP2A genes was correlated with treatment outcomes in AML patients treated with cytosine arabinoside-based chemotherapy in an independent manner. The finding may prove useful in the prediction of treatment responses in M2-subtype AML patients, and also expands the range of possible treatment choices for improved therapeutic efficacy. However, additional confirmatory studies in validation sets will be required in order to actualise the possible benefits illustrated by the results of this study.
Change history
25 June 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Azarova AM, Lyu YL, Lin CP, Tsai YC, Lau JY, Wang JC, Liu LF (2007) Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc Natl Acad Sci USA 104: 11014–11019
Cai J, Damaraju VL, Groulx N, Mowles D, Peng Y, Robins MJ, Cass CE, Gros P (2008) Two distinct molecular mechanisms underlying cytarabine resistance in human leukemic cells. Cancer Res 68: 2349–2357
Carella AM, Berman E, Maraone MP, Ganzina F (1990) Idarubicin in the treatment of acute leukemias. An overview of preclinical and clinical studies. Haematologica 75: 159–169
Chekerov R, Klaman I, Zafrakas M, Könsgen D, Mustea A, Petschke B, Lichtenegger W, Sehouli J, Dahl E (2006) Altered expression pattern of topoisomerase IIalpha in ovarian tumor epithelial and stromal cells after platinum-based chemotherapy. Neoplasia 8: 38–45
Chen CC, Gau JP, You JY, Lee KD, Yu YB, Lu CH, Lin JT, Lan C, Lo WH, Liu JM, Yang CF (2009) Prognostic significance of beta-catenin and topoisomerase IIalpha in de novo acute myeloid leukemia. Am J Hematol 84: 87–92
Chen CF, He X, Arslan AD, Mo YY, Reinhold WC, Pommier Y, Beck WT (2011) Novel regulation of nuclear factor-YB by miR-485-3p affects the expression of DNA topoisomerase IIα and drug responsiveness. Mol Pharmacol 79: 735–741
Coss A, Tosetto M, Fox EJ, Sapetto-Rebow B, Gorman S, Kennedy BN, Lloyd AT, Hyland JM, O'Donoghue DP, Sheahan K, Leahy DT, Mulcahy HE, O'Sullivan JN (2009) Increased topoisomerase IIalpha expression in colorectal cancer is associated with advanced disease and chemotherapeutic resistance via inhibition of apoptosis. Cancer Lett 276: 228–238
Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, Campagne F, Mazumdar M, Greally JM, Valk PJ, Löwenberg B, Delwel R, Melnick A (2010) DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17: 13–27
Friche E, Danks MK, Schmidt CA, Beck WT (1991) Decreased DNA topoisomerase II in daunorubicin-resistant Ehrlich ascites tumor cells. Cancer Res 51: 4213–4218
Galmarini CM, Cros E, Graham K, Thomas X, Mackey JR, Dumontet C (2004) 5'-(3')-nucleotidase mRNA levels in blast cells are a prognostic factor in acute myeloid leukemia patients treated with cytarabine. Haematologica 89: 617–619
Galmarini CM, Thomas X, Graham K, El Jafaari A, Cros E, Jordheim L, Mackey JR, Dumontet C (2003) Deoxycytidine kinase and cN-II nucleotidase expression in blast cells predict survival in acute myeloid leukaemia patients treated with cytarabine. Br J Haematol 122: 53–60
Ge Y, Jensen TL, Stout ML, Flatley RM, Grohar PJ, Ravindranath Y, Matherly LH, Taub JW (2004) The role of cytidine deaminase and GATA1 mutations in the increased cytosine arabinoside sensitivity of Down syndrome myeloblasts and leukemia cell lines. Cancer Res 64: 728–735
Gieseler F, Glasmacher A, Kämpfe D, Wandt H, Nuessler V, Valsamas S, Kunze J, Wilms K (1996) Topoisomerase II activities in AML and their correlation with cellular sensitivity to anthracyclines and epipodophyllotoxines. Leukemia 10: 1177–1180
Greenberg PL, Lee SJ, Advani R, Tallman MS, Sikic BI, Letendre L, Dugan K, Lum B, Chin DL, Dewald G, Paietta E, Bennett JM, Rowe JM (2004) Mitoxantrone, etoposide, and cytarabine with or without valspodar in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome: a phase III trial (E2995). J Clin Oncol 22: 1078–1086
Hubeek I, Stam RW, Peters GJ, Broekhuizen R, Meijerink JP, van Wering ER, Gibson BE, Creutzig U, Zwaan CM, Cloos J, Kuik DJ, Pieters R, Kaspers GJ (2005) The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br J Cancer 93: 1388–1394
Kaufmann SH, Karp JE, Jones RJ, Miller CB, Schneider E, Zwelling LA, Cowan K, Wendel K, Burke PJ (1994) Topoisomerase II levels and drug sensitivity in adult acute myelogenous leukemia. Blood 83: 517–530
Li LC, Dahiya R (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18: 1427–1431
Löwenberg B, Downing JR, Burnett A (1999) Acute myeloid leukemia. N Engl J Med 341: 1051–1062
Mackey JR, Galmarini CM, Graham KA, Joy AA, Delmer A, Dabbagh L, Glubrecht D, Jewell LD, Lai R, Lang T, Hanson J, Young JD, Merle-Béral H, Binet JL, Cass CE, Dumontet C (2005) Quantitative analysis of nucleoside transporter and metabolism gene expression in chronic lymphocytic leukemia (CLL): identification of fludarabine-sensitive and -insensitive populations. Blood 105: 767–774
Mahlknecht U, Dransfeld CL, Bulut N, Kramer M, Thiede C, Ehninger G, Schaich M (2009) SNP analyses in cytarabine metabolizing enzymes in AML patients and their impact on treatment response and patient survival: identification of CDA SNP C-451T as an independent prognostic parameter for survival. Leukemia 23: 1929–1932
McKenna SL, West RR, Whittaker JA, Padua RA, Holmes JA (1994) Topoisomerase II alpha expression in acute myeloid leukaemia and its relationship to clinical outcome. Leukemia 8: 1498–1502
Nakayama M, Wada M, Harada T, Nagayama J, Kusaba H, Ohshima K, Kozuru M, Komatsu H, Ueda R, Kuwano M (1998) Hypomethylation status of CpG sites at the promoter region and overexpression of the human MDR1 gene in acute myeloid leukemias. Blood 92: 4296–4307
Pogliani EM, Carpenedo M, Miccolis I, Belotti D, Corneo GM (2001) P-Glycoprotein expression in acute myeloid leukaemia cells at diagnosis: its relationship to daunorubicin or idarubicin induction therapy and survival; malignancy. Hematology 5: 359–367
Richardson DS, Johnson SA (1997) Anthracyclines in haematology: preclinical studies, toxicity and delivery systems. Blood Rev 11: 201–223
Song JH, Kim SH, Cho D, Lee IK, Kim HJ, Kim TS (2009a) Enhanced invasiveness of drug-resistant acute myeloid leukemia cells through increased expression of matrix metalloproteinase-2. Int J Cancer 125: 1074–1081
Song JH, Kim SH, Kweon SH, Lee TH, Kim HJ, Kim HJ, Kim TS (2009b) Defective expression of deoxycytidine kinase in cytarabine-resistant acute myeloid leukemia cells. Int J Oncol 34: 1165–1171
Stegmann AP, Honders WH, Willemze R, Ruiz van Haperen VW, Landegent JE (1995) Transfection of wild-type deoxycytidine kinase (dck) cDNA into an AraC- and DAC-resistant rat leukemic cell line of clonal origin fully restores drug sensitivity. Blood 85: 1188–1194
Steinbach D, Gillet JP, Sauerbrey A, Gruhn B, Dawczynski K, Bertholet V, de Longueville F, Zintl F, Remacle J, Efferth T (2006) ABCA3 as a possible cause of drug resistance in childhood acute myeloid leukemia. Clin Cancer Res 12: 4357–4363
Ujházy P, Berleth ES, Pietkiewicz JM, Kitano H, Skaar JR, Ehrke MJ, Mihich E (1996) Evidence for the involvement of ecto-5'-nucleotidase (CD73) in drug resistance. Int J Cancer 68: 493–500
Vey N, Mozziconacci MJ, Groulet-Martinec A, Debono S, Finetti P, Carbuccia N, Beillard E, Devilard E, Arnoulet C, Coso D, Sainty D, Xerri L, Stoppa AM, Lafage-Pochitaloff M, Nguyen C, Houlgatte R, Blaise D, Maraninchi D, Birg F, Birnbaum D, Bertucci F (2004) Identification of new classes among acute myelogenous leukaemias with normal karyotype using gene expression profiling. Oncogene 23: 9381–9391
Vos SM, Tretter EM, Schmidt BH, Berger JM (2011) All tangled up: how cells direct, manage and exploit topoisomerase function. Nat Rev Mol Cell Biol 12: 827–841
Wang YH, Takanashi M, Tsuji K, Tanaka N, Shiseki M, Mori N, Motoji T (2009) Level of DNA topoisomerase IIalpha mRNA predicts the treatment response of relapsed acute leukemic patients. Leuk Res 33: 902–907
Wiernik PH, Banks PL, Case DC, Arlin ZA, Periman PO, Todd MB, Ritch PS, Enck RE, Weitberg AB (1992) Cytarabine plus idarubicin or daunorubicin as induction and consolidation therapy for previously untreated adult patients with acute myeloid leukemia. Blood 79: 313–319
Yamauchi T, Negoro E, Kishi S, Takagi K, Yoshida A, Urasaki Y, Iwasaki H, Ueda T (2009) Intracellular cytarabine triphosphate production correlates to deoxycytidine kinase/cytosolic 5'-nucleotidase II expression ratio in primary acute myeloid leukemia cells. Biochem Pharmacol 77: 1780–1786
Acknowledgements
This study was supported by the grant A111218-GM06 of the National Project for Personalized Genomic Medicine, Ministry for Health & Welfare and the National Research Foundation of Korea (NRF) grant 2011-0001388 funded by the Korea government.
Author contributions
JHS designed and performed experiments, analysed data and wrote the manuscript; SHK performed experiments and analysed data; H-JK designed experiments, provided patient samples and clinical expertise; T-HL provided patient samples and performed experiments; W-SM provided patient samples and clinical information; H-JK provided patient samples and clinical data expertise; Y-KK provided valuable reagents; SYH performed experiments and analysed data; TSK designed and supervised the study, and wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies the paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Song, J., Kweon, S., Kim, HJ. et al. High TOP2B/TOP2A expression ratio at diagnosis correlates with favourable outcome for standard chemotherapy in acute myeloid leukaemia. Br J Cancer 107, 108–115 (2012). https://doi.org/10.1038/bjc.2012.206
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.206
Keywords
This article is cited by
-
Association of immunophenotype with expression of topoisomerase II α and β in adult acute myeloid leukemia
Scientific Reports (2020)
-
Targeting of topoisomerases for prognosis and drug resistance in ovarian cancer
Journal of Ovarian Research (2016)
